

Abstract
Oridonin (1) has attracted considerable attention in recent years due to its unique and safe anticancer pharmacological profile. Nevertheless, it exhibits moderate to poor effects against highly aggressive cancers including triple-negative and drug-resistant breast cancer cells. Herein, we report the rational design and synthesis of novel dienone derivatives with an additional α,β-unsaturated ketone system diversely installed in the A-ring based on this class of natural scaffold that features dense functionalities and stereochemistry-rich frameworks. Efficient and regioselective enone construction strategies have been established. Meanwhile, a unique 3,7-rearrangement reaction was identified to furnish an unprecedented dienone scaffold. Intriguingly, these new analogues have been demonstrated to significantly induce apoptosis and inhibit colony formation with superior antitumor effects against aggressive and drug-resistant breast cancer cells in vitro and in vivo, while also exhibiting comparable or lower toxicity to normal human mammary epithelial cells in comparison with 1.
Keywords: oridonin dienone analogs, breast cancer, antiproliferation, apoptosis, drug-resistance
INTRODUCTION
Despite significant advances in the treatment of breast cancer, there are still limited therapeutic interventions available for aggressive and metastatic tumors.1 Particularly, triple-negative breast cancer (TNBC), characterized by lacking the receptors for estrogen and progesterone, and the Her2/neu receptor, remains an unmet medical need and represents an important clinical challenge because these tumors do not respond to endocrine therapy or other available targeted agents.2 In addition, resistance to chemotherapy is another major cause of the ultimate failure of breast cancer treatment.3 Therefore, effective anticancer agents with novel scaffolds or new mechanisms of action are urgently needed for the highly aggressive and drug-resistant breast cancer.
Natural products regularly serve synthetic chemists as a source of inspiration in their search for new molecular entities with unique pharmacological activity.4 Oridonin (1), isolated from the herb Isodon rubescens that is commonly used in Chinese traditional medicine and available over the counter in China, has attracted considerable attention in recent years due to its antitumor, antibacterial, antiviral, and anti-inflammatory activities.5 It has a unique and safe anticancer pharmacological profile. In China, oridonin injection was used alone or in combination with other drugs for the treatment of liver cancer6 and carcinoma of gastric cardia.7 Increasing studies have also demonstrated that 1 exerts extensive anti-neoplastic activities against various cultured human cancer cell lines through a versatile antiproliferative mechanism including regulating the cell cycle, apoptosis, and autophagy.8 While the antitumor activity of 1 was validated in estrogen receptor (ER)-positive breast cancer MCF-7 cells, it failed to reduce the growth of MDA-MB-231, a TNBC cell line, at the same dose range effective for MCF-7 cells,9a suggesting that 1 is ineffective against the growth of highly aggressive breast cancer cells. As part of our ongoing drug discovery program based on natural products, the anticancer profile of 1 intrigued us to take advantage of its unique scaffold as a basic template to synthesize novel natural product-like oridonin derivatives to develop safe and effective anticancer agents. Recently, efficient synthetic methods based on the oridonin scaffold were successfully established by our group to obtain a series of A-ring thiazole-fused or triazole-substituted derivatives with enhanced anticancer activity and improved solubility,10 indicating that A-ring modifications appear to be tolerable for yielding biologically interesting molecules.
Structurally, oridonin is a highly oxygenated 7,20-epoxy-ent-kaurane-type diterpenoid that features a densely functionalized and stereochemistry-rich framework including an exo-methylene cyclopentanone moiety in the D-ring and a 6-hydroxyl-7-hemiacetal group in the Bring (Figure 1). It is well known that the major structural determinant for anticancer activity of 1 is the presence of the α,β-unsaturated ketone (enone) system in the D-ring, and destruction of this enone system could counteract its anticancer activity.5a–b,11 Indeed, the enone system is a common and structurally important functionality which is widespread in various bioactive naturally occurring products such as eriocalyxin B12a,b and plakilactone C12c (Figure 1). Enones have also proven useful as a key pharmacophore existing in synthetic anticancer agents as exemplified by the oleanane tritepenoids CDDO-Me (Phase I/II human clinical trials, Figure 1)13 and brostallicin (Phase II human clinical trials, Figure 1).14 From a biochemical point of view, the α,β-unsaturated carbonyl group, as a Michael acceptor, is an electrophilic center susceptible to nucleophilic attack (Michael addition) by a sulfhydryl group of reduced glutathione or cysteine residues in proteins, leading to the adducts at the β-position.15a Thus, alkylation of crucial cysteine residues can result in a loss of function,15b or activation16 of the target proteins. For instance, eriocalyxin B, a naturally existing enone analogue of 1 isolated from Isodon eriocalyx, has demonstrated significant anticancer effects against various cancer cells likely through this mechanism.12b In addition, a number of α,β-unsaturated ketones have exhibited preferential reactivity toward thiols rather than amino or hydroxyl groups.17 Since thiols are absent in nucleic acids, this enone system may be free of mutagenicity and carcinogenicity caused by some alkylating agents used in cancer chemotherapy.18 Meanwhile, accumulating evidence also demonstrates that dienone compounds with double α,β-unsaturated ketone functionalities, such as curcumin19 (Figure 1), have a capability to undergo two successive alkylations at the β-positions by cellular thiols which interfere with biological cascades at multiple points. This is highly deleterious for malignant cells17a–b,20 and may also permit selective or greater toxicity to malignant cells versus the corresponding normal cells,21 consequently leading to an excellent tolerability in mammal models. Inspired by these advantages, we embarked on constructions of an additional enone functionality in the A-ring of oridonin, and envisioned that the resulting dienone derivatives with α,β-unsaturated ketone substructures present in both the A- and D-rings might display enhanced anticancer activity against drug-resistant ER-positive and triple-negative breast cancer cells relative to 1, while exhibiting less toxicity towards human normal mammary epithelial cells. In our previous work,10 the design of thiazole-fused derivatives was guided by the idea of incorporating nitrogen-containing heterocyclic ring into the A-ring to expand the core scaffold of 1. Different from the previous strategies, the present approach focuses on the diverse construction of the enone functionality at the A-ring within the core template of oridonin. Herein, we disclose our efficient synthetic approaches to generating new oridonin dienone analogues with the enone functionality diversely installed in the A-ring and their marked anti-breast cancer activity.
Figure 1.

Structures of oridonin and representative bioactive enone or dienone compounds
RESULTS AND DISCUSSION
Chemistry
Our synthetic effort was initiated from 1 due to its natural abundance and commercial availability. To date, there is little evidence in pursuit of chemical transformations based on the A-ring of oridonin, probably due to its structural complexity with multiple chemically reactive functionalities. Thus, the goal to diversely assemble an α,β-unsaturated ketone moiety into the tetracyclic ring system of 1 while keeping key functionalities intact posed a formidable synthetic challenge. In developing efficient synthetic strategies, we attempted to employ a protecting protocol to allow regioselective reactions among several functional groups with similar reactivity and avoid the usage of nucleophilic reagents, strong bases and acids, which are chemically reactive with the key functionalities of 1.
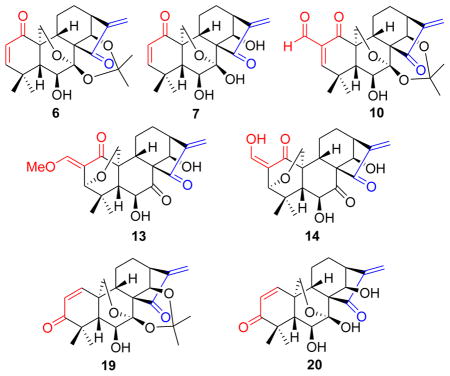
Our approach to synthesize oridonin analogues 6 and 7 with a 1-ketone-Δ2 (1-ketone-2-ene) moiety in the A-ring is outlined in Scheme 1. Oxidation of 1 with Jones reagent selectively afforded the 1-oxo-oridonin derivative 2,22 followed by treatment with 2,2-dimethoxypropane solely leading to the acetonide derivative 3 as a key building block. Although a few methods to introduce unsaturation adjacent to a carbonyl functionality have been developed over the years, the synthesis of α,β-unsaturated carbonyl compounds is often a tedious and sometimes challenging transformation.23a Initially, attempts to achieve a one-step synthesis of 6 from 3 based on reported methods using several oxidizing reagents such as IBX (o-iodoxybenzoic acid),23b 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)/p-toluenesulfonic acid (p-TsOH),23c and activated manganese dioxide (MnO2)23d proved unsuccessful. In addition, although a two-step method for the synthesis of 6 utilizing PhSeCl/LDA at -78 °C followed by selenoxide elimination has been reported,22 this reaction was very complex with several side products and unreacted 3 when the same procedure was carefully tested in our laboratory, and 6 was obtained in only 5% isolated yield. Therefore, a more reliable and efficient synthetic approach for 6 was deemed necessary, and has been achieved herein. Bromination of 2 with PyHBr3 in dry THF at 0 °C,10a followed by treatment with 2,2-dimethoxypropane, afforded 2-bromo oridonin derivative 5 as a mixture of α/β isomers in 63% yield over two steps, which further underwent a DBU-mediated elimination reaction to readily access 6 in 72% yield. It was noteworthy that the protection of the 7,14-dihydroxyl group as an acetonide was critical in this step; otherwise, 6 failed to be generated. Finally, the removal of the acetonide group in 6 with 5% HCl (aq.) successfully provided the dienone compound 7, which could also be viewed as an eriocalyxin B analogue with 14-hydroxyl functionality.
Scheme 1. Synthesis of the dienone analogues 6 and 7a.

