

Abstract
Previous studies with aminothiazolomorphinans suggested that this class of opioid ligands may be useful as a potential pharmacotherapeutic to decrease drug abuse. Novel aminothiazole derivatives of cyclorphan were prepared in order to evaluate a series of aminothiazolomorphinans with varying pharmacological properties at the MOR and KOR. This study was focused on exploring the regioisomeric analogs with the aminothiazole on the C-ring of the morphinan skeleton. Receptor binding and [35S]GTPγS binding assays were used to characterize the affinity and pharmacological properties of the aminothiazolomorphinans. Intracranial self-stimulation (ICSS) was used to compare effects of a representative aminothiazolomorphinan with the morphinan mixed KOR/MOR agonist butorphan (MCL-101) on brain stimulation reward.
INTRODUCTION
Cocaine is a widely abused drug. Mechanistically, cocaine binds to and blocks dopamine reuptake receptors, which results in an increase of dopamine in the central nervous system.1–3 Rapid elevation in dopamine levels within brain regions necessary for reward contribute to the behavioral effects of euphoria, mental alertness and increased energy. Consequently, many attempts have been made to develop drugs that reduce the effects of dopamine with the idea that this would decrease cocaine abuse. However, this strategy has failed to provide an effective drug candidate due to the poor results associated with such drugs.4–6
The opioid system is responsible for the modulation of several key physiological and behavioral processes, such as pain perception, reward function, and stress response.7 Each of the three opioid receptors [κ opioid receptor (KOR), μ opioid receptor (MOR), and δ opioid receptor (DOR)] functions differently within the body. It has been shown that KOR dysregulation can contribute to drug abuse and other psychiatric disorders.8,9 Therefore, KOR agonists/antagonists have become a target for the development of pharmacotherapies for the treatment of addiction; in particular cocaine abuse.10–13 Recent behavioral studies have suggested that mixed MOR and KOR activity may be useful for the treatment of addiction/dependence and cocaine abuse.14–16 We have reported that treatment of rhesus monkeys with cyclorphan (an analog of levorphanol, Figure 1), which has mixed KOR and MOR activity, reduced cocaine self-administration and produced fewer side effects than κ-selective agonists.17 Another analog, butorphan (Figure 1) in contrast to a partial effect with cyclorphan, exhibited full antinociception in the warm-water tail flick test in rats,10b and was shown to be 40 times more potent in suppressing abstinence in morphine dependent monkeys.17 Further analogs were thus evaluated to increase the overall efficacy of these compounds as agents for treating cocaine abuse.
Figure 1.
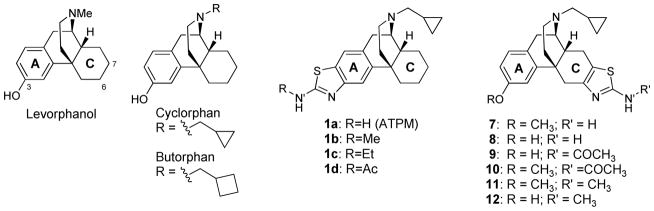
Structure of highly active opioid ligands.
While evaluating several biological isosteres of the phenol moiety to increase the duration of activity of cyclorphan, aminothiazolomorphinans (1) were developed.19–23 These compounds are potent agonists which are highly KOR selective. ATPM, 1a, has been shown to attenuate morphine antinociceptive tolerance, and decreases self-administration of heroin in mice.20 Furthermore, 1c, an N′-alkylated derivative, inhibited morphine sensitization in mice.21 Recent studies of 1a in the 55°C mouse tail flick assay indicate that 1a does not have a longer duration of action than cyclorphan21 and has been shown to inhibit morphine sensitization in mice.22 The regioisomeric aminothiazolomorphinan 8 is also a potent kappa agonist however, 8 is not as selective for the KOR as 1a.23 In fact, there is little selectivity with 8 for either KOR, MOR, or DOR. We were interested in further exploring these regioisomeric analogs with the aminothiazole on the C-ring of the morphinan skeleton (7–12). Herein, we discuss our studies and comparison of 7–12 with the previously reported (1a–d) series of compounds.
CHEMISTRY
Compound 8 (MCL-420)23 was previously prepared in 11 steps and 1.2% overall yield.23 For further in vivo studies we wished to carry out, a modified and scalable synthesis of 8. As depicted in Scheme 1, ring opening/rearrangement proceeded smoothly with 2.5 equivalents of nBuLi.24 Hydrogenation in methanol followed by crystallization as the hydrochloride salt with pyridine-hydrochloride provided pure thebainone 3 in 65% yield. Triflation of 3, in refluxing THF, provided pure 4 after crystallization with ethyl acetate. Reduction of the triflate using a catalytic amount of palladium and 1,3-bis(diphenylphosphino)propane with triethyamine/formic acid as the reductant, produced 5 quantitatively.25 Next, a three step sequence to N-demethylate and alkylate with cyclopropylmethyl bromide resulted in a separable mixture of N-cyclopropylmethyl-O-methyl dihydrothebainone 6a and corresponding phenol 6b. The phenol, 6b, arises from heating the carbamate in acid during the two step sequence to N-demethylate. Careful control of this step allows for a selective formation of the desired compound 6a.
Scheme 1.

