

Abstract
Mutations in human MPV17 cause a hepatocerebral form of mitochondrial DNA depletion syndrome (MDS) hallmarked by early-onset liver failure, leading to premature death. Liver transplantation and frequent feeding using slow-release carbohydrates are the only available therapies, although surviving patients eventually develop slowly progressive peripheral and central neuropathy. The physiological role of Mpv17, including its functional link to mitochondrial DNA (mtDNA) maintenance, is still unclear. We show here that Mpv17 is part of a high molecular weight complex of unknown composition, which is essential for mtDNA maintenance in critical tissues, i.e. liver, of a Mpv17 knockout mouse model. On a standard diet, Mpv17−/− mouse shows hardly any symptom of liver dysfunction, but a ketogenic diet (KD) leads these animals to liver cirrhosis and failure. However, when expression of human MPV17 is carried out by adeno-associated virus (AAV)-mediated gene replacement, the Mpv17 knockout mice are able to reconstitute the Mpv17-containing supramolecular complex, restore liver mtDNA copy number and oxidative phosphorylation (OXPHOS) proficiency, and prevent liver failure induced by the KD. These results open new therapeutic perspectives for the treatment of MPV17-related liver-specific MDS.
Introduction
The term mitochondrial DNA depletion syndrome (MDS) indicates a heterogeneous group of diseases characterized by profound reduction in mitochondrial DNA (mtDNA) copy number in one or several tissues.1 Myopathic, encephalomyopathic, and hepatocerebral forms of MDS are known, due to mutations in gene products involved in mtDNA maintenance, either by controlling the supply of deoxynucleotides for, or by carrying out, the synthesis of mtDNA. MDS is considered rare, with an estimated prevalence of 1:100,000,2 although the number of genes associated with this condition is rapidly expanding, and a systematic diagnostic screening is hampered by tissue specificity. For instance, thymidine kinase 23 and guanosine kinase4 are the two enzymes involved in deoxynucleotide recycling in mitochondria; p53-ribonucleotide reductase subunit 25 and thymidine phosphorylase6 are the two cytosolic enzymes controlling the de novo biosynthesis of deoxynucleotides (p53-ribonucleotide reductase subunit 2) and the catabolism of nucleotides (thymidine phosphorylase), respectively; polymerase γ7 is the mitochondrion-specific DNA polymerase and Twinkle,8 the mtDNA helicase. MPV179,10,11,12,13,14,15,16, a small protein of the inner mitochondrial membrane, is a prominent cause of hepatocerebral MDS, accounting for about 50% of the cases. More than 20 different MPV17 mutations in >70 patients have been reported so far. However, the functional and mechanistic links between Mpv17 and mtDNA maintenance are still missing. Nevertheless, studies on SYM1, the MPV17 yeast ortholog, suggest a role for this protein in controlling the flux of Krebs cycle intermediates across the inner mitochondrial membrane. How this functional data relate to mtDNA maintenance and integrity are unknown. In addition, studies based on blue native gel electrophoresis have demonstrated that SYM1 is present within a high molecular weight complex of ~600 kDa, the composition of which is, however, unknown.17
Liver involvement associated with severe hypoglycemic crises and rapidly progressive deterioration of hepatic function, leading to liver cirrhosis and failure, are early and predominant features of MPV17-dependent MDS. Unlike other MDS, neurological involvement in MPV17-associated hepatocerebral MDS is generally mild or absent at disease onset, but progressive peripheral neuropathy and cerebellar degeneration appear later in those MPV17 mutant patients who survive fatal, early-onset metabolic impairment, and liver failure. Although no cure is currently available for MPV17-related MDS, therapeutic interventions based on liver transplantation13,14 or on frequent meals of a cornstarch-based diet18 have been attempted, to delay disease progression or protect patients from fatal hypoglycemia. Liver transplantation has been carried out in 10 patients but five of them died early thereafter of multiorgan failure or sepsis.14 Prompt-release carbohydrates, such as cornstarch, seem to be effective in preventing fatal hypoglycemic accidents, but the surviving patients invariably progress to hepatic, and eventually neurological, degeneration. Central and peripheral neuropathy is a predominant feature in subjects affected by Navajo neurohepatopathy, a well-known disease of Navajo people from south-east US, recently shown to be caused by a specific mutation in the MPV17 gene.19 Clinical features of Navajo neurohepatopathy/MPV17 syndrome include sensory motor neuropathy with ataxia, leukoencephalopathy, corneal ulcerations, acral mutilation, poor somatic development with sexual infantilism, serious systemic infections, and, of course, liver derangement.
The knockout mouse for Mpv17 (Mpv17−/−) is characterized by profound, early-onset mtDNA depletion in liver,10 degeneration of the inner ear structures, particularly of the organ of Corti and stria vascularis, determining profound hearing loss,20 and late-onset, fatal kidney dysfunction, dominated by proteinuria due to focal segmental glomerulosclerosis.10 Although the molecular and biochemical features in the liver of Mpv17−/− mice closely resemble those of human patients, including severe mtDNA depletion, these animals show hardly any sign of hepatic dysfunction in standard captivity conditions and live well beyond the first year of life, with neither obvious neurological impairment nor neuropathological abnormalities. However, we show here that Mpv17−/− mice fed with a high-fat ketogenic diet (KD) rapidly develop liver cirrhosis and failure. Importantly, treatment with a liver-specific adenoassociated viral vector (serotype 2/8, AAV2/8) expressing human MPV17 (hMPV17) restores mtDNA copy number and prevents liver degeneration. This effect is linked to the formation of the 600 kDa complex containing Mpv17, since Mpv17 variants that do not allow this structure to be formed cannot rescue the liver phenotype in vivo.
