

Abstract
Brain metastasis occurs in 20% to 40% of cancer patients. Treatment is mostly palliative and the inability of most drugs to penetrate the brain presents one of the greatest challenges in the development of therapeutics for brain metastasis. Matrix metalloproteinase-2 (MMP-2) plays important roles in invasion and vascularization of the central nervous system and represents a potential target for treatment of brain metastasis. Carbonate, O-phenyl carbamate, urea, and N-phenyl carbamate derivatives of SB-3CT, a selective and potent gelatinase inhibitor were synthesized and evaluated. The O-phenyl carbamate and urea variants were selective and potent inhibitors of MMP-2. Carbamate 5b was metabolized to the potent gelatinase inhibitor 2, which was present at therapeutic concentrations in the brain. In contrast, phenyl urea 6b crossed the blood-brain barrier, however higher doses would result in therapeutic brain concentrations. Carbamate 5b and urea 6b show potential for intervention of MMP-2-dependent diseases, such as brain metastasis.
INTRODUCTION
Brain metastasis is a significant health problem, occurring in 20% to 40% of patients with cancer.1 Patients with melanoma, lung, breast, and kidney cancer have the highest incidence of brain metastasis,2 occurring in 60% to 80% of patients with metastatic melanoma and representing the major cause of death in patients with melanoma.3 Although standard treatments for brain metastasis include surgery, radiotherapy, and chemotherapy, the prognosis is poor, with survival between 4 to 6 months3 and chemotherapy showing limited or no efficacy.4 A major challenge in the development of therapeutics for brain metastasis is the poor penetration of drugs through the blood-brain barrier (BBB), and >98% of small-molecule drugs do not cross the BBB.5 In addition, the presence of efflux pumps, such as P-glycoprotein, rapidly remove many drugs from the central nervous system (CNS).6 Several drugs are currently being evaluated in clinical trials of patients with brain metastasis, including bevacizumab, a humanized monoclonal anti-vascular endothelial growth factor (VEGF) antibody that inhibits angiogenesis. However, bevacizumab is a large molecule that does not cross the BBB. Trastuzumab, a recombinant humanized human epidermal growth factor receptor 2 (HER2) monoclonal antibody that inhibits epidermal growth factor receptor (EGFR) also does not penetrate the BBB. Lapatinib, a tyrosine kinase inhibitor that targets EGFR and HER2, crosses the BBB, but does not reach therapeutic concentrations in the CNS in mice.7 Crizotinib is an inhibitor of anaplastic lymphoma kinase and tyrosine kinase that is being evaluated in patients with metastatic non-small cell lung cancer (NSCLC); however, crizotinib penetrates the brain poorly, thus hindering efficacy in metastatic brain tumors.8 While strategies to improve BBB permeability,9 including disruption of the BBB, P-glycoprotein inhibitors, transcranial and transnasal delivery, and endogenous BBB transporters, have been explored, the presence of the BBB prevents potential brain metastasis drugs from reaching therapeutic levels in the brain. Thus, novel therapeutics that cross the BBB are urgently needed for the treatment of brain metastasis.
Matrix metalloproteinases (MMPs) are a large group of enzymes that play important roles in many normal physiological functions, such as remodeling of the extracellular matrix during development and reproduction.10 MMPs also degrade the extracellular matrix proteins that regulate processes involved in cancer progression, including cancer cell growth, apoptosis, migration, invasion, and angiogenesis.11 While MMP inhibitors have been evaluated in clinical trials for cancer,12 these studies resulted in failure primarily due to broad-spectrum inhibition of all MMPs, adverse side effects, and insufficient target validation.13 Recent efforts in defining the role of particular MMPs in cancer have correlated increased expression of MMP-2 (also known as gelatinase A) with aggressive breast cancer,14 malignant prostate cancer,15 pancreatic cancer,16 gastric cancer,17 brain metastasis of melanoma,3 and lung cancer brain metastasis.18 Tumors expressing MMP-2 have increased vasculature at the brain-tumor interface, indicating that MMP-2 might play a role in enhancing invasion and vascularization within the CNS.17 Therefore, selective MMP-2 inhibition represents a targeted therapy for the treatment of brain metastasis.