Synthesis of the Aminothiazolomorphinans.a
aReagents and conditions: (i) nBuLi, THF, −78-0°C; (ii) Pd/c, H2, MeOH, RT 62% yield (2 steps); (iii) PhN(Tf)2, K2CO3, THF, 65°C, 85% yield; (iv) Pd(OAc)2, DPPP, Et3N, HCO2H, DMF, 65°C, 99% yield; (v) ClCO2Et, K2CO3, CHCl3, 70°C; (vi) HCl/AcOH, 100°C; (vii) Cyclopropylmethyl Bromide, NaHCO3, EtOH, 70°C 70% yield (3 steps); (viii) Br2, HBr(48% aq.), AcOH, 60°C, then thiourea 100°C 52% yield (ix) BBr3, CH2Cl2, −78°C-RT, 84% yield; (x) Ac2O, Et3N, CH2Cl2, RT 89% yield; (xi) NaOAc, EtOH/H2O, 90°C, 39% yield; (xii) paraformaldehyde, NaOMe, 65°C, then NaBH4, 99% yield; (xiii) BBr3, CH2Cl2, −78 °C, 86% yield.
The synthesis of the aminothiazolomorphinan 8 was completed in two synthetic steps from 6a. The bromine-mediated aminothioazole-cyclocondensation proceeded smoothly under acidic conditions providing 7 in good yield. Finally, O-demethylation using BBr3 provided the aminothiazole 8 as the trihydrobromide salt. N′-methylation of 7 with paraformaldehyde and NaBH4 to afford 11 in quantitative yields. O-Demethylation of 11 using BBr3 provided 12 as the trihydrobromide salt.
Additionally, 9 was prepared by treating 8 with acetic anhydride and triethylamine, which afforded the O,N-bisacylated product. Removal of the O-acyl group with sodium acetate in refluxing ethanol afforded 9.26 The O-methyl analog, 10, was prepared in the same manner as 9; however, only a single step was necessary starting from 7.
RESULTS AND DISCUSSION
All new compounds were evaluated for their binding affinity at each of MOR, KOR, and DOR. For comparison, the binding affinities of other highly active compounds in this series are also included. Aminothiazolomorphinan 8 was the parent compound for all analogs prepared and demonstrated an excellent binding profile (sub-nanomolar affinity) at all three receptors. The lead compound in this series, 8, was not highly selective for the KOR displaying only 2-fold selectivity over MOR and 3-fold over DOR. The O-methyl analog, 7, showed a marked reduction in the binding affinity across all three receptors (~10 fold vs. 8) but was more selective for KOR vs. MOR (3.5-fold) and KOR vs. DOR (7-fold). The N′-methyl analog, 12, was equipotent for KOR and MOR as 8. There was a considerable difference in DOR binding affinities (0.43 nM vs. 0.23 nM) leading to a higher selectivity for the KOR (7-fold). The O,N′-methyl compound, 11, had a significantly lower binding affinity at each MOR, KOR, and DOR, although it is still a potent analog (Ki<10 nM).
The N′-acetyl compounds (9 and 10) were also very potent. The phenolic compound 9, exhibited sub-nanomolar binding affinity at all three MOR, KOR, and DOR. However, unlike the parent aminothiazole 8, compound 9 was not selective for the KOR. Following along with the other compounds in this series, the O-methylated analog 10, had lower affinity than the phenol 9. As seen with compound 11, the methoxy analog 10 was a DOR-preferring ligand, albeit with modest selectivity.
The aminothiazole moiety is well-tolerated in the morphinan skeleton, as evidenced by the binding affinities in Table 1. Both series of compounds, with the aminothiazole on the A (1a–d) or C ring (7–12), similar to levorphanol and cyclorphan, have high affinities to the MOR, KOR, and DOR.
TABLE 1.
Binding Affinities of Aminothiazolomorphinans to MOR, KOR and DOR.a
| Compound | Ki = (nM) ± SEM | [3H]naltrindole (δ) | Selectivity
|
||
|---|---|---|---|---|---|
| [3H]DAMGO (μ) | [3H]U69,593 (κ) | μ/κ | δ/κ | ||
| Levorphanolb | 0.21 ± 0.02 | 2.3 ± 0.3 | 4.2 ± 2.3 | 0.091 | 1.8 |
| Cyclorphanb | 0.062 ± 0.003 | 0.034 ± 0.002 | 1.9 ± 0.1 | 1.8 | 56 |
| Butorphane | 0.23 ± 0.01 | 0.079 ± .003 | 5.9 ± 0.6 | 2.9 | 75 |
| 1a (ATPM)b | 1.5 ± 0.2 | 0.049 ± 0.005 | 29 ± 2 | 31 | 590 |
| 1b (MCL-442)c | 3.0 ± 0.062 | 0.066 ± 0.0037 | 25 ± 1.5 | 45 | 380 |
| 1c (MCL-439)c | 4.7 ± 0.023 | 0.15 ± 0.000 | 43 ± 6.5 | 31 | 290 |
| 1d (MCL-437)c | 57 ± 3.9 | 13 ± 1.1 | 750 ± 64 | 4.4 | 58 |
| 7 (MCL-741)d | 6.7 ± 0.71 | 1.9 ± 0.12 | 13 ± 1.4 | 3.5 | 6.8 |
| 8 (MCL-420)d | 0.12 ± 0.0069 | 0.072 ± 0.0039 | 0.23 ± 0.012 | 1.7 | 3.2 |
| 9 (MCL-744) | 0.085 ± 0.010 | 0.17 ± 0.025 | 0.65 ± 0.043 | 0.50 | 3.8 |
| 10 (MCL-745) | 6.6 ± 0.63 | 9.0 ± 1.2 | 4.6 ± 0.16 | 0.73 | 0.51 |
| 11 (MCL-743) | 9.6 ± 0.39 | 3.6 ± 0.35 | 1.6 ± 0.16 | 2.7 | 0.44 |
| 12 (MCL-742) | 0.14 ± 0.015 | 0.084 ± 0.0026 | 0.43 ± 0.018 | 1.7 | 5.1 |
Membranes from CHO cells, expressing either human KORs, MORs, or DORs, were incubated with 12 different concentrations of the compounds in the presence of receptor-specific radioligands at 25°C, in a final volume of 1 mL of 50 mM Tris-HCl, pH 7.5. Nonspecific binding was determined using 10 μM naloxone.