Results
Mpv17 is part of a high molecular weight complex
In order to investigate the physical status of Mpv17 in vivo, we analyzed isolated mitochondria from Mpv17+/− and Mpv17−/− livers by 2-dimensional blue native gel electrophoresis (2D-BNGE) immunoblot, using an antibody specific against the mammalian Mpv17 protein (αMpv17). Although the antibody did not work in the (native) first dimension, in the (denaturing) second dimension, the αMpv17 was able to visualize Mpv17 crossreacting material in the mitochondria of control, but not of knockout mice. Most of the 20 kDa Mpv17 protein was in fact present in a spot corresponding to a complex of >600 kDa, as suggested by restaining with an αCore1 antibody marking the mitochondrial complex III dimer (Figure 1). These findings are identical to those previously obtained on SYM1.17
Figure 1.
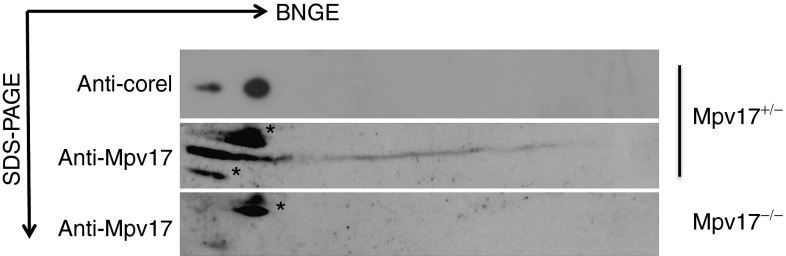
Characterization of the Mpv17-containing macromolecular complex. Liver mitochondria from (a) Mpv17+/− and (b) Mpv17−/− mice were analyzed by 2D BNGE. The blots were immunostained with an αMPV17 antibody; the filter was then stripped and restained with an αCore 1 antibody to determine the molecular weight of the complex. The asterisks (*) mark unspecific bands. BNGE, blue native gel electrophoresis.
AAV2/8-mediated hMPV17 expression rescue the Mpv17−/− mouse liver phenotype
We constructed an AAV2/8 viral vector expressing the hMPV17 cDNA to the liver, under the control of the liver-specific thyroxine-binding globulin (TBG) promoter. The hMPV17 protein is almost identical to the mouse (m)Mpv17 protein, displaying 14 changes, none of which was in highly conserved residues or was predicted to have deleterious consequences by SIFT analysis (Supplementary Figure S1).21 AAV2/8-hMPV17 was administered to groups of 2-month-old Mpv17−/− and Mpv17+/− mice (n = 4 each) by retroorbital injection at a concentration of 4 × 1012 viral genomes (vg)/kg. The mice were sacrificed 3 weeks later. Blood samples drawn before AAV treatment and again just before the sacrifice showed that aspartate aminotransferase and alanine aminotransferase enzymes, two markers of hepatocyte leakage, were high before AAV administration in Mpv17−/− mice, whereas they were normalized after AAV administration (Figure 2a), suggesting correction of liver damage. No change was observed in control animals upon AAV injection, suggesting that AAV has no hepatotoxicity per se. Viral DNA was detected by polymerase chain reaction (PCR) in liver, but not in kidney, skeletal muscle, heart, and brain (data not shown), confirming the specific hepatotropism of AAV2/822; similar amounts of viral DNA per cell were found in Mpv17+/− and Mpv17−/− livers (0.9 ± 0.3 versus 1.2 ± 0.5; Figure 2b). Next, we measured MPV17 mRNA expression by real-time (RT)-qPCR. Since human and mouse MPV17 sequences are almost identical, we were unable to design primers suitable to distinguish endogenous from AAV-mediated expression. However, in both AAV-treated Mpv17−/− and Mpv17+/− mice, Mpv17 mRNA levels were much higher than the levels found in untreated Mpv17+/− mice (Figure 2f). Accordingly, western blot immunovisualization with αMpv17 showed that the amount of Mpv17-crossreacting material in AAV-treated Mpv17−/− liver homogenates (data not shown) and isolated mitochondria (Figure 2d) were much higher than that found in control mice. Analysis by 2D-BNGE immunoblotting demonstrated that hMPV17 was integrated into a high molecular weight complex similar to that detected in control mitochondria (Figure 2e). The mtDNA copy number was markedly increased in livers from AAV-treated Mpv17−/− mice (1282 ± 372 copies/cell) compared to untreated Mpv17−/− littermates (95 ± 34 copies/cell; Whitney–Mann test: P < 0.0001), and similar to that of untreated wild-type livers (1402 ± 192; P = 0.90; Figure 2c). Intriguingly, the AAV-treated Mpv17+/− mice had slightly increased levels of mtDNA as well (1857 ± 144) compared to untreated littermates (P < 0.0001; Figure 2c). Notably, an AAV expressing hMPV17 tagged with a HA epitope, was neither able to enter the complex (Supplementary Figure S2a) nor to complement the mtDNA depletion (Supplementary Figure S2b) in spite of high viral content in liver (Supplementary Figure S2c), suggesting that the HA epitope hampers the function of MPV17, possibly by interfering with the formation of the supramolecular complex, which appears as necessary for mtDNA maintenance. As expected, the mtDNA copy number in skeletal muscle was similar in treated versus untreated Mpv17−/− individuals (data not shown). The increase in mtDNA copy number in AAV-treated versus untreated Mpv17−/− livers, was paralleled by an increase in mtDNA-encoded transcripts (COI, COII, ND5, ND6, and CYTB), and in the enzymatic activities of complexes I, III, and IV (Figure 2f,g). Mitochondrial respiratory chain (MRC) activities were not increased in mice treated with hMPV17-HA-recombinant AAV vectors (Supplementary Figure S2d), and in fact transaminase levels were as high as those found in untreated naive Mpv17−/− littermates (Supplementary Figure S2e).