The search for a selective MMP-2 inhibitor is a challenge due mainly to the high structural similarities of MMPs. We have previously reported the prototype inhibitor, SB-3CT (1),19 which displays a unique slow-binding mechanism of action within the active site of gelatinases (MMP-2 and MMP-9). This inhibition mode leads to a reaction involving deprotonation at the α-methylene to the sulfonyl moiety that results in ring opening of the thiirane to provide picomolar tight-binding inhibition of gelatinases.20 Compound 1 selectively inhibits the gelatinases, while sparing representative members of the other types of MMPs such as MMP-1, −3 and −7.21 Compound 1 is primarily metabolized at the para-position of the terminal phenyl ring to give a more potent gelatinase inhibitor 2.21 We also showed that incorporation of an amino group at the para-position (3) retained potent inhibitory activity against the gelatinases.22 However, in addition to gelatinases, the slow-binding kinetic profile of compounds 2 and 3 was extended to include MMP-14,21, 22 the activator of MMP-2.23 The nanomolar inhibition constants of 2 and 3 for MMP-2, MMP-9 and MMP-14 would not differentiate these three MMPs in targeting.
A structure-activity relationship (SAR) study of 1 revealed that modification at the para-position of the terminal phenyl ring is well tolerated.24 We undertook in the present study the preparation and evaluation of analogs of 2 and 3, using functionalities such as carbonates 4, O-phenyl carbamates 5, ureas 6 and N-phenyl carbamates 7. We report herein that a distinct change in this para substituent led to nanomolar inhibition of only MMP-2, while sparing other MMPs, including MMP-9 and MMP-14.

RESULTS AND DISCUSSION
Chemistry
The syntheses of the carbonate and carbamate derivatives of 2 are outlined in Scheme 1. Carbonate derivatives 4a–d were prepared in good yields by reaction of thiirane 2 with alkyl chloroformates in the presence of triethylamine. On the other hand, the syntheses of target thiiranes 5a–d was initiated by Ullmann coupling of phenyl bromide 8 to phenol 9, using CuI as catalyst, to generate the diphenyl ether 10, which was subsequently MOM-deprotected by acid reflux to give 11. Intermediate O-phenyl carbamates 12 could be easily accessed by nucleophilic addition of phenol 11 to alkyl isocyanates at reflux temperature.25 Since the highly toxic methyl isocyanate is not readily available, this prompted us to explore an alternative route to the preparation of 12a. Activation of phenol 11 with p-nitrophenyl (PNP)-chloroformate, followed by nucleophilic attack by methylamine afforded the N-methyl carbamate 12a in 67% yield over two synthetic steps. The succeeding steps involved oxidation to the corresponding oxiranes 13a–d, and subsequent reaction with thiourea to give the desired 5a–d.21
Scheme 1.

Syntheses of the Carbonate and Carbamate Derivatives of 2a
aReagents and condition: (a) TEA, alkyl chloroformate, MeCN, 76–93%. (b) CuI, Cs2CO3, N,N-dimethylglycine HCl salt, 1,4-dioxane, 110 °C, 24 h, %. (c) HCl, MeOH, reflux, 1 h, 99%. (d) TEA, PNP-chloroformate, CH2Cl2, 0 °C, 30 min. (e) MeNH2, THF, room temperature, 30 min, 67% in two steps. (f) TEA, alkyl isocyanate, dry toluene, reflux, 3 h, 93–98%. (g) m-CPBA, CH2Cl2, 0 °C to room temperature, 6 d, 50–55%. (h) thiourea, MeOH/CH2Cl2, room temperature, 24 h, 76–90%.