Ref. 18.
Ref. 19.
Ref. 23.
The selectivity of the compounds with the aminothiazole on the C-ring (7–12) appears to be lower than the analogous compounds with the aminothiazole on the A-ring (1a–d) but both series demonstrate excellent binding profiles to the MOR, KOR, and DOR. Furthermore, from the data in Table 1, we clearly see the importance of the phenol moiety in the A ring. When comparing methoxy to phenoxy analogs (7, 10, 11, vs. 8, 9, 12 respectively), blockage of the hydrogen bond donor leads to much lower selectivity in all cases. It is clear from the data that the phenol moiety should be present to obtain a sub-nanomolar binding affinity. However, the methoxy group will undoubtedly increase oral bioavailability and the compound may be metabolized to the hydroxyl compound, as well as yield other metabolites.
The N′-acetyl compounds, 9 and 10, are very different from the regioisomeric aminothiazole 1d. Compound 1d demonstrated significantly loweractivity for MOR, KOR, and DOR but showed selectivity for the KOR. This is in stark contrast to compounds 9, which has potent affinity at MOR, KOR, and DOR. The methyl ther 10 however shows less affinity at all three opioid receptors.
The stimulation/inhibition of the [35S]GTPγS binding studies are shown in Table 2. Aminothiazolomorphinan 8 displays both agonist and antagonist properties at both the MOR and KOR as shown by the stimulation and inhibition data. Analogs 7, 10, and 11 were full agonists at the KOR and they both demonstrated agonist and antagonist properties at the MOR. This is in contrast to their phenoxy analogs 8, 9, and 12, which had both agonist and antagonist activities at both μ and κ receptors. The N′-methyl analog, 12 is very similar to 8 and acts as both agonist and antagonist at both receptors with no significant differences in EC50 or IC50 values.
TABLE 2.
Stimulation/Inhibition of [35S]GTPγS Binding Mediated by the MOR and KOR.
| Compound | MOR
|
KOR
|
||||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | EC50 (nM) | Imax (%) | IC50 (nM) | Emax (%) | EC50 (nM) | Imax (%) | IC50 (nM) | |
| 1ab | 45 ± 4 | 73 ± 5 | NI | NA | 80 ± 6 | 2.4 ± 0.6 | NI | NA |
| 1bc | 40 ± 0.87 | 47 ±9.0 | NI | NA | 82 ± 10 | 14 ± 3.1 | NI | NA |
| 1cc | 83 ± 2.5 | 29 ± 4.6 | NI | NA | 190 ± 6.4 | 22 ± 5.1 | NI | NA |
| 7d | 24 ± 0.90 | 39 ±7.8 | 69 ± 2.4 | 640 ± 76 | 140 ± 4.9 | 49 ± 1.3 | NI | NA |
| 8d | 34± 2.3 | 0.41 ± 0.021 | 64 ± 3.6 | 3.0 ± 0.56 | 59 ± 2.5 | 0.61 ± 0.049 | 49 ± 3.7 | 1.8 ± 0.20 |
| 9 | 34 ± 2.6 | 0.52 ± 0.086 | 70 ± 2.1 | 1.8 ± 0.38 | 72 ± 4.2 | 3.3 ± 0.27 | 44 ± 0.25 | 12 ± 1.4 |
| 10 | 47 ± 3.6 | 80 ± 13 | 60 ±0.55 | 500 ± 58 | 120 ± 2.4 | 140 ± 6.7 | NI | NA |
| 11 | 28 ± 1.7 | 51 ± 6.6 | 76 ± 0.68 | 650 ± 100 | 140 ± 5.8 | 97 ± 6.9 | NI | NA |
| 12 | 25 ± 1.7 | 0.52 ±0.076 | 76 ± 2.3 | 2.5 ± 0.26 | 85 ± 10.0 | 1.3 ± 0.10 | 27 ± 2.7 | 2.8 ± 0.42 |
Interestingly, 1a was a full agonist at both MOR and KOR and did not display any inhibition at either receptor. Altering the position of the aminothiazole to the C-ring, as in 8 however, demonstrates both agonist/antagonist properties and stimulates both receptors with a much lower EC50 than 1a (MOR: 0.41 nM vs. 73 nM and KOR: 0.61 nM vs. 2.4 nM). This trend is also apparent between the N′-methylated compounds 12 and 1b further illustrating the significant pharmacological differences by altering the position of the aminothiazole.
Of more interest, the acylated analog 1d, did not display high enough binding affinities to determine the [35S]GTPγS assay. The regioisomeric analogs were analyzed due to their high binding affinities. Aminothiazolomorphinan 9 demonstrated both agonist and antagonist properties at each receptor. The O′-methylated analog 10 exhibited agonist and antagonist properties at the MOR but was a full agonist at the KOR.