a Reagents and conditions: a) Jones reagent, acetone, 2 h, rt, 82%; b) PyHBr3, THF, 0 °C, 3 h, 66%; (c) 2,2-dimethoxylpropane, cat. p-TsOH, acetone, 1 h, rt, 95%; (d) DBU, toluene, reflux, 4 h, 72%; (e) 5% HCl (aq), MeOH/CH2Cl2, 0.5 h, 89%; (f) PhSeCl, LDA, THF, −78 °C; (g) 10% H2O2 (aq), pyridine, CH2Cl2, rt, 5% (2 steps).
Since the electrophilic β-carbon of α,β-unsaturated ketone moiety might dictate the biological effects via nucleophilic addition, it is likely that chemically altering the reactivity of this carbon toward nucleophiles would have a profound effect on activity.24 Based on this hypothesis, it was expected that introduction of an electron-withdrawing substituent such as a formyl group at the α-position of the enone system in the A-ring would increase the electrophilicity of the β-carbon, henceforth enhancing the bioactivity, in a similar fashion to that of oleanane tritepenoids (CDDO)13 and punaglandins.25 Scheme 2 illustrates the synthesis of the dienone analogues 10 and 12 with an α-formyl enone moiety in the A-ring. It was initially intended to prepare 10 directly from 6 through a Baylis-Hillman reaction followed by oxidation of the resulting 2-hydroxymethyl group. Unfortunately, all attempts to obtain the 2-hydroxymethylated compound under several standard conditions26 failed to produce the desired products.27 Therefore, we pursued an alternative route to the α-formyl enone moiety through α-formylation of 3 followed by successive selenenylation and selenoxide elimination. Typically, installation of a formyl group at the α-position of the ketone can be realized by reaction with ethyl formate in the presence of strong base,13a,28a but 3 immediately decomposed upon addition of a strong base such as NaOMe and t-BuOK. Therefore, a circuitous strategy was employed to introduce α-formyl group. Treatment of 3 with N,N-dimethylformamide dimethyl acetal in refluxing toluene readily afforded the enamine derivative 8 in 60% yield, which was hydrolyzed with 5% HCl aqueous solution for 15 min leading to the 2-formyl derivative 9 in 83% yield.28b It should be noted that longer hydrolysis reaction time resulted in removal of the acetonide group to give 11. Selenenylation of 9 with PhSeCl in the presence of pyridine at RT followed by 30% H2O2-mediated oxidation and elimination successfully provided the desired analogue 10 with an α-formyl enone in the A-ring in 70% yield for two steps. Unexpectedly, the removal of the acetonide group in 10 with 5% HCl (aq.) in MeOH/CH2Cl2 failed to give the desired product 12. Instead, a 3,20-epoxy-ent-kaurane diterpenoid 13 with 2-exo-E-methoxymethylene moiety in the A-ring was obtained, the structure of which was unambiguously confirmed by the single crystal X-ray crystallographic analysis.29 Interestingly, when THF was used as the solvent, a similar 3,20-epoxy product 14 with 2-exo-Z-hydroxymethylene moiety was also found, and further treatment of the isolated 14 with 5% HCl aqueous solution in methanol afforded 13. These results suggested 14 was resulted from 3,7-rearrangement of 12, and subsequent enol etherification of 14 led to 13. Initially, we assumed that the rearrangement reaction was triggered by acid to result in the hydrolysis of 7-hemiacetal group to form a free 20-methylol group, which further underwent an intramolecular 1,4-conjugated Michael addition towards the unsaturated ketone moiety of the A-ring from the α-face followed by enolization or enol etherification leading to the 3,20-epoxy products 13 and 14. Based on this assumption, to avoid the presence of an acid, we attempted to use the 7,14-diol derivative 11 without a protecting group as the substrate to synthesize 12 through the sequential selenenylation and selenoxide elimination reactions in the same fashion. To our surprise, the 3,20-epoxy product 14 instead of 12 was obtained again under these conditions, although no acid was involved in this reaction. These results strongly indicated that 12 endowed with both α-formyl enone moiety and 7-hemiacetal group was unstable, and could automatically undergo 3,7-rearrangement reaction without the aid of acid, presumably owing to the increased electrophilicity of the β-carbon in the α-formyl enone system.
Scheme 2. Synthesis of the dienone analogues 10, 13 and 14a.

aReagents and conditions: a) DMF-DMA, toluene, reflux, 24 h, 60%; b) 5% HCl (aq), THF, rt, 10 min, 83%; c) PhSeCl, pyridine, CH2Cl2, 0 °C, 1 h; d) 30% H2O2 (aq.), CH2Cl2, rt, 0.5 h, 70% (two steps) for 10 from 9, 60% (two steps) for 14 from 11; e) 5% HCl (aq), MeOH/CH2Cl2, rt, 1 h, 80–82%; (f) 5% HCl (aq), THF, rt, 2 h, 80% for 14, 73% for 11.
Transposition of a functional group from one carbon to another often offers a wide degree of diversity and flexibility in natural product synthesis and related drug design.30a We initially considered the 1,3-enone transposition strategy in the A-ring via direct Wharton carbonyl transposition30b of 6 to generate Δ1-3-ketone (1-ene-3-ketone) analogues 19 and 20. Nevertheless, this approach was not feasible due to the harsh reaction conditions and the lack of regioselectivity in the enone formation. We thus developed an alternative and efficient synthetic approach in a controlled regioselective manner (Scheme 3). The synthesis of analogues 19 and 20 started with the protection of the 7,14-dihydroxyl group of 1 as an acetonide. The 1-hydroxyl group of the acetonide was then selectively activated as a mesylate 16, which further underwent an elimination reaction31 in the presence of Li2CO3 at 110 °C to provide the 1-ene analogue 17 in 84% yield.10b To introduce a hydroxyl group to the 3-position of the A-ring, we initiated a key allylic oxidation by the treatment of 17 with selenium dioxide32 in refluxing 1,4-dioxane to stereoselectively produce the 1-ene-3β-hydroxyl analogue 18 in an excellent yield;10b however, prolonged reaction time failed to give the enone product 19. Having completed the synthesis of 18, our attention was focused on the oxidation of the allylic alcohol. To our disappointment, neither activated MnO2 nor Dess-Martin reagent promoted this transformation. Finally, the goal was realized by using pyridinium dichromate (PDC) to furnish the 1-ene-3-ketone analog 19 in 80% yield, followed by the removal of the protecting group to provide the desired analogue 20 bearing a 1-ene-3-ketone moiety in the A-ring.
Scheme 3. Synthesis of the dienone analogues 19 and 20a.

aReagents and conditions: a) MeCH(OMe)2, cat. p-TsOH, acetone, rt, 2 h, 93%; b) MsCl, Et3N, CH2Cl2, rt, overnight, 80%; c) LiBr, Li2CO3, DMF, 110 °C, 2 h, 84%; d) SeO2, 1,4-dioxane, 110 °C, 16 h, 76%; e) PDC, CH2Cl2, rt, 4 h, 80%; f) 5% HCl (aq), MeOH, CH2Cl2, rt, 1 h, 85%.
In Vitro Antiproliferative Activity
With seven novel dienone analogues including 6, 7, 10, 13, 14, 19 and 20 in hand, their antiproliferative activities were evaluated against two breast cancer cell lines, MCF-7 (ER-positive) and MDA-MB-231 (triple-negative), with the data summarized in Table 1. 1 was also tested for comparison. The results showed that five 7,20-epoxy dienone analogues (6, 7, 10, 19 and 20) not only exhibited significantly improved antiproliferative activity relative to 1 against ER-positive breast cancer MCF-7 cells with IC50 values varying from low micromolar to submicromolar range (0.56 ± 0.31 μM ~ 3.48 ± 0.19 μM), but also displayed good growth inhibitory effects on triple-negative MDA-MB-231 cells with low micromolar IC50, for which 1 had only modest activity with an IC50 value of 28.0 ± 1.40 μM. For two 3,20-epoxy dienone compounds 13 and 14, no obvious antiproliferative activities were observed, indicating the biological importance of the oridonin core ring system.
Table 1.
Antiproliferative effects of oridonin and the dienone analogues against human breast cancer cell lines.
| ||
|---|---|---|
| Compounds | IC50 (μM)a
|
|
| MCF-7 | MDA-MB-231 | |
| 1 | 4.36 ± 1.41 | 28.0 ± 1.40 |
| 6 | 0.56 ± 0.31 | 3.49 ± 1.21 |
| 7 | 1.31 ± 0.25 | 2.23 ± 0.68 |
| 10 | 1.28 ± 0.47 | 3.46 ± 1.33 |
| 13 | 10.2 ± 3.07 | > 30b |
| 14 | > 30 | > 30 |
| 19 | 0.98 ± 0.19 | 5.6 ± 1.06 |
| 20 | 3.48 ± 2.16 | 9.39 ± 0.48 |
Breast cancer cell lines: MCF-7 and MDA-MB-231. Software: MasterPlex ReaderFit 2010, MiraiBio, Inc. Values are mean ± SE of three independent experiments.
If a specific compound is given a value > 30, it indicates that a specific IC50 cannot be calculated from the data points collected, meaning ‘no effect’.
In Vitro Growth Inhibitory Activity against Drug-Resistant Breast Cancer Cells
Resistance to chemotherapy is a major cause of the ultimate failure of breast cancer treatment. To investigate whether these dienone analogues are still effective on drug-resistant breast cancer cells, compounds 6, 7, 10 and 19 with potent antiproliferative effects against both MCF-7 and MDA-MB-231 cells were selected for further evaluation of growth inhibitory effects on ADR (adriamycin, a.k.a. doxorubicin)-resistant breast cancer cell MCF-7 clone (Figure 1S in Supporting Information). As shown in Figure 2, 1 displayed no growth inhibitory activity at concentrations from 1 μM to 10 μM with an IC50 value higher than 30 μM, while new compounds 6, 7, 10 and 19 were found to dose-dependently suppress the growth of MCF-7/ADR cells with IC50 values of 5.03 ± 1.91 μM, 5.82 ± 2.12 μM, 6.55 ± 0.96 μM, and 6.02 ± 1.28 μM, respectively (Table 2).