The N-methyl urea 6a was prepared in a similar fashion as N-phenyl carbamate 12a. An activated carbamate intermediate was first synthesized by direct coupling of thiirane 3 with PNP-chloroformate, followed by selective nucleophilic addition of methylamine to the activated carbamate to generate 6a in 53% yield (Scheme 2). A more convenient route to access asymmetric ureas is by reaction of amines with isocyanates.26 Such a method was utilized to prepare urea derivatives 6b–d in acceptable yields through a one-step reaction of thiirane 3 with the corresponding commercially available alkyl isocyanates, in the presence of triethylamine. Reaction of 3 with alkyl chloroformates, using N-methylmorpholine as a base, readily furnished N-phenyl carbamates 7a–d in 49–80% yield.
Scheme 2.

Syntheses of the Urea and Carbamate Derivatives of 3a
a Reagents and condition: (a) PNP-chloroformate, CH2Cl2, 0 °C, 30 min. (b) MeNH2, THF, room temperature, 15 min, 53% in two steps. (c) TEA, alkyl isocyanate, THF, room temperature, 24 h, 55–61%. (d) NMM, alkyl chloroformate, THF, 0 °C, 20 min, 49–80%.
MMP Inhibition
The results of the MMP kinetics of compounds 4–7 are given in Table 1. Compounds 4–7 exhibited slow-binding inhibition of MMP-2, −9cat (catalytic domain) and −14cat, which is a distinguishing feature of the thiirane class of inhibitors.20 In addition, slow-binding albeit poor inhibition of MMP-8cat was observed. On the other hand, 4–7 showed marginal to no inhibition of MMP-1cat, −3cat and −7. A general observation was that the entire set of inhibitors listed in Table 1 exhibited comparable nanomolar potencies against MMP-2, while displaying poorer inhibition of MMP-9cat and MMP-14cat. Compounds 4a and 7a were the more potent MMP-9cat inhibitors among the derivatives, with inhibition constants of 0.23 ± 0.06 µM and 0.18 ± 0.03 µM, respectively. Analogs 4b and 7b showed comparable inhibition constants against MMP-9cat as ND-322 (3, Ki value 0.87 µM).22 Carbonates 4 and N-phenyl carbamates 7 were 2–5-fold less potent against MMP-9cat, even less so against MMP-14cat, than MMP-2. The selectivity of inhibition of MMP-2 was more pronounced for the O-phenyl carbamates 5 and ureas 6, which were at least 20-fold more active against MMP-2 than MMP-8cat, MMP-9cat or MMP-14cat. The inhibition constants of 5 and 6 against MMP-9cat or MMP-14cat were at best 10 µM, a concentration that is less likely to be attained in vivo, while retaining their nanomolar potencies against MMP-2. Hence, these compounds would find utility where MMP-2 selectivity is desired.
Table 1.
Kinetic Parameters for Inhibition of MMPs
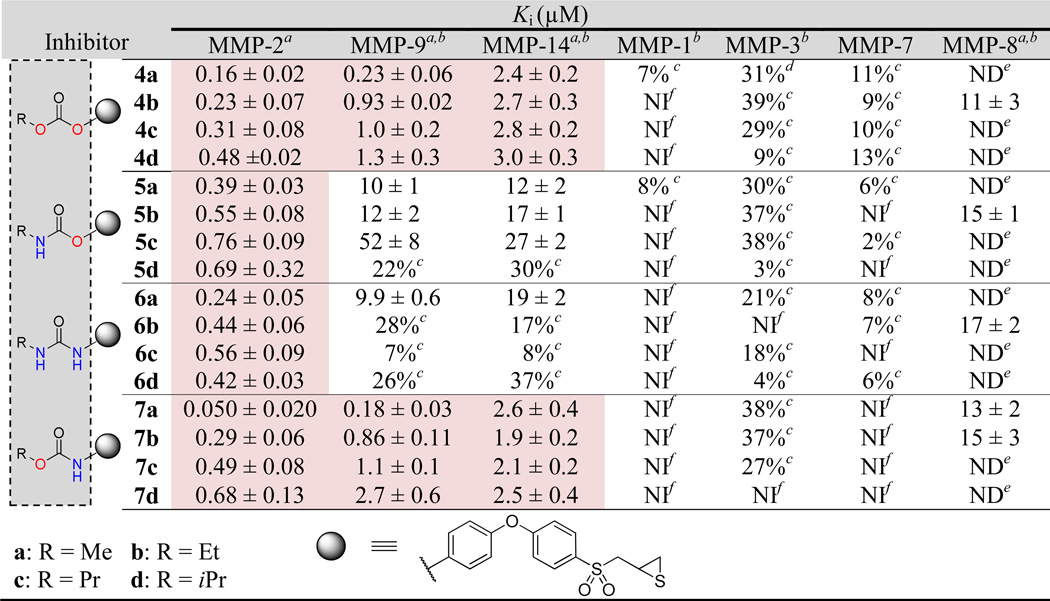 |
Kiis calculated from the ratio of koff/kon. Slow-binding kinetic parameters (kon and koff values) are given in the Supporting Information (Table S1).