There was no significant effect of butorphan or 8 on ICSS thresholds at the doses tested (Butorphan Treatment: F(4,96) = 0.524, not significant; 8 Treatment: F(4,80) = 1.091, not significant Fig. 2A, D). However, there was a significant effect of butorphan treatment on maximum rates of responding [F(5, 120) = 6.845, <0.001]: 8 (4.0 mg/kg) completely suppressed responding from 15–60 min, making it impossible to determine stimulation thresholds for this dose at these times. In experiments examining the effects of butorphan and 8 on the reward-potentiating effect of cocaine on ICSS, there were significant effects of treatment [butorphan, F(4,20) = 3.89, p<0.05; 8, F(5,20) = 4.51, p<0.01]. Cocaine decreased ICSS thresholds (Fig. 2C, 2F), indicative of increased reward function. Butorphan, but not 8, dose-dependently blocked the threshold-decreasing (reward-enhancing) effect of cocaine on ICSS thresholds (Fig. 2C). There were no significant effects of treatment on maximum rates of responding (data not shown). Previously it has been shown that selective KOR agonists increase ICSS thresholds, have anxiogenic effects in the elevated plus maze, and produce conditioned place aversions.27 These results are consistent with the neurochemical finding that KOR agonists profoundly suppress dopamine release into reward-related brain regions28 and have anti-cocaine behavioral effects.29 In contrast, KOR antagonists have anxiolytic and antidepressant-like effects,27 and the reward-related effects of cocaine are prolonged in rats treated with the selective KOR antagonist norBNI.30 Thus, the ability of butorphan to block the effects of cocaine in ICSS without having aversive effects on its own is likely due to its KOR agonist activity combined with its MOR partial agonist activity. 8 May not have sufficient KOR activity to decrease the effects of cocaine on ICSS, but it may turn out to attenuate the aversive effects of cocaine withdrawal.
FIGURE 2. Effects of Butorphan and 8 on basal and cocaine-potentiated reward function.

Butorphan or 8 was injected intraperitoneally to adult male rats, and ICSS thresholds (A, D) and maximum rates of responding (B, E) were measured for 1 hr. Neither compound altered ICSS thresholds, but butorphan significantly decreased rates of responding at 4.0 mg/kg. In separate groups of rats, butorphan or 8 was injected, followed 15-min later by vehicle or cocaine (5.0 mg/kg, i.p.). ICSS thresholds were measured for 15-min post-dose and then for 1-hr post-cocaine (C, F). Cocaine significantly decreased ICSS thresholds on its own, and butorphan (C) but not 8 (F) dose-dependently blocked the reward-potentiating effects of cocaine. Data are expressed as 100 x post-treatment/pre-treatment thresholds or max rates of responding +SEM. **p<0.01, *p<0.05 compared to Veh + Veh; #p<0.01 comparing groups indicated with bracket. N=6 rats/treatment.
CONCLUSIONS
We have extended the SAR investigations of C-ring aminothiazolomorphinans. A surprising finding was that the methoxy derivatives were full agonists at the κ receptor, while the analogs with a hydroxyl group were partial agonists at the κ receptor. It has previously been suggested that high-efficacy KOR agonists with MOR receptor activity may be promising as candidates for treatment of cocaine intoxication.31 Results from the current study support this suggestion since butorphan, which has full KOR agonist activity, decreases the rewarding effects of cocaine in ICSS whereas 8, which has only partial KOR activity has no effect. Recent studies have demonstrated that KOR activation contributes to cocaine withdrawal-induced depressive-like states32 and can actually facilitate reinstatement of drug seeking and relapse.33 Taken together, these findings suggest that compounds with high KOR activity may be useful for blocking acute cocaine intoxication whereas partial agonists or antagonists at the KOR may be useful for treating effects of cocaine dependence, including withdrawal and relapse. Further in vivo studies of these aminothiazolomorphinan compounds are necessary to test this hypothesis.
EXPERIMENTAL SECTION
General information and materials
All reactions were magnetically stirred and monitored by analytical thin-layer chromatography (TLC) Silica gel 60 F254 plates using UV light to visualize the compounds. Column chromatography was carried out on SiliaFlash F60 (230–400 mesh, Silicycle). 1H and 13C NMR spectra were recorded on a Varian 300 MHz spectrometer using tetramethylsilane (TMS) as an internal reference. All target compounds were determined to be >95% pure by HPLC analysis, using a Varian Prostar HPLC apparatus equipped with a Varian Microsorb C18 100A analytical column and gradient solvent system of 0.1% trifluoroacetic acid in water and acetonitrile, detected at a wavelength of 254nM. Melting points were obtained using a Thomas-Hoover capillary melting point apparatus and are uncorrected. Reagents and solvents were purchased from commercial suppliers and used without further purification.
6,7-(2-N-Methyl)aminothiazolo-N-cyclopropylmethyl-morphin-3-ol (9)
To a cooled solution (0 °C) of 8 (40 mg, 0.109 mmol) and triethylamine (150 μL, 1.09 mmol) in CH2Cl2 (1 mL) was added acetic anhydride (51.5 μL, 0.545 mmol). The solution was stirred and allowed to warm to room temperature and stirring was continued for 2 hours. At this time, TLC analysis showed the starting material was consumed. The mixture was diluted with 28% NH4OH aqueous solution (3 mL) and was extracted with CH2Cl2 (3 x 10 mL). The organic extracts were combined, dried over Na2SO4, and concentrated. The crude yellow oil was purified by filtration through a silica plug with EtOAc/MeOH/Et3N (50/3/1) as eluent. To a stirred solution of this product (23 mg, 0.05mmol) in ethanol/H2O (400 μL, 2/1) was added solid sodium acetate (42.6 mg, 0.52 mmol) and the mixture was heated at 90 °C for 16 hours. The mixture was diluted with water (1 mL) and extracted with CH2Cl2 (3 x 5 mL). The organic layers were combined, dried over Na2SO4, and concentrated. The crude product was purified by filtration through a silica plug eluting with EtOAc/MeOH/Et3N (50/3/1) to provide the product 9 as a white solid in 39% yield (8 mg). mp (HCl salt)= 231°C (d) 1H NMR (300 MHz, CDCl3) δ 6.89 (d, J = 8.3 Hz, 1H), 6.79 (s, 1H), 6.60 (d, J = 8.2 Hz, 1H), 3.65 – 3.32 (m, 1H), 3.17 (d, J = 7.1 Hz, 0H), 3.04–2.70 (m, 2H), 2.71 – 2.27 (m, 6H), 2.12 (s, 4H), 1.96 (d, J = 6.0 Hz, 2H), 1.47 (d, J = 12.1 Hz, 1H), 1.34 – 1.06 (m, 2H), 1.01 – 0.81 (m, 1H), 0.53 (d, J = 5.9 Hz, 2H), 0.16 (d, J = 4.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 167.82, 158.27, 156.49, 142.07, 140.14, 129.22, 128.88, 121.84, 111.21, 110.90, 60.08, 55.22, 54.58, 45.14, 41.52, 41.42, 37.69, 37.30, 24.42, 23.74, 23.38, 9.55, 4.05.