Figure 2.
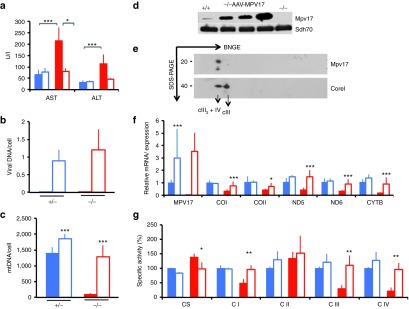
Molecular and clinical characterization of AAV2/8-TBG-h.MPV17-treated mice. A single retroorbital injection of 4 × 1012 vg/kg was performed in 2-month-old Mpv17+/− and Mpv17−/− mice. (a) Analysis of AST and ALT plasma levels in mice before and after AAV administration. The Asterisks (*) indicate significance (P), calculated by either the Wilcoxon test to compare mice pre- and post-AAV treatment, or the Mann–Whitney test to compare different groups of animals: *P < 0.05; ***P < 0.0001. (b) Viral DNA content in livers from Mpv17+/− and Mpv17−/− mice. No amplification was obtained in AAV-untreated mice. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/−; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the standard deviation (SD). (c) Mitochondrial DNA (MtDNA) content in livers from AAV-treated and untreated Mpv17+/− and Mpv17−/− mice. Note that mtDNA content in AAV-treated animals is comparable to that of naive Mpv17+/− animals, and that a slight, but significant, increase is also present in the AAV-treated versus untreated controls. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/−; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the SD. Asterisks (*) indicate significance (P) calculated by the Mann–Whitney test: *P < 0.05; **P < 0.01; ***P < 0.0001. (d) Western blot analysis of the h.MPV17 protein: the amount detected in isolated liver mitochondria from three AAV-treated Mpv17−/− mice is higher than that detected in Mpv17-/+ littermates, whereas the protein is absent in a mitochondrial sample from a Mpv17−/− naive individual. (e) Two-dimensional blue native gel electrophoresis on liver mitochondria from AAV-treated Mpv17−/− mice. Note that the spot has a molecular weight similar to the CIII2+CIV supercomplex, as determined by staining with an αCore1 antibody. (f) mRNA expression analysis. Note that AAV-treated Mpv17−/− mice show normal levels of transcripts. All values are normalized to the untreated control. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/−; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the SD. Asterisks (*) indicate significance (P) calculated by the Mann–Whitney test: *P < 0.05; ***P < 0.0001. (g) Mitochondrial respiratory chain activities in treated versus untreated Mpv17+/− and Mpv17−/− mice. CI, CIII, and CIV activities are normalized in AAV-treated Mpv17−/− animals. Note that CS activity, which is increased in Mpv17−/− mice, is normal in the AAV-treated group. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/−; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the SD. Asterisks (*) indicate significance (P) calculated by the Mann–Whitney test: *P < 0.05; **P < 0.01.
KD induces severe cirrhosis in Mpv17−/− mice
KD supplies ≈80% of the energy from fat. Although KD has been suggested to induce mitochondrial biogenesis in skeletal muscle,23 it seems to act detrimentally in OXPHOS-defective mouse models unable to consume the excess of NADH derived from fat metabolism.24 We investigated the effects of KD administered for 2 months to 2-month-old groups of Mpv17−/− and Mpv17+/− mice (n = 4 individuals each). Both Mpv17−/− and Mpv17+/− KD-treated animals showed initial weight loss, which was progressively recovered by Mpv17+/− but not Mpv17−/− individuals (Supplementary Figure S3a). After 2 months, KD-treated mice showed marked increase of plasma alanine aminotransferase and aspartate aminotransferase levels, which was much higher in the Mpv17−/− than in the Mpv17+/− group (Supplementary Figure S3b). At necroscopy, the Mpv17−/− livers were markedly enlarged and yellowish (Supplementary Figure S3c). Histologic examination revealed diffuse micro- and macrovacuolar steatosis, hepatocellular necrosis with inflammatory infiltrates, and lipogranulomas. In addition, picrosirius red, which selectively stains collagen fibers, showed fibrous septa surrounding nodules of regenerating hepatocytes, consistent with cirrhosis (Figure 3a,b). Contrariwise, Mpv17+/− livers displayed only a mild, albeit variable, micro- and macrovacuolar steatosis, and perisinusoidal collagen deposition (Figure 3c,d), as expected by prolonged exposure to a high-fat diet. The mtDNA content was unchanged in both KD-fed genotypes compared to standard diet (SD)-fed littermates, indicating that KD failed to induce mitochondrial biogenesis in liver (Supplementary Figure S4a), as confirmed by histochemical staining for cytochrome c oxidase (Supplementary Figure S4b). Indeed, transcript analysis showed a reduction of some MRC subunits in KD- versus SD-fed mice (Supplementary Figure S4c,d). Massive steatosis prevented us to perform spectrophotometric assays of MRC activities in liver homogenates.