Figure 2.
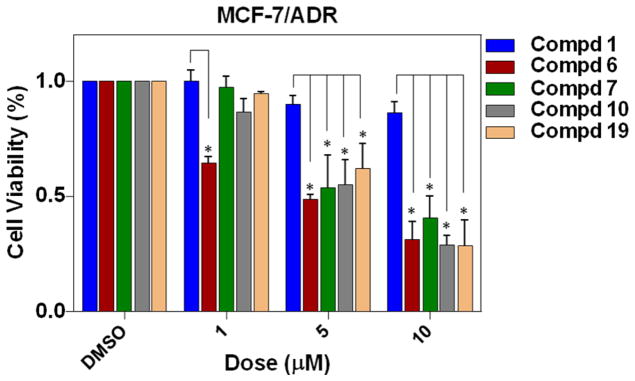
Growth inhibitory effects of compounds 1, 6, 7, 10 and 19 on drug-resistant breast cancer cells. MCF-7/ADR cells were treated with varying concentrations of drugs for 48 h. Values are mean ± SE of three independent experiments. Statistical significance was determined using Student’s t-test. *p < 0.05, in comparison with corresponding value of oridonin treatment at the same concentration.
Table 2.
Growth inhibitory effects of oridonin and the selected dienone analogues against drug-resistant breast cancer MCF-7/ADR cells.
| Compounds | IC50 (μM)a |
|---|---|
| 1 | >30 |
| 6 | 5.03 ± 1.91 |
| 7 | 5.82 ± 2.12 |
| 10 | 6.55 ± 0.96 |
| 19 | 6.02 ± 1.28 |
Breast cancer cell line: MCF-7/ADR. Software: MasterPlex ReaderFit 2010, MiraiBio, Inc. Values are mean ± SE of three independent experiments.
If a specific compound is given a value > 30, it indicates that a specific IC50 cannot be calculated from the data points collected, meaning ‘no effect’.
In Vitro Growth Inhibitory Activity on Human Normal Mammary Epithelial Cells (HMEC)
Selective toxicity for cancer, but not normal cells, is essential in the development of targeted cancer experimental therapeutics. To investigate whether the improved antiproliferative effects of analogs 6, 7, 10, 19 and 20 against breast cancer cells were attributed to the undesired cell toxicities, we further examined their inhibitory effects on the growth of HMEC, and 1 was also tested for comparison. As shown in Figure 3, all of these dienone analogues exhibited comparable or lower growth inhibitory activity againstHMEC cells at all tested concentrations, albeit displaying markedly enhanced anticancer activities against drug-resistant ER-positive MCF-7 and triple-negative MDA-MB-231 cancer cells when compared with 1. Particularly, analogue 19 displayed lower toxicity at 10 μM than oridonin (*p < 0.05), and the IC50 values of analogues 19 and 20 are much higher than that of oridonin (Table 3), indicating their lower toxicities to HMEC cells.
Figure 3.
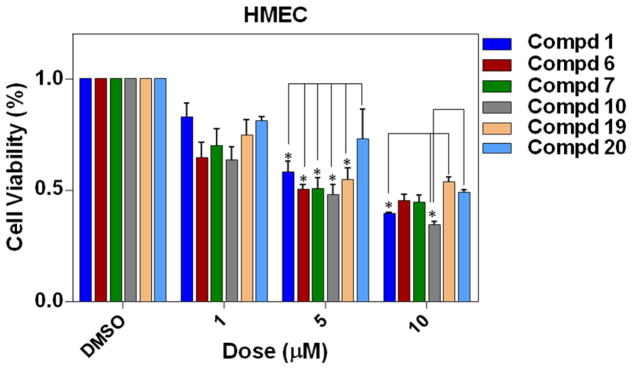
Effects of compounds 1, 6, 7, 10, 19 and 20 on normal human mammary epithelial cell (HMEC) proliferation. Human mammary epithelial cells (HMEC) were treated with varying concentrations of drugs for 24 h. Values are mean ± SE of three independent experiments. Statistical significance was determined using Student’s t-test. *p < 0.05, in comparison with corresponding value of oridonin treatment at the same concentration.
Table 3.
Effects of oridonin and the selected dienone analogues on normal human mammary epithelial cell (HMEC) proliferation.
| Compounds | IC50 (μM)a |
|---|---|
| 1 | 5.60 ± 0.38 |
| 6 | 5.11 ± 0.18 |
| 7 | 4.95 ± 0.63 |
| 10 | 4.51 ± 0.54 |
| 19 | 10.2 ± 0.24 |
| 20 | 10.1 ± 0.68 |
Cell line: HMEC. Software: MasterPlex ReaderFit 2010, MiraiBio, Inc. Values are mean ± SE of three independent experiments.
Compounds 10 and 19 Inhibited Colony Formation of Breast Cancer Cells
Considering their potent antiproliferative activities against MDA-MB-231 cells, two structually representative dienone analogues 10 (CYD0692) and 19 (CYD0686) were selected for colony formation assay. Both of these two compounds have demonstrated to inhibit the colony formation of highly invasive triple-negative breast cancer cells MDA-MB-231 as shown in Figure 4, and the results are consistent with their antiproliferative activity. Especially, the most promising compound 19 significantly blocked the colony formation of MDA-MB-231 cells at a submicromolar concentration.
Figure 4.

Inhibitory effects of compounds 10 and 19 on colony formation of breast cancer cells. Colony formation assay was performed to measure the capacity of MDA-MB-231 cells to form colonies. (A) 10 and 19 were used to treat the cells at various concentrations as indicated. Culture areas were scanned and digitally quantified with GelCount™ instrument. (B) Density data from scanning the stained culture areas were graphed as shown. Error bars represent standard deviations (p < 0.0001). Experiments were performed in triplicate, and the statistical significance was obtained with one-way ANOVA.
Compounds 10 and 19 Induced Apoptosis of Breast Cancer Cells
On the basis of their promising anti-proliferative effects and their potent activities in the colony formation assay, compounds 10 and 19 were selected for further mechanistic studies to determine whether the growth inhibition induced by them in human breast cancer cells was due to apoptosis. MDA-MB-231 cells were treated with vehicle alone as control and also with 10 or 19 at different concentrations (1.0 μM, 5.0 μM or 10 μM) for 24 h and stained with FITC-Annexin V and propidium iodide (PI). The percentages of apoptotic MDA-MB-231 cells were determined by flow cytometry. As shown in Figure 5, both compounds 10 and 19 displayed significant effects to induce apoptosis of MDA-MB-231 cells in a dose-dependent manner. The findings support that the apoptosis of MDA-MB-231 cells mediated by these two compounds contributes to their antiproliferative effects.
Figure 5.
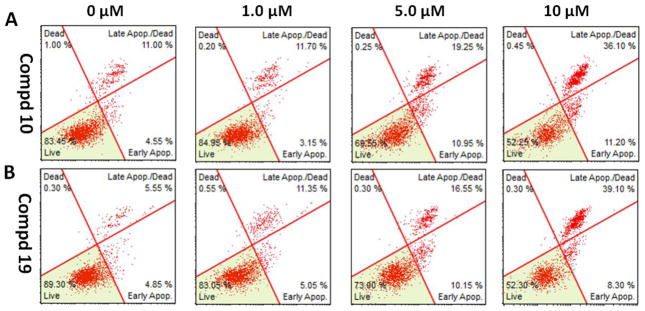
Induction of apoptosis on MDA-MB-231 cells by compounds 10 and 19. (A) Flow cytometry analysis of apoptotic MDA-MB-231 cells induced by 10 at different concentrations. (B) Flow cytometry analysis of apoptotic MDA-MB-231 cells induced by 19 at different concentrations. Cells were treated with vehicle, 10 or 19 at 1.0 μM, 5.0 μM, and 10 μM concentrations, respectively, for 24 h. The values are means ±SE of at least three independent experiments.
Compounds 10 and 19 Regulated Apoptotic Related Proteins
Previous studies have demonstrated that 1 induces apoptosis of cancer cells by modulating a series of transcription factors, protein kinases as well as pro- and/or anti-apoptotic proteins such as NF-kB,9 MAPK,33a Bax and Bcl-2.33b To elucidate the potential mechanisms contributing to apoptosis induction by the new derivatives 10 and 19, several proteins related to apoptosis were determined by Western blot assay. As shown in Figure 6, treatment of MDA-MB-231 cells with compounds 10 and 19, respectively, at low concentrations (2.5 μM-10 μM) led to the down-regulation of antiapoptotic protein Bcl-2 levels and the up-regulation of the pro-apoptotic protein Bax. In addition, they also induced a significant decrease of NF-κB (p65) protein expression, suggesting that NF-κB inhibition might contribute to the reduction of Bcl-2/Bax ratio. Meanwhile, compounds 10 and 19 also triggered PARP cleavage from its full-length form (116 kDa) to the cleaved form (25 kDa) as indicated by appearance of PARP fragments and activated caspase-3 in a dose-dependent manner, which might be either partially or totally responsible for the proteolytic cleavage of PARP. For comparison, exposure to high doses of 1 (10 μM-30 μM) also led to down-regulation of NF-κB (p65), Bcl-2, and PARP (116 kDa), and up-regulation of Bax and cleaved PARP (25 KDa); nevertheless, it did not activate caspase-3 cleavage from the concentrations of 10 μM to 30 μM. These preliminary results indicated that the dienone derivatives 10 and 19 induced the apoptosis in MDA-MB-231 cells likely by regulating unique apoptotic pathways. Other than apoptosis, oridonin has also been found to suppress tumor cell proliferation and induce cancer cell death though cell cycle arrest,9a,33b autophagy,9b and necrosis.33a Therefore, more extensive mechanism studies on the new dienone analogues are ongoing.