6,7-(2-N-Acetyl)aminothiazolo-N-cyclopropylmethyl-3-methoxymorphinan (10)
To a cooled solution (0 °C) of 7 (HCl salt, 40 mg, 0.0816 mmol) and triethylamine (114 μL, 0.82 mmol) in CH2Cl2 (0.5 mL) was added acetic anhydride (38.7 μL, 0.41 mmol). The solution was stirred and allowed to warm to room temperature, and stirred for 3 hours. TLC analysis showed the starting material was consumed. The mixture was diluted with 28% aqueous NH4OH solution (3 mL) and was extracted with CH2Cl2 (3 x 10 mL). The organic extracts were combined, dried over Na2SO4, and concentrated. The crude yellow oil was purified by filtration through a silica plug with EtOAc/MeOH/Et3N (50/3/1) as eluent to provide pure 10 as a white solid (30.9 mg) in 89% yield. mp (2HCl salt) = 231°C (d) 1H NMR (300 MHz, CDCl3) δ 6.99 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 2.2 Hz, 1H), 6.64 (dd, J = 8.4, 2.5 Hz, 1H), 3.67 (s, 3H), 3.45 (dd, J = 19.5, 11.3 Hz, 2H), 2.98 (d, J = 18.5 Hz, 1H), 2.88 – 2.53 (m, 4H), 2.43 (dd, J = 15.3, 9.1 Hz, 4H), 2.20 (s, 3H), 2.03 (ddd, J = 15.5, 10.4, 4.3 Hz, 3H), 1.58 (d, J = 12.4 Hz, 1H), 1.34 – 1.03 (m, 1H), 1.02 – 0.77 (m, 1H), 0.53 (d, J = 8.0 Hz, 2H), 0.25 – 0.04 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 168.13, 156.50, 155.74, 141.90, 139.56, 129.25, 127.36, 121.64, 114.53, 111.79, 59.65, 58.91, 54.53, 45.29, 40.66, 37.26, 36.90, 24.32, 23.69, 23.30, 8.84, 8.44, 4.29, 4.13.
6,7-(2-N-Methyl)aminothiazolo-N-cyclopropylmethyl-3-methoxymorphinan (11)
To a stirred solution of 7 (161.3 mg, 0.423 mmol) and paraformaldehyde (63.6 mg, 2.12 mmol) in methanol (1.5 mL) was added sodium methoxide (114.5 mg, 2.12 mmol) at room temperature. The resulting mixture was heated to 65°C for 2 hours. The solution was then cooled to 0°C and sodium borohydride (80.5 mg, 2.12 mmol) was added slowly. The resulting mixture was then heated to 65°C for 4 hours. The reaction mixture was then cooled to room temperature and diluted with water (10 mL) and CH2Cl2 (10 mL). The aqueous layer was extracted with CH2Cl2 (3 x 10 mL), the extracts were combined, dried over Na2SO4, and concentrated to yield 11 in 84% yield (140 mg). The product was confirmed pure by NMR and HPLC analysis and used without further purification. MP (HCl salt) = 228°C (d) 1H NMR (300 MHz, CDCl3) δ 7.00 – 6.92 (m, 1H), 6.85 (d, J = 2.6 Hz, 1H), 6.64 (dd, J = 8.4, 2.6 Hz, 1H), 3.70 (s, 3H), 3.40 (t, J = 12.0 Hz, 2H), 3.05 – 2.90 (m, 1H), 2.86 (s, 3H), 2.80 – 2.63 (m, 2H), 2.63 – 2.20 (m, 5H), 2.20 – 1.81 (m, 2H), 1.55 (d, J = 12.4 Hz, 1H), 1.00 – 0.77 (m, 1H), 0.52 (dd, J = 8.1, 1.6 Hz, 2H), 0.22 – 0.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 163.66, 153.28, 139.15, 135.67, 124.27, 123.68, 109.87, 106.67, 105.49, 55.14, 50.30, 49.70, 40.21, 36.66, 36.52, 32.62, 32.54, 27.25, 19.80, 18.89, 4.61, −0.92, −1.00.