Catalytic domains.
Inhibition at 50 µM.
Inhibition at 30 µM.
ND = not determined.
NI = no inhibition at 50 µM.
Computational Analyses
We conducted computational docking of thiiranes 4–7 into the active sites of MMP-2, −9 and −14, to gain insight into the differences in activities against these MMPs. The S1' cavities of these MMPs vary by the non-conserved amino-acid residues lining up the pocket, which in turn alter the conformational state of the S1' loop. Additionally, the amino acids that reside deep in the pocket have been shown to undergo significant conformational adjustment upon inhibitor binding.27 The para-substituents of thiiranes 4–7 extend into the pocket, interacting differentially with the loop residues of MMP-2, MMP-9 and MMP-14. In MMP-2, the presence of relatively smaller amino acids Thr220 and Thr222 renders a sufficiently accessible S1' pocket to accommodate para-substitutions of 4–7 (Figure 1A). Thus, compounds 4–7 show similar nanomolar range of inhibition irrespective of the size of the para-substituents (Table 1). In comparison, the bulkier loop residue Arg424 in MMP-9 disfavors larger functional group substitutions at the para position. The SAR trend of MMP-9 inhibition shows that the larger size propyl and isopropyl variants produced weaker inhibition, than methyl substituted analogs. In general, the weakest inhibition was observed for MMP-14 compared to MMP-2 and MMP-9, due to sterically unfavorable loop residues Gln262 and Met264 (Figure 1B). Although the computational analysis could rationalize the general trend in the SAR, the more pronounced loss of potency for compounds 5 and 6 against MMP-9 and MMP-14 compared to 4 and 7 could not be readily explained.
Figure 1.

(A) Stereo view of the thiirane analogs (4b, 5b, 6b, 7b) docked to MMP-2 using Glide SP methodology. The ligands are represented in grey carbon capped stick representation while MMP-2 carbon atoms and ribbons are colored aquamarine. Zn2+ is depicted as a gray sphere with coordinating histidines and glutamic acid as capped sticks. Connolly surface in green was generated for MMP-2 except for the loop and zinc coordinating residues for clarity. (B) Compounds superposed to the MMP-14 crystal structure catalytic site (PDB ID: 3MA2) show the bulkier amino acids Gln262 and Met264 substituted at the S1′ selectivity loop region in comparison to Thr426 and Thr428 in MMP-2.
Microsomal Stability and Metabolite Identification
Select compounds that showed good inhibition of MMP-9cat (4b, 7a, 7b) as well as highly MMP-2-selective analogs (5b, 6b) were evaluated for metabolic stability in rat liver S9, as summarized in Table 2. Carbonate 4b completely hydrolyzed within 5 min to form compound 2, which in turn had a half-life of 23.5 ± 0.5 min. Functionalization with a more stable carbamate moiety (5b) resulted in a half-life of 22.8 ± 1.5 min; 5b was further converted to 2. Three metabolites of 5b were found: hydrolysis of the carbamate linkage to give M1 (or compound 2), the sulfinic acid M2, and thiirane S-oxide M3 (Scheme 3). The structures of the metabolites were proposed based on their product-ion spectra (Figure 2); the metabolites were similar to the sulfoxides and sulfinic acids previously observed for other thiirane compounds.21, 28
Table 2.