Figure 6.
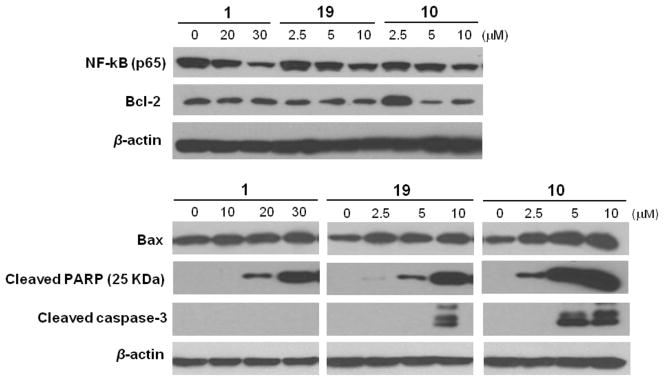
Western blot analysis of biological markers for apoptosis induction by compounds 10 and 19 and 1 in the MDA-MB-231 cells at different concentrations (48 h).
Compound 19 Suppressed Growth of Xenograft Tumors in Mice
In our in vivo studies, dienone analogue 19 was further evaluated for its antitumor activity in suppression of tumor growth in the triple-negative MDA-MB-231 xenograft model due to its potent antiproliferative and colony formation inhibitory effects in MDA-MB-231 cells as well as lower toxicity in HMEC cells. Meanwhile, compound 19 was selected for further in vivo efficacy studies because of its good in vitro dose-response relationship. As shown in Figure 7A, compound 19 at 5.0 mg/kg was much more efficacious in suppressing xenograft tumor growth as compared to oridonin at the same dosage (p < 0.0001). Meanwhile, compound 19 was also found to be well tolerated during experiments and showed no significant loss of body weight (Figure 7B). These results suggest that compound 19 is a promising anticancer drug candidate with potent antitumor activity and good tolerability for further clinical development.
Figure 7.
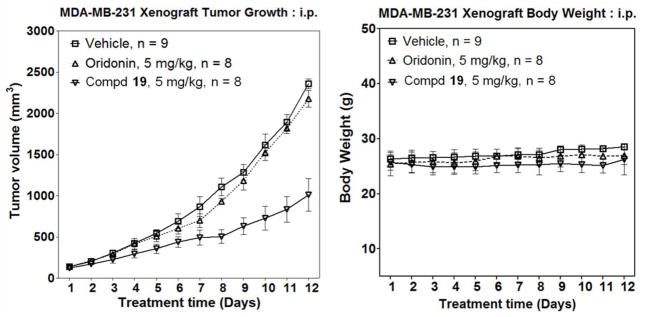
In vivo therapeutic efficacy of compound 19 compared to 1 in inhibiting growth of xenograft tumors (triple-negative breast cancer cell line MDA-MB-231) in mice (i.p.) at 5 mg/kg: (A) average tumor size changes; (B) average body weight changes. Values are mean ± SE of three independent experiments. Statistical significance was determined using one-way ANOVA (p < 0.0001).
CONCLUSIONS
In summary, our efficient synthetic methodologies to access several kinds of oridonin analogues with diverse enone functionality presented in the A-ring have been achieved in moderate to good yields through regioselective enone construction strategies starting from oridonin. A key α-bromination/HBr elimination sequence was applied to introduce a double bond to the carbonyl functionality to achieve analogues 6 and 7. The α-formyl enone analogue 10 was prepared through the hydrolysis of enamine 8 followed by sequential selenenylation and selenoxide elimination, while analogue 12 with both an α-formyl enone system and a 7-hemiacetal group proven to be unstable, and spontaneously underwent a novel 3,7-rearrangement reaction to give unprecedented 3,20-epoxy products 13 and 14. Different from the conventional protocols, the goal to generate the 1-ene-3-ketone analogues 19 and 20 was realized through 1-ene functionality formation with subsequent successive oxidations of allylic methylene. Intriguingly, dienone analogues 6, 7, 10 and 19 have demonstrated enhanced antiproliferative effects against ER-positive MCF-7 and TNBC MDA-MB-231 cells as well as drug-resistant MCF-7/ADR clones, while exhibiting comparable or lower toxicity to normal cells relative to 1. In our preliminary mechanism studies, dienone analogues 10 and 19 were found to significantly inhibit colony formation and induce apoptosis of MDA-MB-231 cells in a dose-dependent manner through regulating a series of apoptotic related proteins. Meanwhile, analogue 19 has demonstrated more efficacious antitumor activity than oridonin and excellent tolerability in MDA-MB-231 xenograft-bearing nude mice, indicating the potential of these new dienone analogues for the treatment of highly aggressive triple negative and drug-resistant breast cancers.
EXPERIMENTAL SECTION
General
All commercially available starting materials and solvents were reagent grade, and used without further purification. Oridonin was purchased from Shanxi Huike, China. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Brucker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer or Brucker-300 (1H, 300 MHz; 13C, 75 MHz). 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and uncorrected. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 30% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. All biologically evaluated compounds are > 96% pure.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11aS,11bS)-7-hydroxy-5,5,8,8-tetramethyl-15-methylene-3,3a,7,7a,8,11b-hexahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxine-11,14(2H)-dione (6)
To a solution of 4 (80 mg, 0.18 mmol) in acetone (4 mL) was added p-TsOH (5 mg) and 2,2-dimethoxypropane (0.32 mL) at rt. The resulting mixture was stirred at rt for 2 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to afford compound 5 (83 mg, 95%) as a colorless gel. To a solution of 5 (50 mg, 0.10 mmol) in toluene (5 mL) was added DBU (20 mg, 0.13 mmol) at rt. The resulting mixture was stirred at 110 °C for 4 h, and diluted with water and extracted with EtOAc. The organic extract was washed with 3 N HCl aqueous solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue, which was purified using preparative TLC developed by 30% EtOAc in hexane to afford the desired product 6 as a colorless amorphous gel (30 mg, 72%). [α]25D −54 (c 0.10, CH2Cl2); HPLC purity 98.7% (tR = 19.78 min); 1H NMR (600 MHz, CDCl3) δ 6.80 (d, 1H, J = 9.6 Hz), 6.17 (s, 1H), 5.84 (d, 1H, J = 10.2 Hz), 5.59 (s, 1H), 5.41 (d, 1H, J = 12.0 Hz), 4.88 (s, 1H), 4.24 (dd, 1H, J = 1.2 Hz, 10.2 Hz), 4.08 (m, 2H), 3.08 (d, 1H, J = 9.0 Hz), 2.53 (m, 1H), 2.00 (m, 3H), 1.67 (s, 3H), 1.62 (m, 3H), 1.42 (s, 3H), 1.36 (s, 3H), 1.27 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.7, 196.5, 162.1, 150.4, 126.6, 120.8, 101.3, 95.7, 71.7, 69.9, 65.1, 56.5, 55.9, 47.4, 45.8, 40.1, 35.9, 30.4, 30.2, 30.1, 25.4, 25.0, 19.3. HRMS Calcd for C23H29O6: [M + H]+ 401.1959; found 401.1957.