Figure 3.

Histological analysis of livers from ketogenic diet (KD)-fed mice. Hematoxylin and eosin (a and c) and picrosirius red (b and d) staining in KD-fed Mpv17−/− and Mpv17+/− livers. See text for details. Scale bars: 150 µm (a–c); 300 µm (b–d).
AAV-mediated Mpv17 expression corrects liver damage of KD
We next investigated whether treatment by AAV2/8-hMPV17 was able to prevent and/or correct the severe liver derangement induced in Mpv17−/− mice by KD. In a first protocol, aiming at preventing damage, KD administration for 2 months was started 3 weeks after the injection of 4 × 1012 (n = 4) or 4 × 1013 (n = 3) vg/kg AAV2/8-hMPV17 into 2-month-old Mpv17+/− and Mpv17−/− mice. Here, we show the data referred to the higher viral dose, but similar results were also obtained using the lower dose (Supplementary Figure S5). Body weights were monitored throughout the experiment, and hepatic enzyme levels were measured at the end of the treatment. As mentioned before, untreated Mpv17−/− mice showed progressive loss of body weight, whereas AAV-treated Mpv17−/− individuals recovered their starting weight after initial loss, and gained weight over time with a trend similar to that of AAV-treated and untreated Mpv17+/− animals (Supplementary Figure S6a). In contrast to untreated KD-fed Mpv17−/− mice, AAV-treated KD-fed Mpv17−/− mice showed plasmatic levels of hepatic enzymes similar to those of control littermates in KD (Supplementary Figure S6b). Livers from KD-fed AAV-treated Mpv17−/− animals were significantly less enlarged (9.9 ± 2.6 versus 7.0 ± 1.0; Mann–Whitney test: P < 0.01; Supplementary Figure S6c), and remarkably less damaged upon histological examination than those from untreated Mpv17−/− littermates (Figure 4). Eventually, while AAV-untreated Mpv17−/− individuals invariably developed overt cirrhosis and necrosis by 2 months of KD, AAV-treated Mpv17−/− animals showed only variable degrees of steatosis and some inflammatory infiltrates, comparable to those of control animals exposed to the same diet (Figure 4). These results clearly indicate that AAV2/8-hMPV17 treatment prevents KD-induced liver damage in Mpv17−/− mice.
Figure 4.

Histological analysis of livers from ketogenic diet (KD)-fed mice pretreated with AAV. A single retroorbital injection of 4 × 1013 vg/kg was performed in 2 months old Mpv17+/− and Mpv17−/− mice. KD was started 3 weeks later. (a–d) Hematoxylin and eosin. (e–h) Picrosirius red staining. (a–e) Untreated Mpv17+/−. (b–f) AAV-treated Mpv17+/−. (c–g) Untreated Mpv17−/−. (d–h) AAV-treated Mpv17−/−. See text for details. Scale bars: a–d: 150 µm, e–h: 300 µm.
We observed that the viral DNA content was much lower in KD-fed than in SD-fed livers (1.2 ± 0.2 versus 12.8 ± 2.3 in Mpv17+/−, P < 0.05; 0.4 ± 0.3 versus 14.8 ± 1.0 copies/cell in Mpv17−/−, P < 0.05; Figure 5a), suggesting that KD induced strong dilution of viral DNA. Since the DNA of recombinant AAV vectors remains episomal25 and is replication-defective, a possible explanation for this phenomenon is increased cell proliferation. To confirm this hypothesis, we performed quantitative analysis of proliferating versus nonproliferating hepatocytes by measuring proliferating cell nuclear antigen, a marker of cell proliferation.26 The nuclear staining index for proliferating cell nuclear antigen was 18.2 ± 13.6% in SD versus 74.2 ± 7.7% in KD in Mpv17+/− and 20.9 ± 15.1% in SD versus 81.7 ± 12.5 in KD in Mpv17−/− mice (Student t-test: P < 0.005 for both groups; Supplementary Figure S7a). Similar results were obtained in AAV-treated mice (Supplementary Figure S7b). This result highlights the effect of KD in promoting hepatocyte proliferation in mouse liver and may explain the decrease of viral content observed during the KD treatment. Nevertheless, the mtDNA content increased from 49 ± 8 copies/cell in naive Mpv17−/− to 576 ± 211 copies/cell in AAV-treated Mpv17−/− littermates (Figure 5b). This increase was paralleled by robust expression of mtDNA transcripts, becoming quantitatively comparable to that of untreated KD-fed Mpv17+/−littermates (Figure 5c), and led to significant recovery of cytochrome c oxidase histochemical activity (Figure 5d).