6,7-(2-N-Acetyl)aminothiazolo-N-cyclopropylmethyl-morphin-3-ol (12)
To a −78°C solution of 11 (100 mg, 0.253 mmol) in CH2Cl2 (5 mL) was added BBr3 (1M in CH2Cl2, 1.27 mL, 1.27 mmol). The resulting mixture was stirred for 2.5 hours and allowed to warm slowly to room temperature. The solvent was removed under reduced pressure. Methanol (5 mL) was added carefully and the mixture was concentrated again. This process was repeated two more times and the reaction mixture was dried in an Abderhalden drying pistol to yield 12 as an orange solid in 86% yield (135.5 mg). mp=239°C (d) 1H NMR (300 MHz, CD3OD) δ 7.10 (s, 1H), 6.79 (d, J = 2.2 Hz, 1H), 6.76 – 6.64 (m, 1H), 4.28 (d, J = 3.3 Hz, 1H), 3.66 – 3.34 (m, 3H), 3.31 (s, 3H), 3.28 – 3.09 (m, 3H), 2.80 (ddd, J = 25.4, 12.5, 6.6 Hz, 5H), 2.45 – 2.20 (m, 2H), 2.03 – 1.79 (m, 1H), 1.23 (d, J = 5.1 Hz, 1H), 0.79 (dd, J = 7.7, 3.4 Hz, 2H), 0.66 – 0.37 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 169.41, 157.25, 136.51, 132.02, 130.06, 123.97, 115.18, 113.57, 110.86, 58.99, 56.60, 45.86, 38.47, 37.94, 35.67, 32.17, 31.47, 23.35, 22.97, 5.85, 4.08, 3.93.
PHARMACOLOGICAL ASSAYS
Opioid Binding to the Human KOR, DOR, and MOR
Chinese hamster ovary (CHO) cells stably transfected with the human KOR (Dr. Lee-Yuan Liu-Chen; Temple University, Philadelphia, PA), the human DOR (Dr. Larry Toll; Torrey Pines Institute for Molecular Studies, Port St. Lucie, FL), and the human MOR (Dr. George Uhl; NIDA Intramural Program, Baltimore, MD) were used in the experiments. The cells were grown in 100-mm dishes in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin (10,000 U/mL) at 37°C in a 5% CO2 atmosphere. The affinity and selectivity of the compounds for MOR, KOR, and DOR were determined by incubating the membranes with radiolabeled ligands and 12 different concentrations of the compounds at 25°C in a final volume of 1 mL of 50 mM Tris–HCl, pH 7.5. Incubation times of 60 min were used for the μ-selective peptide [3H]DAMGO at a final concentration of 0.25 nM, and the κ-selective ligand [3H]U69,593 at a concentration of 0.4 nM. A 3-hr incubation was used with the δ-selective ligand [3H]naltrindole at a concentration of 0.2 nM.
[35S]GTPγS Binding Studies to Measure Receptor Coupling to G Proteins
Membranes from CHO cells stably expressing either the human KOR or MOR were used in the experiments. Cells were scraped from tissue culture plates and then centrifuged at 1000g for 10 min at 4°C. The cells were resuspended in phosphate-buffered saline, pH 7.4, containing 0.04% EDTA. After centrifugation at 1000g for 10 min at 4 °C, the cell pellet was resuspended in membrane buffer, which consisted of 50 mM Tris–HCl, 3 mM MgCl2, and 1 mM EGTA, pH 7.4. The membranes were homogenized with a Dounce homogenizer, followed by centrifugation at 40,000g for 20 min at 4 °C. The membrane pellet was resuspended in membrane buffer, and those transfected with the centrifugation step was repeated. The membranes were then resuspended in assay buffer, which consisted of 50 mM Tris–HCl, 3 mM MgCl2, 100 mM NaCl, and 0.2 mM EGTA, pH 7.4. The protein concentration was determined by the Bradford assay using bovine serum albumin as the standard. The membranes were frozen at −80°C until used.
CHO cell membranes expressing either the human KOR (15 μg of protein per tube) or MOR (7.5 μg of protein per tube) were incubated with 12 different concentrations of the compounds in assay buffer for 60 min at 30 °C in a final volume of 0.5 mL. The reaction mixture contained 3 μM GDP and 80 pmol of [35S]GTPγS. Basal activity was determined in the presence of 3 μM GDP and in the absence of an agonist, and nonspecific binding was determined in the presence of 10 μM unlabeled GTPγS. Then, the membranes were filtered onto glass fiber filters by vacuum filtration, followed by three washes with 3 mL of ice-cold 50 mM Tris–HCl, pH 7.5. Samples were counted in 2 mL of ScintiSafe 30% scintillation fluid. Data represent the percent of agonist-stimulation [35S]GTPγS binding over the basal activity, defined as [(specific binding/basal binding) x 100] − 100. All experiments were repeated at least three times and were performed in triplicate. To determine antagonist activity of a compound at the MOR, CHO membranes expressing the MOR were incubated with the compound in the presence of 200 nM of the agonist DAMGO. To determine antagonist activity of a compound at the KOR, CHO membranes expressing the KOR were incubated with the compound in the presence of 30 nM of the κ agonist U50,488.
Intracranial self-stimulation (ICSS)
Male Sprague Dawley rats (n = 24) were implanted with stainless steel monopolar electrodes (0.25 mm diameter; Plastics One, Roanoke, VA) aimed at the medial forebrain bundle at the level of the lateral hypothalamus (2.8 mm posterior to bregma, 1.7 mm lateral to midline, 7.8 mm below dura). After one week of recovery from surgery, rats were trained to respond for brain stimulation using a continuous reinforcement schedule (FR1) at 141 Hz, where each lever press earned a 500 ms train of square wave cathodal pulses (100 ms per pulse). Stimulation current was adjusted (final range: 140 – 240 XA) for each rat to the lowest value that would sustain a reliable rate of responding (average of 40 responses per 50 s). After the minimal effective current was found for each rat, it was kept constant throughout the remainder of training and testing. Rats were trained using the rate-frequency method, which allows for calculation of the threshold frequency (Hz) at which the stimulation first sustains responding on the operant lever. These procedures have been described in detail.30,34 Rats were trained until mean stimulation thresholds remained stable (± 10% for 4 consecutive days).