Half-lives of Selected Compounds in Male Rat Liver S9
| Compound | t½ (min) |
|---|---|
| 2 | 23.5 ± 0.5 |
| 3 | 14.0 ± 2.4; 59.0 ± 3.0 |
| 5b | 22.8 ± 1.5 |
| 6b | 40.7 ± 2.2 |
| 7a | 17.6 ± 0.1 |
| 7b | 24.8 ± 1.7 |
Scheme 3.

Metabolic pathways of selected compounds
Figure 2.

Product-ion mass spectra of (A) parent compound 5b, and metabolites (B) M1, (C) M2, and (D) M3.
The half-life of 3 was observed to be biphasic, with an initial half-life of 14.0 ± 2.4 min and a 4-fold longer second-phase half-life (Table 2). The urea derivative 6b proved to be more metabolically stable among the derivatives, with a half-life of 40.7 ± 2.2 min. The O-methyl carbamate 7a had a lower metabolic stability, compared to the corresponding O-ethyl derivative 7b. Carbamates 7a and 7b had relatively shorter half-lives of 17.6 ± 0.1 and 24.8 ± 1.7, respectively. Carbamate 7a had similar metabolic pathways as 5b as illustrated in Scheme 3. Metabolism of 7a led to hydrolysis of the carbamate linkage to release M4 (or 3), oxidation at the α-methylene to the sulfonyl group to give sulfinic acid M5, and thiirane sulfur oxidation to produce sulfoxide M6. The structures of the metabolites were proposed based on their production mass spectra, as shown in Figure 3.
Figure 3.

Product-ion mass spectra of (A) parent compound 7a, and metabolites (B) M4, (C) M5, and (D) M6.
Pharmacokinetics and Brain Distribution of Compounds 5b and 6b
Compounds 5b and 6b were selected for investigation of their pharmacokinetic properties. Since compounds 1 and 2 are readily distributed to the brain,29 the ability of compounds 5b and 6b to cross the BBB was also investigated. After a single subcutaneous (sc) dose of 5b to mice, the compound was not quantifiable in plasma or brain at all time points as it was converted to 2, an active gelatinase inhibitor. Plasma concentrations of 2 were maximal at 10 min (1.24 ± 0.676 µM), the first time point collected, and decreased to 0.114 ± 0.0147 µM at 4 h (Figure 4A and Table S2 in the Supporting Information). Levels of compound 2 in brain were maximal at 20 min (1.35 ± 1.10 pmol/mg tissue, equivalent to 1.35 ± 1.10 µM assuming a density of 1.0 g/mL) and declined to 0.255 ± 0.0154 pmol/mg at 4 h. Brain levels remained above the Ki for MMP-2 of 6 nM at all time points. Systemic exposure (AUC0-∞) was 69.9 µM·min in plasma and 129 pmol·min/mg in brain (Table 3); the brain to plasma AUC ratio was 1.64, indicating that compound 2, derived from 5b, crossed the BBB readily.
Figure 4.

Brain and plasma concentration-time curves of (A) compound 2 after a single 25 mg/kg sc dose of compound 5b to mice and (B) compound 6b after a single 25 mg/kg sc dose of compound 6b to mice. Concentrations in pmol/mg tissue for brain and in µM for plasma.
Table 3.