Synthesis of (4aR,5S,6S,6aR,9S,11aS,11bS,14R)-5,6,14-trihydroxy-4,4-dimethyl-8-methylene-4,4a,5,6,9,10,11,11a-octahydro-1H-6,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalene-1,7(8H)-dione (7)
To a solution of 6 (8.0 mg, 0.02 mmol) in a mixture of MeOH (2 mL) and CH2Cl2 (0.5 mL) was added 5% HCl aqueous solution (0.5 mL) at rt. The resulting mixture was stirred at rt for 4 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 50% EtOAc in hexane to afford the desired product 7 as a colorless amorphous gel (6.5 mg, 89%). [α]25D −56 (c 0.10, CH2Cl2); HPLC purity 99.0% (tR = 16.02 min); 1H NMR (600 MHz, CDCl3/CD3OD = 5:1) δ 6.88 (d, 1H, J = 9.6 Hz), 6.21 (s, 1H), 5.87 (d, 1H, J = 10.2 Hz), 5.63 (s, 1H), 4.97 (s, 1H), 4.27 (m, 2H), 4.06 (dd, 1H, J = 1.2 Hz, 10.2 Hz), 3.96 (d, 1H, J = 8.4 Hz), 3.04 (d, 1H, J = 9.6 Hz), 2.52 (m, 1H), 2.10 (m, 2H), 2.03 (d, 1H, J = 8.4 Hz), 1.62 (m, 1H), 1.48 (m, 1H), 1.39 (s, 3H), 1.27 (s, 3H); 13C NMR (150 MHz, CDCl3/CD3OD = 5:1) δ 206.7, 197.3, 161.8, 150.8, 126.8, 121.2, 97.9, 72.3, 72.2, 65.2, 61.4, 56.8, 50.0, 45.9, 42.7, 35.7, 29.8, 29.4, 23.9, 18.9; HRMS Calcd for C20H25O6: [M + H]+ 361.1646; found 361.1544.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11aS,11bS,Z)-10-((dimethylamino)methylene)-7-hydroxy-5,5,8,8-tetramethyl-15-methyleneoctahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxine-11,14(2H)-dione (8)
To a solution of 2 (250 mg, 0.68 mmol) in acetone (10 mL) was added p-TsOH (20 mg) and 2,2-dimethoxypropane (1.0 mL) at rt. The resulting mixture was stirred at rt for 2 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to afford compound 3 as a colorless gel (263 mg, 95%). To a solution of 3 (230 mg, 0.57 mmol) in DMF (4 mL) was added DMF-DMA (136 mg, 1.14 mmol) at rt. The resulting mixture was refluxed at 110 °C for 36 h. The solvent was then removed under vacuum to give a brown oily residue, which was further purified using preparative TLC developed by 66% EtOAc in hexane to afford the desired product 8 as a brown gel (156 mg, 60%). 1H NMR (600 MHz, CDCl3) δ 7.42 (s, 1H), 6.14 (s, 1H), 5.55 (s, 1H), 5.20 (d, 1H, J = 12.0 Hz), 4.87 (s, 1H), 4.31 (d, 1H, J = 10.2 Hz), 4.04 (d, 1H, J = 10.2 Hz), 3.87 (m, 1H), 3.07 (s, 6H), 3.04 (d, 1H, J = 9.6 Hz), 2.47 (m, 3H), 1.97 (m, 2H), 1.66 (s, 3H), 1.62 (m, 1H), 1.56 (m, 2H), 1.34 (s, 3H), 1.23 (s, 3H), 1.00 (s, 3H); HRMS Calcd for C26H36NO6: [M + H]+ 458.2537; found 458.2541.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11aS,11bS,Z)-7-hydroxy-10-(hydroxymethylene)-5,5,8,8-tetramethyl-15-methyleneoctahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxine-11,14(2H)-dione (9)
To a solution of 8 (200 mg, 0.43 mmol) in THF (5 mL) was added 5% HCl aqueous solution (0.5 mL) at rt. The resulting mixture was stirred at rt for 15 min. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was further purified using preparative TLC developed by 60% EtOAc in hexane to afford the desired product 9 (100 mg, 83%) as a pale pink gel. 1H NMR (300 MHz, CDCl3) δ 14.72 (d, 1H, J = 3.3 Hz), 8.39 (s, 1H), 6.19 (s, 1H), 5.60 (s, 1H), 5.29 (d, 1H, J = 12.0 Hz), 4.90 (s, 1H), 4.30 (dd, 1H, J = 1.2 Hz, 9.9 Hz), 4.09 (dd, 1H, J = 0.9 Hz, 9.9 Hz), 3.92 (m, 1H), 3.09 (d, 1H, J = 9.6 Hz), 2.55 (m, 1H), 2.29 (d, 1H, J = 15.0 Hz), 2.05 (m, 3H), 1.84 (m, 1H), 1.67 (s, 3H), 1.60 (m, 2H), 1.37 (s, 3H), 1.29 (s, 3H), 1.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 204.6, 185.4, 184.8, 150.4, 120.7, 109.2, 101.2, 95.7, 71.6, 70.0, 64.4, 58.1, 56.0, 48.3, 43.7, 40.1, 39.9, 33.2, 30.5, 30.3, 30.0, 25.3, 20.6, 19.8; HRMS Calcd for C24H31O7: [M + H]+ 431.2064; found 431.2063.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11aS,11bS)-7-hydroxy-5,5,8,8-tetramethyl-15-methylene-11,14-dioxo-2,3,3a,7,7a,8,11,11b-octahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxine-10-carbaldehyde (10)
To a stirring solution of phenylselenyl chloride (33.6 mg, 0.175 mmol) in CH2Cl2 (3 mL) at 0 °C was added pyridine (0.017 mL, 0.208 mmol). The solution was stirred for 45 min, and then a solution of α-keto aldehyde 9 (60 mg, 0.139 mmol) in CH2Cl2 (2 mL) was added. The mixture was stirred at 0 °C for 15 min and at rt for 45 min. It was then extracted twice with 1 N HCl (aq.). The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was further purified using the preparative TLC developed by hexane/EtOAc (1:1) to afford the selenide as a yellow gel (60.0 mg, 74%). To a stirring solution of the above selenide (60.0 mg, 0.102 mmol) in CH2Cl2 (5.8 mL) was added 35% H2O2 (aq.) solution (0.10 mL, 1.2 mmol). The mixture was vigorously stirred for 5 min, followed by the addition of another portion of 35% H2O2 (aq.) solution (0.10 mL, 1.2 mmol) with vigorous stirring for another 5 min. The reaction mixture was then extracted twice with water. The extract was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was further purified using the preparative TLC developed by hexane/EtOAc (1:1) to afford 10 (43 mg, 97%) as a pale yellow gel. [α]25D −102 (c 0.10, CH2Cl2); HPLC purity 98.1% (tR = 18.33 min); 1H NMR (600 MHz, CDCl3) δ 9.83 (s, 1H), 7.59 (s, 1H), 6.18 (s, 1H), 5.61 (s, 1H), 5.42 (d, 1H, J = 12.6 Hz), 4.89 (s, 1H), 4.33 (dd, 1H, J = 1.2 Hz, 10.2 Hz), 4.09 (m, 2H), 3.10 (d, 1H, J = 9.0 Hz), 2.56 (m, 1H), 2.06 (m, 2H), 2.00 (d, 1H, J = 8.4 Hz), 1.67 (s, 3H), 1.56 (m, 3H), 1.52 (s, 3H), 1.36 (s, 3H), 1.32 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.5, 195.2, 188.0, 168.4, 150.0, 133.0, 121.3, 101.5, 95.8, 71.3, 69.8, 64.9, 56.1, 55.7, 47.0, 46.5, 40.0, 36.2, 30.0, 29.7, 25.3, 24.2, 19.0; HRMS Calcd for C24H29O7: [M + H]+ 429.1908; found 429.1897.
Synthesis of (3S,4aR,5S,6aR,9S,11aS,11bS,14R,E)-5,14-dihydroxy-2-(methoxymethylene)-4,4-dimethyl-8-methyleneoctahydro-1H-3,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalene-1,6,7(2H,8H)-trione (13)
To a solution of 10 (5 mg, 0.011 mmol) in a mixture of MeOH (2 mL) and CH2Cl2 (0.5 mL) was added 5% HCl aqueous solution (0.2 mL) at rt. The resulting mixture was stirred at rt for 2 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by EtOAc to afford the desired product 13 as a colorless amorphous gel (3.5 mg, 78%). [α]25D −110 (c 0.10, CH2Cl2); HPLC purity 98.3% (tR = 14.58 min); 1H NMR (300 MHz, CDCl3) δ 7.59 (s, 1H), 6.18 (s, 1H), 5.47 (s, 1H), 4.67 (m, 2H), 4.43 (d, 1H, J = 0.9 Hz), 4.33 (s, 1H), 4.22 (m, 1H), 3.94 (s, 3H), 3.91 (m, 1H), 3.09 (m, 1H), 2.92 (m, 1H), 1.62 (m, 3H), 1.57 (m, 1H), 1.52 (s, 3H), 0.99 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 205.5, 201.4, 196.7, 156.3, 146.0, 118.6, 115.4, 75.1, 74.2, 71.7, 66.3, 62.1, 61.5, 51.7, 51.0, 45.3, 42.0, 38.2, 30.9, 28.5, 21.8, 20.1; HRMS Calcd for C22H27O7: [M + H]+ 403.1751; found 403.1768.