Four experiments were conducted using separate cohorts of rats (N=6/cohort). (1) A dose-effect function of butorphan (0.0, 0.25, 0.5, 1.0, 2.0, and 4.0 mg/kg, i.p. dissolved in 20% DMSO in water) on baseline brain stimulation reward. (2) A dose-effect function of butorphan (0.0, 0.5, 1.0, and 2.0 mg/kg, i.p.) on potentiation of brain stimulation reward by cocaine (Mallinckrodt; 5.0 mg/kg, i.p.). (3) A dose-effect function of 8 (1.0, 3.0, 10.0, and 30.0 mg/kg, i.p. in 0.9% saline) on baseline brain stimulation reward. (4) A dose-effect function of 8 (0.0, 3.0, 10.0, and 30.0 mg/kg, i.p.) on potentiation of brain stimulation reward by cocaine (5.0 mg/kg, i.p.). Drug doses were administered in increasing concentrations. For all experiments, three rate-frequency functions (3 × 15 min) were determined for each rat immediately before the initial drug injection. ICSS thresholds and maximum rates of responding for the second and third functions were averaged and served as baseline parameters. For experiments 1 and 3 (dose-effect function on baseline brain stimulation reward), rats received an injection of drug and 4 more 15-min rate frequency trials. For experiments 2 and 4 (effects of MCL compounds on cocaine reward), rats received an injection of butorphan or 8 and were placed back in the ICSS chambers for one 15- min rate frequency trial. Rats were then injected with cocaine and placed back in the ICSS chambers for 4 more 15-min rate frequency trials. Data are expressed as a percent of pre-drug baseline threshold values. Drug treatment days were separated by 2–3 non-drug days during which six 15-min rate frequency trials were performed to maintain baseline ICSS thresholds. The time course of drug effects on ICSS thresholds and maximum rates were analyzed using two-way (drug dose x time) ANOVAs with repeated measures on time. The effects of butorphan and 8 on cocaine-induced decreases in ICSS thresholds were measured with a one-way ANOVA. All significant effects and interactions were analyzed further using Tukey’s multiple comparison tests.
Acknowledgments
This work was supported by NIH Grants R01-DA14351 (J.L.N.) and K05-DA00360 (J.M.B.) W.L. was supported as a Visiting Scholar by the China Scholarship Council.
ABBREVIATIONS
- SAR
structure-activity relationship
- ICSS
intracranial self-stimulation
- KOR
κ-opioid receptor
- MOR
μ-opioid receptor
- DOR
δ-opioid receptor
- ATPM
aminothiazolo-N-cyclopropylmorphinan
- CHO
Chinese Hamster Ovaries
References
- 1.Johanson CE, Fischman MW. The Pharmacology of Cocaine Related to its Abuse. Pharmacol Rev. 1989;41:3–52. [PubMed] [Google Scholar]
- 2.Kuhar MJ, Ritz MC, Boja JW. The Dopamine Hypothesis of the Reinforcing Properties of Cocaine. Trends Neurosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 3.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine Receptors on Dopamine Transporters are Related to Self-Administration of Cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 4.Mendelson JH, Mello NK. Management of Cocaine Abuse and Dependence. N Engl J Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 5.Kleven M, Woolverton W. Effects of Continuous Infusions of SCH-23390 on Cocaine- or Food-Maintained Behavior in Rhesus Monkeys. Behav Pharmacol. 1990;1:365–373. doi: 10.1097/00008877-199000140-00010. [DOI] [PubMed] [Google Scholar]
- 6.Negus SS, Mello NK, Lamas X, Mendelson JH. Acute and Chronic Effects of Flupenthixol on the Discriminative Stimulus and Reinforcing, Effects of Cocaine in Rhesus Monkeys. J Pharmacol Exp Ther. 1996;278:879–890. [PubMed] [Google Scholar]
- 7.Aldrich JV, Vigil-Cruz SC. Narcotic Analgesics. In: Abraham D, editor. Burger’s Medicinal Chemistry and Drug Discovery. Vol. 6. John Wiley & Sons; New York: 2003. pp. 329–481. Chapter 7. [Google Scholar]
- 8.Shippenberg TS. The dynorphin/kappa opioid receptor system: a new target for the treatment of addiction and affective disorders? Neuropsychopharmacology. 2009;34:247. doi: 10.1038/npp.2008.165. [DOI] [PubMed] [Google Scholar]
- 9.Tejeda HA, Shippenberg TS, Henriksson R. The Dynorphin/κ-opioid Receptor System and its Role in Psychiatric Disorders. Cell Mol Life Sci. 2012;69:857–896. doi: 10.1007/s00018-011-0844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Neumeyer JL, Negus SS, Bidlack JM. Kappa Opioid Agonists as Targets for Pharmacotherapies in Cocaine Abuse. Pharm Acta Helv. 2000;74:337–344. doi: 10.1016/s0031-6865(99)00044-8. [DOI] [PubMed] [Google Scholar]; (b) Neumeyer JL, Bidlack JM, Zong R, Bakthavachalam V, Gao P, Cohen DJ, Negus SS, Mello NK. Synthesis and Opioid Receptor Affinity of Morphinan and Benzomorphan Derivatives: Mixed κ and μ Agonists/Antagonists as Potential Pharacotherapeutics for Cocaine Dependence. J Med Chem. 2000;42:114–122. doi: 10.1021/jm9903343. [DOI] [PubMed] [Google Scholar]
- 11.Schenk S, Partridge B, Shippenberg T. Effects of the Kappa-Opioid Receptor Agonist, U69593 on the Development of Sensitization and on the Maintenance of Cocaine Self-Administration. Neuropsychopharmacology. 2001;24:441–450. doi: 10.1016/S0893-133X(00)00190-1. [DOI] [PubMed] [Google Scholar]