Figure 5.
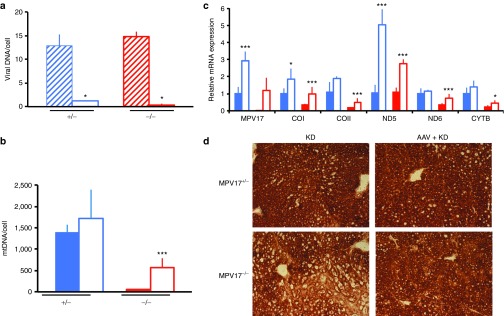
Molecular and biochemical characterization of ketogenic diet (KD)-fed mice pretreated with AAV. (a) Viral DNA content in livers from AAV-treated Mpv17+/− and Mpv17−/− mice in standard diet (SD) versus KD. Blue stripes: AAV-treated Mpv17+/− mice in SD; blue outline: AAV-treated Mpv17+/− mice in KD; red stripes: AAV-treated Mpv17−/− mice in SD; red outline: AAV-treated Mpv17−/− mice in KD. Bars indicate the standard deviation (SD). (b) Mitochondrial DNA (MtDNA) analysis in AAV-treated versus untreated, KD-fed mice. Note that mtDNA content in AAV-treated Mpv17−/− is higher than in untreated littermates, but remains lower than in control littermates. (c) Mitochondrial transcripts analysis in AAV-treated versus untreated, KD-fed mice. All values are normalized to the KD-fed untreated controls. (d) Cytochrome c oxidase histochemical staining. In b and c, the asterisks (*) indicate significance (P) calculated by the Mann–Whitney test for independent samples: *P < 0.05; ***P < 0.0001. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/−; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the standard deviation (SD).
In a second protocol, aiming at correcting damage, we tested whether AAV-based hMPV17 gene replacement therapy can stop or reverse an already ongoing liver derangement, as typically found in patients. We first KD-fed for 1 month two groups of 2-month-old Mpv17−/−and Mpv17+/−mice. This treatment caused liver damage, as clearly indicated by increased hepatic-enzyme levels in plasma of both groups (Supplementary Figure S8a). We then administered 4 × 1013 vg/kg of AAV-hMPV17 to both groups of animals, while KD was continued for one additional month before sacrifice, so that this group of mice was overall exposed to KD as long as the first group. Livers from Mpv17−/− animals showed a statistically nonsignificant trend toward reduced hepatomegaly compared to untreated littermates, whereas livers from Mpv17+/− mice were unaffected (Supplementary Figure S8b).
Again, the livers from Mpv17−/− mice were protected from developing extensive KD-induced liver damage, showing moderate hepatocyte steatosis, in the absence of necrosis and fibrosis (Figure 6a,b), while livers from Mpv17+/− exposed to this protocol displayed only mild and focal steatosis (Figure 6c,d). Viral DNA quantification showed 5.6 ± 3.1 vg/cell in Mpv17−/−and 5.0 ± 2.5 vg/cell in Mpv17+/− mice (Figure 7a), leading to high levels of MPV17 mRNA in both Mpv17−/−and Mpv17+/−livers (Figure 7b). The mtDNA copy number was significantly increased in AAV-treated versus untreated Mpv17−/− individuals (501 ± 263 versus 49 ± 8; Mann–Whitney test: P < 0.01) although remained lower than in treated (1387 ± 331 copies/cell) and untreated (1384 ± 194 copies/cell) Mpv17+/−littermates (Figure 7c). In parallel, mitochondrial transcripts were significantly increased in the Mpv17−/− livers, up to levels comparable to KD-fed control littermates (Figure 7b), and cytochrome c oxidase histochemical activity was increased as well (Figure 7d). Nevertheless, the overall clinical conditions were clearly worse in post-KD AAV-treated than in pre-KD AAV–treated Mpv17−/− mice. The hepatic enzyme levels increased, whereas the body weight decreased (Supplementary Figure S8c,d), suggesting that our “late” AAV treatment can possibly slow the evolution of preexisting liver damage, but is unable to reverse the downhill clinical progression of the disease.
Figure 6.

Histological analysis of livers from AAV posttreated, ketogenic diet (KD)-fed mice. A single retroorbital injection of 4 × 1013 vg/kg was performed in two-month old Mpv17+/− and Mpv17−/− mice one month after starting KD. (a and c) Hematoxylin and eosin and (b and d) picrosirius red staining in AAV posttreated Mpv17−/− and Mpv17+/− livers. See text for details. Scale bars: a–c: 150 µm, b–d: 300 µm.
Figure 7.