Synthesis of (3S,4aR,5S,6aR,9S,11aS,11bS,14R,Z)-5,14-dihydroxy-2-(hydroxymethylene)-4,4-dimethyl-8-methyleneoctahydro-1H-3,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalene-1,6,7(2H,8H)-trione (14)
To a solution of 10 (15 mg, 0.035 mmol) in THF (2 mL) was added 5% HCl (aq.) solution (0.3 mL) at rt. The resulting mixture was stirred at rt for 4 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 5% methanol in dichloromethane to afford the desired product 14 as a pale pink amorphous gel (11 mg, 80%). [α]25D −104 (c 0.1, CH2Cl2); HPLC purity 96.6% (tR = 4.47 min); 1H NMR (300 MHz, CDCl3) δ 7.99 (br s, 1H), 7.13 (s, 1H), 6.26 (s, 1H), 5.49 (s, 1H), 4.61 (d, 1H, J = 6.0 Hz), 4.54 (d, 1H, J = 4.5 Hz), 4.47 (s, 1H), 3.83 (m, 4H), 3.11 (d, 1H, J = 1.2 Hz), 3.05 (d, 1H, J = 3.9 Hz), 1.89 (m, 1H), 1.75 (m, 2H), 1.57 (m, 2H), 1.48 (s, 3H), 0.96 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 205.1, 200.4, 199.4, 160.1, 144.8, 120.3, 113.2, 71.6, 65.8, 60.9, 51.4, 50.6, 45.6, 41.2, 38.7, 31.1, 29.6 (2C), 22.5, 20.0. HRMS Calcd for C21H25O7: [M + H]+ 389.1595; found 389.1591.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11S,11aS,11bS)-7-hydroxy-5,5,8,8-tetramethyl-15-methylene-14-oxodecahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxin-11-yl methanesulfonate (16)
To a solution of oridonin (500 mg, 1.36 mmol) in acetone (20 mL) was added p-TsOH (20 mg) and 2,2-dimethoxypropane (3.0 mL) at rt. The resulting mixture was stirred at rt for 2 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to afford compound 15 as a colorless gel (520 mg, 93%); To a solution of 15 (277 mg, 0.68 mmol) in dichloromethane was added Et3N (138 mg, 1.37 mmol) and MsCl (94 mg, 0.82 mmol) dropwise at 0 °C. The mixture was stirred at rt overnight, diluted with water, and extracted with dichloromethane. The organic extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The crude residue was further purified by silica gel column; elution with 50% EtOAc in hexane afforded the desired product 16 as a colorless amorphous gel (264 mg, 80%). 1H NMR (600 MHz, CDCl3) δ 6.16 (s, 1H), 5.82 (d, 1H, J = 12.0 Hz), 5.58 (s, 1H), 4.76 (dd, 1H, J = 6.0 Hz, 12.0 Hz), 4.14 (d, 1H, J = 4.2 Hz), 3.92 (m, 1H), 3.07 (d, 1H, J = 9.0 Hz), 2.99 (s, 3H), 2.51 (m, 1H), 2.05 (m, 1H), 1.89 (m, 2H), 1.77 (m, 3H), 1.63 (s, 3H), 1.52 (m, 1H), 1.38 (d, 1H, J = 7.2 Hz), 1.33 (s, 3H), 1.19 (s, 3H), 1.18 (s, 3H), 1.16 (d, 1H, J = 7.2 Hz); 13C NMR (150 MHz, CDCl3) δ 205.1, 150.4, 120.5, 101.0, 94.6, 84.7, 72.9, 69.9, 62.3, 59.5, 55.9, 50.0, 40.6, 40.1, 38.2, 33.2, 33.0, 31.5, 30.1 (2C), 26.4, 25.4, 22.4, 18.8; HRMS Calcd for C24H36O8S: [M+H]+ 483.2047; found 483.2052.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aR,11aR,11bS)-7-hydroxy-5,5,8,8-tetramethyl-15-methylene-2,3,3a,7,7a,8,9,11b-octahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxin-14-one (17)
To a solution of 16 (34 mg, 0.07 mmol) in DMF (5 mL) was added LiBr (18 mg, 0.21 mmol) and Li2CO3 (15 mg, 0.21 mmol) at rt. The resulting mixture was stirred at 115 °C for 2 h. The reaction mixture was then diluted with water and extracted with EtOAc. The organic extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The crude residue was further purified by silica gel column; elution with 25% EtOAc in hexane afforded the desired product 17 as a colorless powder (25 mg, 84%). HPLC purity 99.8% (tR = 18.30 min). 1H NMR (600 MHz, CDCl3) δ 6.16 (s, 1H), 5.77 (m, 1H), 5.56 (s, 1H), 5.41 (d, 1H, J = 12.0 Hz), 5.19 (dd, 1H, J = 3.0 Hz, 10.2 Hz), 4.82 (s, 1H), 3.99 (d, 1H, J = 10.2 Hz), 3.90 (dd, 1H, J = 8.4 Hz, 12.0 Hz), 3.81 (d, 1H, J = 9.6 Hz), 3.06 (d, 1H, J = 9.0 Hz), 2.53 (m, 1H), 1.95 (d, 1H, J = 17.4 Hz), 1.76 (m, 4H), 1.65 (s, 3H), 1.56 (m, 1H), 1.50 (d, 1H, J = 8.4 Hz), 1.35 (s, 3H), 1.18 (s, 3H), 1.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.5, 150.5, 130.3, 124.1, 120.4, 101.2, 95.4, 72.0, 70.1, 64.8, 57.9, 56.3, 49.1, 41.1, 40.3, 38.1, 32.2, 31.1, 30.3, 30.1, 25.5, 22.1, 17.3; HRMS Calcd for C23H31O5: [M + H]+ 387.2166; found 387.2169.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aS,9S,11aR,11bS)-7,9-dihydroxy-5,5,8,8-tetramethyl-15-methylene-2,3,3a,7,7a,8,9,11b-octahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxin-14-one (18)
A mixture of 17 (20 mg, 0.05 mmol) and SeO2 (16 mg, 0.15 mmol) in 1,4-dioxane (4 mL) was stirred at 100 °C for 16 h. The reaction mixture was then filtered, and the filtrate was diluted with water and extracted with dichloromethane. The extract was washed with brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 50% EtOAc in hexane to afford the desired product 18 as a colorless amorphous gel (16 mg, 76%). HPLC purity 99.7% (tR = 17.31 min); 1H NMR (600 MHz, CDCl3) δ 6.16 (s, 1H), 6.00 (dd, 1H, J = 6.0 Hz, 10.2 Hz), 5.56 (s, 1H), 5.42 (m, 2H), 4.82 (s, 1H), 3.94 (m, 2H), 3.84 (d, 1H, J = 9.6 Hz), 3.06 (d, 1H, J = 9.6 Hz), 2.53 (m, 1H), 1.87 (d, 1H, J = 9.0 Hz), 1.82 (m, 2H), 1.71 (m, 2H), 1.65 (s, 3H), 1.57 (m, 1H), 1.35 (s, 1H), 1.24 (s, 3H), 1.01 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.1, 150.3, 130.9, 128.6, 120.6, 101.2, 95.5, 72.6, 71.5, 70.0, 64.5, 56.1, 51.0, 48.8, 40.3, 38.2, 36.7, 30.2, 30.1, 25.9, 25.4, 21.9, 17.3; HRMS Calcd for C23H31O6: [M + H]+ 403.2115; found 403.2118.
Synthesis of (3S,3aR,3a1R,6aR,7S,7aS,11aR,11bS)-7-hydroxy-5,5,8,8-tetramethyl-15-methylene-3,3a,7,7a,8,11b-hexahydro-1H-6a,11a-(epoxymethano)-3,3a1-ethanophenanthro[1,10-de][1,3]dioxine-9,14(2H)-dione (19)
To a solution of 18 (10 mg, 0.025 mmol) in dichloromethane (2 mL) was added PDC (11.2 mg, 0.03 mmol) at rt. The resulting mixture was stirred at rt for 4 h. The reaction mixture was then filtered, and the filtrate was diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 50% EtOAc in hexane to afford the desired product 19 as a colorless amorphous gel (8.0 mg, 80%). [α]25D −100 (c 0.10, CH2Cl2); HPLC purity 97.5% (tR = 18.62 min); 1H NMR (600 MHz, CDCl3) δ 6.29 (d, 1H, J = 10.2 Hz), 6.19 (s, 1H), 6.00 (d, 1H, J = 10.2 Hz), 5.60 (s, 1H), 5.54 (d, 1H, J = 12.0 Hz), 4.84 (s, 1H), 4.16 (d, 1H, J = 10.2 Hz), 4.07 (m, 1H), 4.01 (d, 1H, J = 10.2 Hz), 3.10 (d, 1H, J = 8.4 Hz), 2.58 (m, 1H), 1.93 (d, 1H, J = 7.8 Hz), 1.88 (m, 2H), 1.76 (m, 1H), 1.66 (s, 3H), 1.60 (m, 2H), 1.37 (s, 3H), 1.36 (s, 3H), 1.27 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.1, 203.2, 149.9, 142.6, 129.8, 121.2, 101.5, 95.4, 70.9, 69.8, 64.1, 56.2, 55.7, 48.3, 44.6, 40.1, 38.8, 30.1, 29.9, 25.4, 23.9, 22.4, 17.1; HRMS Calcd for C23H29O6: [M + H]+ 401.1959; found 361.1962.
Synthesis of (4aS,5S,6S,6aR,9S,11aS,11bR,14R)-5,6,14-trihydroxy-4,4-dimethyl-8-methylene-4,4a,5,6,9,10,11,11a-octahydro-3H-6,11b-(epoxymethano)-6a,9-methanocyclohepta[a]naphthalene-3,7(8H)-dione (20)
To a solution of 19 (15 mg, 0.037 mmol) in a mixture of MeOH (2 mL) and CH2Cl2 (0.5 mL) was added 5% HCl aqueous solution (0.5 mL) at rt. The resulting mixture was stirred at rt for 4 h. The reaction mixture was then diluted with water and extracted with dichloromethane. The extract was washed with saturated NaHCO3 (aq.) solution and brine, dried over anhydrous Na2SO4, filtered, and evaporated to give an oily residue. The residue was purified using preparative TLC developed by 66% EtOAc in hexane to afford the desired product 20 as a colorless amorphous gel (11.5 mg, 85%). [α]25D −128 (c 0.10, CH2Cl2); HPLC purity 98.2% (tR = 14.87 min); 1H NMR (600 MHz, CDCl3) δ 6.31 (d, 1H, J = 10.2 Hz), 6.22 (s, 1H), 6.13 (d, 1H, J = 11.4 Hz), 6.02 (d, 1H, J = 10.8 Hz), 5.63 (s, 1H), 4.92 (s, 1H), 4.17 (d, 1H, J = 10.2 Hz), 4.06 (dd, 1H, J = 1.8 Hz, 10.2 Hz), 3.98 (m, 1H), 3.10 (d, 1H, J = 9.0 Hz), 2.58 (m, 1H), 1.95 (d, 1H, J = 9.0 Hz), 1.91 (m, 2H), 1.65 (m, 3H), 1.34 (s, 3H), 1.26 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 206.0, 202.8, 150.5, 142.7, 130.0, 122.2, 97.7, 72.4, 72.1, 64.8, 61.7, 55.6, 51.4, 44.4, 42.5, 39.2, 29.4, 23.6, 22.0, 17.5; HRMS Calcd for C20H25O6: [M + H]+ 361.1646; found 361.1651.
In Vitro Determination of Effects of Synthesized Compounds on Cancer Cell Proliferation
Breast cancer cell lines MCF-7 and MDA-MB-231 were seeded in 96-well plates at a density of 1 × 104 cells/well and treated with DMSO, 0.01 μM, 0.1 μM, 1 μM, 5 μM, 10 μM, and 100 μM of individual compound for 48 h, and then 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/mL in PBS) was added to each well and further incubated for another 4 h. Then, MTT solution was removed and 150 μL of DMSO was added to each well. Absorbance of all wells was determined by measuring OD at 550 nm after 10 min incubation on a 96-well GlowMaxate Absorbance Reader (Promega, Madison, WI). Each individual compound was tested in quadruplicate wells for each concentration.