- 12.Chefer VI, Moron JA, Hope B, Rea W, Shippenberg TS. Kappa-Opioid Receptor Activation Prevents Alterations in Mesocortical Dopamine Neurotransmission That Occur during Abstinence from Cocaine. Neuroscience. 2000;101:619–627. doi: 10.1016/s0306-4522(00)00417-6. [DOI] [PubMed] [Google Scholar]
- 13.Lutz P-E, Kieffer BL. The Multiple Facets of Opioid Receptor Function: Implications for Addiction. Curr Opin Neurobiol. doi: 10.1016/j.conb.2013.02.005. Online Early Access. Published online Feb. 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MA, Cole KT, Iordanou JC, Kerns DC, Newsome PC, Peitz GW, Schmidt KT. The Mu/Kappa Agonist Nalbuphine Attenuates Sensitization to the Behavioral Effects of Cocaine. Pharmacol, Biochem Behav. 2013;104:40–46. doi: 10.1016/j.pbb.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mello NK, Negus SS. Interactions between Kappa Opioid Agonists and Cocaine. Preclinical Studies. Ann N Y Acad Sci. 2000;909:104–132. doi: 10.1111/j.1749-6632.2000.tb06678.x. [DOI] [PubMed] [Google Scholar]
- 16.Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of Kappa Opioids on Cocaine Self-Administration by Rhesus Monkeys. J Pharmacol Exp Ther. 1997;282:44–55. [PubMed] [Google Scholar]
- 17.Bowen CA, Negus SS, Zong R, Neumeyer JL, Bidlack JM, Mello NK. Effects of Mixed-Action Kappa/Mu Opioids on Cocaine Self-Administration and Cocaine Discrimination by Rhesus Monkeys. Neuropsychopharmacol. 2003;28:1125–1139. doi: 10.1038/sj.npp.1300105. [DOI] [PubMed] [Google Scholar]
- 18.Zhang A, Xiong W, Hilbert JE, DeVita EK, Bidlack JM, Neumeyer JL. 2-Aminothiazole-Derived Opioids. Bioisosteric Replacement of Phenols. J Med Chem. 2004;47:1886–1888. doi: 10.1021/jm049978n. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Yan Z, Sromek A, Knapp BI, Scrimale T, Bidlack JM, Neumeyer JL. Aminothiazolomorphinans with Mixed κ and μ Opioid Activity. J Med Chem. 2011;54:1903–1913. doi: 10.1021/jm101542c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YJ, Tao YM, Li FY, Wang YH, Xu XJ, Chen J, Cao YL, Chi ZL, Neumeyer JL, Zhang A, Liu J-G. Pharmacological Characterization of ATPM [(−)-3-Amino-thiazolo- [5, 4-b]-N-cyclopropylmorphinan hydrochloride] a Novel Mixed κ-Agonist and μ-Agonist/Antagonist that Attenuates Morphine Antinociceptive Tolerance Heroin Self-Administration Behavior. J Pharmacol Exp Ther. 2009;329:306–313. doi: 10.1124/jpet.108.142802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang A, Yan Z-H, Neumeyer JL, Hilbert JE, DeVita EK, Bidlack JM. Further Modification of Aminothiazolomorphinans. Abstracts of Papers, 232nd ACS National Meeting; San Francisco, CA, United States. Sept. 10–14: 2006; p. MEDI-459. [Google Scholar]
- 22.Sun J-f, Wang Y-h, Lu G, Tao Y-m, Cheng Y, Chen J, Xu X-j, Chi Z-j, Neumeyer JL, Zhang A, Liu J-g. Effects of ATPM-ET a Novel κ Agonist with Partial μ Activity on Morphine-Induced Physical Dependence and Behavior Sensitization in Mice. Acta Pharmacol Sin. 2010;31:1547–1552. doi: 10.1038/aps.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang A, Li F, Ding C, Yao Q, Knapp BI, Bidlack JM, Neumeyer JL. Synthesis and Pharmacological Evaluation of 6,7-Indolo/Thiazolo-Morphinans-Further SAR of Levorphanol. J Med Chem. 2007;50:2747–2751. doi: 10.1021/jm0701674. [DOI] [PubMed] [Google Scholar]
- 24.Coop A, Rice KC. Direct and Simple Conversion of Codeine to Thebainone-A and Dihydrothebainone. Heterocycles. 1999;50:39–42. [Google Scholar]
- 25.Hupp CD, Neumeyer JL. Rapid Access to Morphinones: Removal of 4,5-Ether Bridge with Pd-Catalyzed Triflate Reduction. Tetrahedron Lett. 2010;51:2359–2361. doi: 10.1016/j.tetlet.2010.02.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narender T, Reddy KP, Madhur G. NaOAc-Mediated Selective Deprotection of Aromatic Acetates and its Application in the Synthesis of Natural Products. Synth Commun. 2009;39:1949–1956. [Google Scholar]
- 27.Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology. 2010;210:241–252. doi: 10.1007/s00213-010-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA., Jr The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry. 2008;64:982–988. doi: 10.1016/j.biopsych.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potter DN, Damez-Werno D, Carlezon WA, Jr, Cohen BM, Chartoff EH. Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry. 2011;70:744–753. doi: 10.1016/j.biopsych.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowen CA, Negus SS, Zong R, Neumeyer JL, Bidlack JM, Mello NK. Effects of mixed-action kappa/mu opioids on cocaine self-administration and cocaine discrimination by rhesus monkeys. Neuropsychopharmacology. 2003;28:1125–1139. doi: 10.1038/sj.npp.1300105. [DOI] [PubMed] [Google Scholar]
- 32.Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA. Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology. 2012;62:167–176. doi: 10.1016/j.neuropharm.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nature Protocols. 2007;2:2987– 2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]