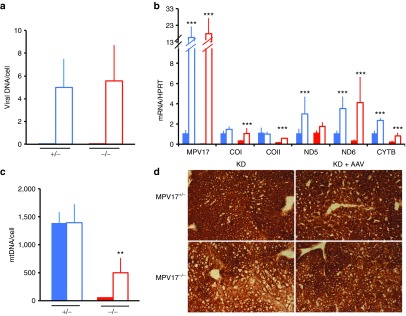
Molecular and biochemical characterization of livers from AAV posttreated, ketogenic diet (KD)-fed mice. (a) Viral DNA content in livers. (b) Mitochondrial DNA (MtDNA) analysis. Note that mtDNA content in AAV-treated Mpv17−/− is higher than in untreated littermates, but remains lower than in control littermates. (c) Mitochondrial transcripts analysis. (d) Cytochrome c oxidase histochemical staining in b and c. The asterisks (*) indicate significance (P) calculated by the Mann–Whitney test for independent samples; **P < 0.001; ***P < 0.001. Color codes: solid blue: untreated Mpv17+/−; blue outline: AAV-treated Mpv17+/; solid red: Mpv17−/−; red outline: AAV-treated Mpv17−/−. Bars indicate the standard deviation (SD).
Discussion
We here provide novel information on the connection between MPV17 and mtDNA maintenance, although the complete elucidation of the physiological role of this elusive, medically relevant, and biologically intriguing protein is still missing. First, we have demonstrated that, like the yeast ortholog SYM1, mammalian MPV17 forms a high molecular weight complex in both cell cultures and mouse tissues. By reconstitution into lipid bilayers, Sym1 has recently been shown to aggregate as a high molecular complex to form a functional pore possibly a cation channel.27 Similar to mitochondrial carrier proteins, both Mpv17 and SYM1 are imported into the inner mitochondrial membrane via the TIM23 complex without cleavage of a leader peptide. A channel activity has not been (yet) demonstrated for the mammalian Mpv17, but we have shown here that Mpv17 participates in a high molecular weight complex necessary for its function, since variants that fail to be incorporated do not rescue the liver phenotype of Mpv17−/− mice, including mtDNA depletion.
A second goal of our work consisted in the development of therapeutic strategies for liver-specific MDS. Although many mitochondrial abnormalities cause multisystem disorders, some affect only or predominantly a single organ, being thus amenable to treatment by tissue-specific gene replacement. Most of the MPV17 mutant cases are characterized by early onset, often fatal liver failure, associated with profound tissue-specific mtDNA depletion, whereas other tissues are relatively (e.g., skeletal muscle) or completely (e.g., heart) spared. We used recombinant AAV vectors to target the therapeutic gene to the liver, because this approach is relatively safe, noninvasive, and translatable to humans, we clearly showed that AAV-mediated expression of human MPV17 in Mpv17−/− mouse livers can correct the molecular phenotype, i.e., severe mtDNA depletion, in standard conditions, thus normalizing the MRC defects. We then showed that KD causes Mpv17−/− mice to rapidly develop severe hepatomegaly and macrovacuolar steatosis, which evolves into cirrhosis and overt liver failure within 2 months. By contrast, Mpv17+/− littermates exposed to the same diet displayed only mild steatosis, mostly of the microvacuolar type. Administration of AAV2/8-hMPV17 before starting KD effectively protected Mpv17−/− mice from liver derangement.
Partial protection from KD-induced liver failure was also obtained in mice exposed to KD before AAV treatment, that is when liver damage was already ongoing, as indicated by elevated plasma levels of hepatic enzymes. However, in this case, AAV treatment was able to halt the progression of the disease, but not to revert already established liver damage. This result is in agreement with the evidence that the AAV1 serotype is able to efficiently transduce both normal and cirrhotic livers,28 with no significant dilution of the viral load over time.
We propose AAV2/8-based therapy as a realistic strategy to prevent liver failure in patients with mutations in MPV17 and eventually in other forms of liver MDS. AAV2/8 serotype can remain for a long time in the liver of mice,29,30,31 cats,32 and humans,33 with no significant side effects. While targeting of AAV-mediated therapeutic protein is still challenging for large-size tissues such as skeletal muscle or impermeable organs such as the brain, this approach is becoming a realistic option for gene conditions affecting a single, compliant organ, such as the liver in case of hepatic MDS, for proteins that are secreted by a targetable organ, such as the liver again for factor IX, the circulating enzyme missing in hemophilia B,33 or in disorders that can be effectively cured by using liver to clear the bloodstream from toxic substances by making liver competent for clearing the bloodstream from toxic compounds. The latter strategy has proven successful for ethylmalonic encephalopathy, a mitochondrial disease due to the accumulation of toxic free sulphide22 and can in principle be applied to mitochondrial neurogastrointestinal encephalomyopathy, due to accumulation of thymidine to toxic levels in plasma and tissues.
Materials and Methods
Construction of AAV2/8 vectors. AAV2/8-TBG-h.MPV17 and AAV2/8-TBG-h.MPV17HA vectors were produced by the AAV Vector Core of the Telethon Institute of Genetics and Medicine (Naples, Italy) by triple transfection of 293 cells and purified by CsCl gradients.34 Physical titers of the viral preparations (genome copies per ml) were determined by real-time PCR35 (Applied Biosystems, Foster City, CA) and dot-blot analysis.