Colony Formation Assay
Breast cancer MDA-MB-231 cells were seeded in 6-well tissue culture plates with a density of 800 cells/per well and maintained in regular culture media. After 24 h, the cells were treated with compounds 19 and 10 at different concentrations (0.625 μM, 1.25 μM, 2.5 μM, 5 μM, and 10 μM, respectively) or DMSO as the vehicle. The culture media with the compounds were changed every 72 h. At the end of two weeks, the wells were washed twice with PBS buffer and 2 mL of 0.01% crystal violet staining buffer was added to each well and incubated for 10 min. The wells were then washed with PBS for 5 min for three times, and allowed to dry. Photographs were then taken and the density of the entire culture well area was digitally measured using the GelCount™ instrument (Oxford Optronix, UK). Experiments were performed in triplicate and the density data were analyzed with one-way ANOVA using GraphPad Prizm 5 software package. Error bars represent standard deviation.
Cell Apoptosis Assay
Breast cancer MDA-MB-231 cells were incubated in 6-well plates (2.5 × 105 cells/well). Cells were then treated with DMSO, oridonin or new compounds at different concentrations for 24 h, and then both adherent and floating cells were collected, washed once with PBS. Resuspended cells were incubated with 100 μL PBS containing 1% BSA and 100 μL Annexin V and dead cell detection reagent at room temperature for 20 min. Apoptosis was measured immediately using the Muse Cell Analyzer with the Muse™ Apoptosis Kit (Catalog No. MCH100105).
Western Blot Analysis
Breast cancer MDA-MB-231 cells were treated with DMSO, oridonin or compounds 10 and 19, respectively. After 48 h of treatment, cells were harvested and lysed. Protein concentrations were quantified by the method of Bradford with bovine serum albumin as the standard. Equal amounts of total cellular protein extract (30 μg) was separated by electrophoresis on SDS-polyacrylamide gels and transferred to PVDF membranes. After blocking with 5% non-fat milk, the membrane was incubated with the desired primary antibody overnight at the following dilution: anti-Bcl-2 (1:200), anti-Bax (1:1000), anti-PARP (1:10000), anti-NF-κB (1:2000), anti-caspase-3 (1:1000) and β-actin (1:20000). Subsequently, the membrane was incubated with appropriate secondary antibody. The immunoreactive bands were visualized by enhanced chemiluminescence as recommended by the manufacturer.
In Vivo Antitumor Efficacy Determination
All drugs were dissolved in 50% DMSO with 50% polyethylene glycol for in vivo administration. Body weights and tumors volume were measured daily and tumor volume was calculated according to the formula V = 0.5 × L × W2, where L = length (mm) and W = width (mm). All procedures including mice and in vivo experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of UT M. D. Anderson Cancer Center (MDACC). 25 female nude mice were obtained from MDACC and were used for orthotopic tumor studies at 4 to 6 weeks of age. The mice were maintained in a barrier unit with 12 h light-dark switch. Freshly harvested MDA-MB-231 cells (2.5 × 106 cells per mouse, resuspended in 100 μL PBS) were injected into the fat pad of the 3rd mammary gland of mice, and then randomizedinto 3 groups. The mice were treated 5 days/week for 10 days with 5 mg/kg of compound 19, 1 or vehicle through intraperitoneal injection, when the tumor volume reached 200 mm3.
Statistical Analysis
Statistical significance was determined using Student’s t-test in drug-resistant breast cancer cell and HMEC viability assay or one way ANOVA in in vivo experiments. * represents a p value less than 0.05.
Supplementary Material
Acknowledgments
This work was supported by grants P50 CA097007, P30 DA028821, R21 MH093844 (JZ) from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia (GCC), a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the GCC (NIGMS grant T32 GM089657-03), Sealy and Smith Foundation grant (to the Sealy Center for Structural Biology and Molecular Biophysics), John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at UTMB. We thank Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
ABBREVIATIONS USED
- TNBC
triple-negative breast cancer
- SAR
Structure-Activity Relationships
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- IC50
half maximal inhibitory concentration
- PI
propidium iodide
- HRMS
High-resolution mass spectrometry
- HPLC
high performance liquid chromatography
- TFA
trifluoroacetic acid
- DMSO
dimethyl sulfoxide
- TLC
thin layer chromatography
- NMR
nuclear magnetic resonance
- TMS
tetramethylsilane
- THF
tetrahydrofuran
- EtOAc
ethyl acetate
- DMF
N,N-dimethylformamide
- p-Ts
4-toluenesulfonyl
- Py
pyridine
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- LDA
lithium diisopropylamide
- DMF-DMA
N,N-dimethylformamide dimethyl acetal
- Ms
methanesulfonyl
- PDC
pyridinium dichromate
- PBS
phosphate-buffered saline
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- SDS
sodium dodecyl sulfate
- PVDF
polyvinylidene difluoride
- HMEC
human normal mammary epithelial cell
Footnotes
Author Contributions
These authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
1H and 13C NMR spectra for the compounds described in this paper, and X-ray CIF files for compounds 13 and 18. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fernández Y, Cueva J, Palomo AG, Ramos M, de Juan A, Calvo L, Garcia-Mata J, Garcia-Teijido P, Peláez I, Garcia-Estevez L. Novel therapeutic approaches to the treatment of metastatic breast cancer. Cancer Treat Rev. 2010;36:33–42. doi: 10.1016/j.ctrv.2009.10.001. [DOI] [PubMed] [Google Scholar]; (b) Beaumont T, Leadbeater M. Treatment and care of patients with metastatic breast cancer. Nurs Stand. 2011;25:49–56. doi: 10.7748/ns2011.06.25.40.49.c8566. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]; (b) McAllister SD, Murase R, Christian RT, Lau D, Zielinski AJ, Allison J, Almanza C, Pakdel A, Lee J, Limbad C, Liu Y, Debs RJ, Moore DH, Desprez PY. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res Treat. 2011;129:37–47. doi: 10.1007/s10549-010-1177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Simstein R, Burow M, Parker A, Weldon C, Beckman B. Apoptosis, chemoresistance, and breast cancer: insights from the MCF-7 cell model system. Exp Biol Med (Maywood, NJ, U S) 2003;228:995–1003. doi: 10.1177/153537020322800903. [DOI] [PubMed] [Google Scholar]; (b) Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V, Consoli DP, Cabot MC. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008;22:2541–2551. doi: 10.1096/fj.07-092981. [DOI] [PubMed] [Google Scholar]
- 4.(a) Vuorelaa P, Leinonenb M, Saikkuc P, Tammelaa P, Rauhad JP, Wennberge T, Vuorela H. Natural products in the process of finding new drug candidates. Curr Med Chem. 2004;11:1375–1389. doi: 10.2174/0929867043365116. [DOI] [PubMed] [Google Scholar]; (b) Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Fujita E, Nagao Y, Kaneko K, Nakazawa S, Kuroda H. The antitumor and antibacterial activity of the Isodon diterpenoids. Chem Pharm Bull. 1976;24:2118–2127. doi: 10.1248/cpb.24.2118. [DOI] [PubMed] [Google Scholar]; (b) Fujita E, Nagao Y, Kohno T, Matsuda M, Ozaki M. Antitumor activity of acylated oridonin. Chem Pharm Bull. 1981;29:3208–3213. doi: 10.1248/cpb.29.3208. [DOI] [PubMed] [Google Scholar]; (c) Li B, Li N, Wang S, Gao J, Fang S. Pharmacokinetics of injectable beta-cyclodetrin inclusion complex in wistar rats. Int J Pharm Pharm Sci. 2012;4:92–95. [Google Scholar]; d) Yin F, Liang JY, Liu J. Chemical constitutes of “Dong ling cao”. J Chin Pharm Univ. 2003;34:302–304. [Google Scholar]
- 6.(a) Guan YZ, Wei TH. Clinical research on the oridonin injection for the interventional therapy of liver cancer. J Med Radiol Technol. 2005;236:43–45. [Google Scholar]; (b) Wang RL. Clinical evaluation of Rabdosia rubescences for 31 patients with liver cancer. Chin J Cancer. 1984;8:50–52. [Google Scholar]
- 7.Henan Research Group for Rabdosia rubescences. Conclusion of clinical evaluation of Rabdosia rubescences for the treatment of 68 patients with esophageal carcinoma and carcinoma of gastric cardia. Cancer Res Prev Treat. 1976;3:32–33. [Google Scholar]
- 8.Li C, Wang E, Cheng Y, Bao J. Oridonin: An active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int J Biochem Cell Biol. 2011;43:701–704. doi: 10.1016/j.biocel.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hsieh TC, Wijeratne EK, Liang JY, Gunatilaka AL, Wu JM. Differential control of growth, cell cycle progression, and expression of NF-kappaB in human breast cancer cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin, diterpenoids from the chinese herb Rabdosia rubescens. Biochem Biophys Res Commun. 2005;337:224–231. doi: 10.1016/j.bbrc.2005.09.040. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Wu Y, Wu D, Tashiro S, Onodera S, Ikejima T. NF-kappab facilitates oridonin-induced apoptosis and autophagy in HT1080 cells through a p53-mediated pathway. Arch Biochem Biophys. 2009;489:25–33. doi: 10.1016/j.abb.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 10.(a) Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: Protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J Med Chem. 2013;56:5048–5058. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ding C, Zhang Y, Chen H, Wild C, Wang T, White MA, Shen Q, Zhou J. Overcoming Synthetic Challenges of Oridonin A-ring structural diversification: Regio- and stereoselective installation of azides and 1,2,3-triazoles at the C-1, C-2, or C-3 position. Org Lett. 2013;15:3718–3721. doi: 10.1021/ol4015865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Huang Q, Hua Z. Oridonin: A promising anticancer drug from China. Front Biol. 2010;5:540–545. [Google Scholar]