Genomic DNA extraction, PCR, and quantitative PCR. Total DNA was extracted from frozen tissues using the Maxwell apparatus (Promega, Madison, WI) following the instructions of the manufacturer. AAV-derived DNA was detected by standard PCR amplification using primer pairs specific to the hMPV17 gene. SYBR-GREEN–based real-time quantitative PCR was carried out for mtDNA and AAV-copy number analysis as previously described10,22; the RNAseP gene was used as a reference. Total RNA was extracted from liquid nitrogen snap frozen liver by Total RNA Mini Kit (tissue), according to the manufacturer's instructions (Geneaid, Taipei, Taiwan). Of total RNA, 2 µg was treated with Rnase-free DNase and retrotranscribed using the GoTaq 2-Step RT-qPCR System (Promega). Approximately 25 ng of cDNA was used for real-time PCR assay using primers specific for amplification of several genes.36 Oligonucleotide sequences are available on request.
Biochemical analysis of MRC complexes. Liver samples stored in liquid nitrogen were homogenized in 10 mmol/l phosphate buffer (pH 7.4), and the spectrophotometric activity of cI, cII, cIII, and cIV, as well as citrate synthase, was measured as described.37
Morphological analysis. Histochemical analyses were performed as described.38 Hematoxylin and eosin and picrosirius red staining were performed according to standard protocols. Livers were formalin fixed and paraffin embedded. Consecutive sections (5–6 µm thick) were stained with hematoxylin and eosin and picrosirius red for histologic examination. Immunostaining with antiproliferating cell nuclear antigen monoclonal antibodies (Abcam, Cambridge, UK; dilution 1:6,000) was performed for quantitative evaluation of proliferating hepatocytes. Two-hundred cells per slide were manually counted in four high-power fields (original magnification: ×40), and the number of proliferating hepatocytes (in all cell cycle stages) was expressed as a ratio over the total cell number.
Immunoblotting. Ten percent weight per volume homogenates in 10 mmol/l phosphate buffer pH 7.4 were prepared from liver and centrifuged at 800g for 10 minutes to eliminate cellular debris. Total protein extracts were run through a 12% sodium dodecyl sulfate–polyacrylamide gel (40 µg/sample) and electroblotted onto nitrocellulose filters. The filters were immunostained with specific antibodies against MPV17 (Proteintech, Manchester, UK), Core1, and the 70 kDa subunit of succinate dehydrogenase (Molecular Probes; Invitrogen, Paisley, UK), and protein bands were visualized using the ECL chemioluminscence kit (Amersham, Milan, Italy). Twenty microgram of proteins from isolated liver mitochondria39 were run on a 12% sodium dodecyl sulfate–polyacrylamide gel.
BNGE was performed in isolated liver mitochondria as described.40 Fifteen microliters of sample were run through a 5–13% nondenaturating gradient (1D-BNGE). For denaturing two-dimensional BNGE electrophoresis, the one-dimensional BNGE lane was excised, treated for 1 hour at room temperature (20 °C) with 1% sodium dodecyl sulfate and 1% β-mercaptoethanol and then run through a 16.5% tricine-sodium dodecyl sulfate–polyacrylamide gel using a 10% spacer gel.
Analysis of body fluids. aspartate aminotransferase and alanine aminotransferase were determined in blood samples by standard methods.
Experimental ethics policy. Animal studies were approved by the Ethics Committee of the Foundation ‘Carlo Besta' Neurological Institute, in accordance with the guidelines of the Italian Ministry of Health. The use and care of animals followed the Italian Law D.L. 116/1992 and the EU directive 86/609/CEE. The mice were kept on a C57Bl6/129Sv mixed background, and wild-type littermates were used as controls. Standard food and water were given ad libitum. KD was administered for 2 months (SSNIFF, Germany) ad libitum. Body weight and food consumption were monitored twice a week.
SUPPLEMENTARY MATERIAL Figure S1. Alignment of eukaryotic Mpv17 orthologues. Figure S2. AAV-mediated expression of hMPV17-HA in Mpv17−/− mice. Figure S3. Effects of KD on Mpv17−/− and control littermates. Figure S4. KD does not induce mitochondrial biogenesis in liver. Figure S5. AAV2/8-hMPV17 4 × 1012 vg/kg partially rescues KD-induced liver damage inMpv17−/− mice. Figure S6. AAV2/8-hMPV17 4 × 1013 vg/kg rescues KD-induced liver damage in Mpv17−/− mice. Figure S7. Effect of KD on hepatocytes proliferation. Figure S8. Effects of administration of AAV-hMPV17 in mice pretreated with KD.
Acknowledgments
This work was supported by the Pierfranco and Luisa Mariani Foundation Italy, Telethon-Italy GPP10005 and GGP11011 (to M.Z.), Cariplo2011-0526 (to M.Z.), European Research Council Grant ERC-322424 (to M.Z.), andThe Italian Ministry of Health Research Grant, GR-2010-2306-756 (to C.V.). We thank Daniele Ghezzi for the helpful discussion.
Supplementary Material
References
- Suomalainen A, Isohanni P. Mitochondria DNA depletion syndromes – many genes, common mechanisms. Neuromusc Disorders. 2010;20:429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Cohen BH, Naviaux RK. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods. 2010;51:364–373. doi: 10.1016/j.ymeth.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA. Brain. 2005;128 Pt 4:723–731. doi: 10.1093/brain/awh410. [DOI] [PubMed] [Google Scholar]
- Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lönnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130 Pt 11:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D'Adamo P, Calvo S, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Spinazzola A, Maggioni M, Fernandez-Vizarra E, Massa V, Pagano C, et al. Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum Mol Genet. 2009;18:12–26. doi: 10.1093/hmg/ddn309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, Dahms BB, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–1227. doi: 10.1002/hep.21799. [DOI] [PubMed] [Google Scholar]
- Navarro-Sastre A, Martín-Hernández E, Campos Y, Quintana E, Medina E, de Las Heras RS, et al. Lethal hepatopathy and leukodystrophy caused by a novel mutation in MPV17 gene: description of an alternative MPV17 spliced form. Mol Genet Metab. 2008;94:234–239. doi: 10.1016/j.ymgme.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Santer R, Akman OH, Tsiakas K, Schaefer H, Ding X, et al. Hepatocerebral form of mitochondrial DNA depletion syndrome: novel MPV17 mutations. Arch Neurol. 2008;65:1108–1113. doi: 10.1001/archneur.65.8.1108. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, Wong LJ. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations. Mol Genet Metab. 2010;99:300–308. doi: 10.1016/j.ymgme.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Nogueira C, de Souza CF, Husny A, Derks TG, Santorelli FM, Vilarinho L. MPV17: fatal hepatocerebral presentation in a Brazilian infant. Mol Genet Metab. 2012;107:764. doi: 10.1016/j.ymgme.2012.10.010. [DOI] [PubMed] [Google Scholar]
- AlSaman A, Tomoum H, Invernizzi F, Zeviani M. Hepatocerebral form of mitochondrial DNA depletion syndrome due to mutation in MPV17 gene. Saudi J Gastroenterol. 2012;18:285–289. doi: 10.4103/1319-3767.98439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallabona C, Marsano RM, Arzuffi P, Ghezzi D, Mancini P, Zeviani M, et al. Sym1, the yeast ortholog of the MPV17 human disease protein, is a stress-induced bioenergetic and morphogenetic mitochondrial modulator. Hum Mol Genet. 2010;19:1098–1107. doi: 10.1093/hmg/ddp581. [DOI] [PubMed] [Google Scholar]
- Parini R, Furlan F, Notarangelo L, Spinazzola A, Uziel G, Strisciuglio P, et al. Glucose metabolism and diet-based prevention of liver dysfunction in MPV17 mutant patients. J Hepatol. 2009;50:215–221. doi: 10.1016/j.jhep.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, Johnsen SD, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zum Gottesberge AM, Reuter A, Weiher H. Inner ear defect similar to Alport's syndrome in the glomerulosclerosis mouse model Mpv17. Eur Arch Otorhinolaryngol. 1996;253:470–474. doi: 10.1007/BF00179952. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med. 2012;4:1008–1014. doi: 10.1002/emmm.201201433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahola-Erkkilä S, Carroll CJ, Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, et al. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–1984. doi: 10.1093/hmg/ddq076. [DOI] [PubMed] [Google Scholar]
- Wenz T, Luca C, Torraco A, Moraes CT. mTERF2 regulates oxidative phosphorylation by modulating mtDNA transcription. Cell Metab. 2009;9:499–511. doi: 10.1016/j.cmet.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard MT, Malinak RN, Nagy LE. Early growth response (EGR)-1 is required for timely cell-cycle entry and progression in hepatocytes after acute carbon tetrachloride exposure in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1124–G1131. doi: 10.1152/ajpgi.00544.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold R, Krüger V, Meinecke M, Schulz C, Schmidt B, Grunau SD, et al. The channel-forming Sym1 protein is transported by the TIM23 complex in a presequence-independent manner. Mol Cell Biol. 2012;32:5009–5021. doi: 10.1128/MCB.00843-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevals L, Enguita M, Rodriguez C, Gonzalez-Rojas J, Alzaguren P, Razquin N, et al. AAV vectors transduce hepatocytes in vivo as efficiently in cirrhotic as in healthy rat livers. Gene Ther. 2012;19:411–417. doi: 10.1038/gt.2011.119. [DOI] [PubMed] [Google Scholar]
- Tessitore A, Faella A, O'Malley T, Cotugno G, Doria M, Kunieda T, et al. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- Chandler RJ, Venditti CP. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol Ther. 2010;18:11–16. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscioni D, Morizono H, McCarter RJ, Stern A, Cabrera-Luque J, Hoang A, et al. Long-term correction of ammonia metabolism and prolonged survival in ornithine transcarbamylase-deficient mice following liver-directed treatment with adeno-associated viral vectors. Mol Ther. 2006;14:25–33. doi: 10.1016/j.ymthe.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Cotugno G, Annunziata P, Tessitore A, O'Malley T, Capalbo A, Faella A, et al. Long-term amelioration of feline Mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol Ther. 2011;19:461–469. doi: 10.1038/mt.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Qu G, Burnham MS, Huang J, Chirmule N, Joshi B, et al. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum Gene Ther. 2000;11:2079–2091. doi: 10.1089/104303400750001390. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1a axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Invernizzi F, Alberio S, Briem E, Lamantea E, Carrara F, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Sciacco M, Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- Fernández-Vizarra E, López-Pérez MJ, Enriquez JA. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.