- 12.(a) Niu XM, Li SH, Li ML, Zhao QS, Mei SX, Na Z, Wang SJ, Lin ZW, Sun HD. Cytotoxic ent-kaurane diterpenoids from Isodon eriocalyx var. laxiflora. Planta Med. 2002;68:528–533. doi: 10.1055/s-2002-32551. [DOI] [PubMed] [Google Scholar]; (b) Wang L, Zhao WL, Yan JS, Liu P, Sun HP, Zhou GB, Weng ZY, Wu WL, Weng XQ, Sun XJ, Chen Z, Sun HD, Chen SJ. Eriocalyxin B induces apoptosis of t(8;21) leukemia cells through NF-kB and MAPK signaling pathways and triggers degradation of AML1-ETO oncoprotein in a caspase-3-dependent manner. Cell Death Differ. 2007;14:306–317. doi: 10.1038/sj.cdd.4401996. [DOI] [PubMed] [Google Scholar]; (c) Festa C, Lauro G, De Marino S, D’Auria MV, Monti MC, Casapullo A, D’Amore C, Renga B, Mencarelli A, Petek S, Bifulco G, Fiorucci S, Zampella A. Plakilactones from the marine sponge Plakinastrella mamillaris. Discovery of a new class of marine ligands of peroxisome proliferator-activated receptor γ. J Med Chem. 2012;55:8303–8317. doi: 10.1021/jm300911g. [DOI] [PubMed] [Google Scholar]
- 13.(a) Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro F, Suh N, Wang Y, Sporn MB, Gribble GW. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem. 2000;43:4233–4246. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]; (b) Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]; (c) Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. New Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 14.Lorusso D, Mainenti S, Pietragalla A, Ferrandina G, Foco G, Masciullo V, Scambia G. Brostallicin (PNU-166196), a new minor groove DNA binder: preclinical and clinical activity. Expert Opin Investig Drugs. 2009;18:1939–1946. doi: 10.1517/13543780903401284. [DOI] [PubMed] [Google Scholar]
- 15.(a) Shiraki T, Kamiya N, Shiki S, Kodama TS, Kakizuka A, Jingami H. Alpha,beta-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor gamma. J Biol Chem. 2005;280:14145–14153. doi: 10.1074/jbc.M500901200. [DOI] [PubMed] [Google Scholar]; (b) Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 16.(a) Gayarre J, Stamatakis K, Renedo M, Perez-Sala D. Differential selectivity of protein modification by the cyclopentenone prostaglandins PGA1 and 15-deoxy-Delta12,14-PGJ2: role of glutathione. FEBS Lett. 2005;579:5803–5808. doi: 10.1016/j.febslet.2005.09.069. [DOI] [PubMed] [Google Scholar]; (b) Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 17.(a) Das U, Sharma RK, Dimmock JR. 1,5-diaryl-3-oxo-1,4-pentadienes: a case for antineoplastics with multiple targets. Curr Med Chem. 2009;16:2001–2020. doi: 10.2174/092986709788682218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mutus B, Wagner JD, Talpas CJ, Dimmock JR, Phillips OA, Reid RS. 1-p-chlorophenyl-4,4-dimethyl-5-diethylamino-1-penten-3-one hydrobromide, a sulfhydryl-specific compound which reacts irreversibly with protein thiols but reversibly with small molecular weight thiols. Anal Biochem. 1989;177:237–243. doi: 10.1016/0003-2697(89)90045-6. [DOI] [PubMed] [Google Scholar]; (c) Lee KH, Huang ES, Piantadosi C, Pagano JS, Geissman TA. Cytotoxicity of sesquiterpene lactones. Cancer Res. 1971;31:1649–1654. [PubMed] [Google Scholar]
- 18.(a) Lee KH, Hall IH, Mar EC, Starnes CO, El Gebaly SA, Waddell TG, Hadgraft RI, Ruffner CG, Weidner I. Sesquiterpene antitumor agents: inhibitors of cellular metabolism. Science. 1977;196:533–536. doi: 10.1126/science.191909. [DOI] [PubMed] [Google Scholar]; (b) Benvenuto JA, Connor TH, Monteith DK, Laidlaw JA, Adams SC, Matney TS, Theiss JC. Degradation and inactivation of antitumor drugs. J Pharm Sci. 1993;82:988–991. [PubMed] [Google Scholar]
- 19.Ohori H, Yamakoshi H, Tomizawa M, Shibuya M, Kakudo Y, Takahasji A, Takahashi S, Kato S, Suzuki T, Ishioka C, Iwabuchi Y, Shibata H. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol Cancer Ther. 2006;5:2563–2571. doi: 10.1158/1535-7163.MCT-06-0174. [DOI] [PubMed] [Google Scholar]
- 20.Dimmock JR, Raghavan SK, Logan BM, Bigam GE. Antileukemic evaluation of some Mannich bases derived from 2-arylidene-1,3-diketones. Eur J Med Chem. 1983;18:248–254. [Google Scholar]
- 21.Chen G, Waxman DJ. Role of cellular glutathione and glutathione S-transferase in the expression of alkylating agent cytotoxicity in human breast cancer cells. Biochem Pharmacol. 1994;47:1079–1087. doi: 10.1016/0006-2952(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 22.Zhou W, Cheng Y. The chemoselective synthesis for eriocalyxin B and its analogues. Acta Chim Sinica. 1990;48:1185–1190. [Google Scholar]
- 23.(a) Reich HJ, Renga JM, Reich IL. Organoselenium chemistry. conversion of ketones to enones by selenoxide syn-elimination. J Am Chem Soc. 1975;97:5434–5447. [Google Scholar]; (b) Nicolaou KC, Monagnon T, Baran PS, Zhong YL. Iodine(V) reagents in organic synthesis. Part 4. o-Iodoxybenzoic acid as a chemospecific tool for single electron transfer-based oxidation processes. J Am Chem Soc. 2002;124:2245–2258. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]; (c) Chai KB, Sampson P. Macrolactonization-transannular aldol condensation approach to the taxane AB ring system. J Org Chem. 1993;58:6807–6813. [Google Scholar]; (d) Takahito I, Kyoko T, Takahiro U, Masataka K, Norimitsu T, Mikiji M. Topochemical Reaction of 7-bromoethoxycarbonyl-7-cyano-1,4-benzoquinone methide in the solid state. Chem Lett. 2006;35:918–919. [Google Scholar]
- 24.(a) Henschler D, Eder E. Structure-activity relationships of alpha, beta-unsaturated carbonylic compounds. IARC Sci Publ. 1986;70:197–205. [PubMed] [Google Scholar]; (b) Rosen JD, Segall Y, Casida JE. Mutagenic potency of haloacroleins and related compounds. Mutat Res. 1980;78:113–119. doi: 10.1016/0165-1218(80)90090-7. [DOI] [PubMed] [Google Scholar]
- 25.Verbitski SM, Mullally JE, Fitzpatrick FA, Ireland CM. Punaglandins, chlorinated prostaglandins, function as potent Michael receptors to inhibit ubiquitin isopeptidase activity. J Med Chem. 2004;47:2062–2070. doi: 10.1021/jm030448l. [DOI] [PubMed] [Google Scholar]
- 26.(a) Basavaiah D, Rao AJ, Satyanarayana T. Recent advances in the Baylis-Hillman reaction and applications. Chem Rev. 2003;103:811–892. doi: 10.1021/cr010043d. [DOI] [PubMed] [Google Scholar]; (b) Ito H, Takenaka Y, Fukunishi S, Iguchi K. A practical preparation of 2-hydroxymethyl-2-cyclopenten-1-one by Morita-Baylis-Hillman reaction. Synthesis. 2005;18:3035–3038. [Google Scholar]
- 27.Baylis-Hillman reaction was initially attempted to synthesize 10 from compound 6, but it was proven unsuccessful to form the 2-hydroxymethylated intermediate.
- 28.Liotta D, Barnum C, Puleo R, Zima G, Bayer C, Kezar HS., III A simple method for the efficient sysnthesis of unsaturated β-dicarbonyl compunds. J Org Chem. 1981;46:2920–2923.(b) Actually, compound 8 was unstable and a minor amount of 9 was found when purified with silica gel column or preparative thin layer chromatography (PTLC).
- 29.CCDC 919314 (13) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 30.(a) Jana CK, Hoecker J, Woods TM, Jessen HJ, Neuburger M, Gademann K. Synthesis of withanolide A, biological evaluation of its neuritogenic properties, and studies on secretase inhibition. Angew Chem Int Ed. 2011;50:8407–8411. doi: 10.1002/anie.201101869. [DOI] [PubMed] [Google Scholar]; (b) Wharton PS, Bohlen DH. Hydrazine reduction of α,β-epoxy ketones to allylic alcohols. J Org Chem. 1961;26:3615–3616. [Google Scholar]
- 31.Muller R, Ruedi P. An unexpected (3→2)-hydride shift in phyllocladane (=13β-kaurane) diterpenoids and in related trimethyl-substituted bi- and tricyclic compounds. Helv Chim Acta. 2003;86:439–456. [Google Scholar]
- 32.Hirai S, Nakada M. An enantioselective approach to (−)-platencin via catalytic asymmetric intramolecular cyclopropanation. Tetrahedron Lett. 2010;51:5076–5079. [Google Scholar]
- 33.(a) Zhang CL, Wu LJ, Tashiro S, Onodera S, Ikejima T. Oridonin induces a caspase-independent but mitochondria- and MAPK-dependent cell death in the murine fibrosarcoma cell line L929. Biol Pharm Bull. 2004;27:1527–1531. doi: 10.1248/bpb.27.1527. [DOI] [PubMed] [Google Scholar]; (b) Chen S, Gao J, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. The cytostatic and cytotoxic effects of oridonin (Rubescenin), a diterpenoid from Rabdosia rubescens, on tumor cells of different lineage. Int J Oncol. 2005;26:579–588. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.