Pharmacokinetic Parameters after Single Subcutaneous Dose Administration of Compound 5b or 6b at 25 mg/kg to Mice
| Parameter | Compound 2 after dose of 5b |
Compound 6b after dose of 6b |
||
|---|---|---|---|---|
| Brain | Plasma | Brain | Plasma | |
| AUC0-lasta | 88.8 | 54.0 | 9.86 | 181 |
| AUC0-∞a | 129 | 69.9 | 13.8 | 187 |
| t½α (min) | -- | 41.3 | 18.2 | 8.38 |
| t½β (min) | 533 | 462 | 204 | 61.9 |
| brainAUC/plasmaAUC | 1.64 | 0.0738 | ||
AUC in pmol·min/mg for brain and in µM·min for plasma
The urea derivative 6b proved to be more stable and was quantifiable both in plasma and brain. Plasma and brain levels of 6b were maximal at 10 min, and were quantifiable up to 5 h, with concentrations of 0.0704 ± 0.0506 µM in plasma and 0.0133 ± 0.00337 pmol/mg in brain (Figure 4B and Table S3 in the Supporting Information). Brain AUC0-∞ was 13.8 pmol·min/mg while that in plasma was 187 µM·min (Table 3). The brain to plasma AUC0-∞ ratio of 0.0738 indicates that 6b crosses the BBB. Levels of 6b in brain were at all times below the Ki for MMP-2 of 0.44 µM. However, the thiirane class of inhibitors binds to MMP-2 as a thiolate generated at the active site with Ki values in the picomolar range.15 Thus, brain levels of 6b are likely to be above the thiolate Ki, but the concentrations are not measurable by the analytical methodology. Alternatively, a dose of 100 mg/kg of compound 6b given sc would result in brain levels above the thiirane Ki. Compound 6b had a 3.3-fold longer elimination half-life in brain than in plasma. Hydrolysis of 6b to give compound 3 was not observed both in plasma and brain.
CONCLUSION
We synthesized and evaluated carbonate, O-phenyl carbamate, urea, and N-phenyl carbamate derivatives of 1. The O-phenyl carbamate and urea variants were selective and potent inhibitors of MMP-2, and poorly inhibited or showed no inhibition of MMP-1cat, MMP-3cat, MMP-7, MMP-8cat, MMP-9cat, and MMP-14cat. The ethyl O-phenyl carbamate 5b was metabolized to the potent gelatinase inhibitor 2, which was readily absorbed and distributed to the brain. Levels of compound 2 in the brain ranged from 13- to 73-fold higher than Ki for MMP-2, indicating that therapeutic concentrations were attained in the CNS. The terminal half-lives of 2 were 7.7 h in plasma and 8.9 h in brain. The phenyl urea 6b was not hydrolyzed to compound 3, however 6b crossed the BBB. However, higher doses of 6b are required to attain therapeutic levels in the CNS. Carbamate 5b and urea 6b show promise for the treatment of MMP-2-dependent diseases, such as brain metastases.
EXPERIMENTAL SECTION
Chemistry
All reactions were performed under an atmosphere of nitrogen, unless noted otherwise. 1H and 13C NMR spectra were recorded on a Varian INOVA-500 (Varian Inc., Palo Alto, CA, USA), or Varian UnityPlus 300 spectrometer (Varian Inc., Palo Alto, CA, USA). Thin-layer chromatography was performed with Whatman reagents 0.25 mm silica gel 60-F plates. Flash chromatography was carried out with silica gel 60, 230–400 mesh (0.040–0.063 mm particle size) purchased from EM Science (Gibbstown, NJ, USA). High-resolution mass spectra were obtained at the Department of Chemistry and Biochemistry, University of Notre Dame by ESI ionization, using Bruker micrOTOF/Q2 mass spectrometer (Bruker Daltonik, Bremen, Germany). Purity of the prepared compounds was generally >95%, confirmed by UPLC. Detailed conditions are given in the UPLC section.
Syntheses of the Carbonate Derivatives of 2 (4a–d)
General Procedure for the Syntheses of Thiiranes 4a–d
A solution of thiirane 2 (60 mg, 0.19 mmol) in acetonitrile (3 mL) was mixed with TEA (52 µL, 0.37 mmol) at ice-water temperature, followed by dropwise addition of the alkyl chloroformate (0.37 mmol). The reaction was stirred for 15 min, concentrated in vacuo and reconstituted in EtOAc. The resulting mixture was washed with water, dried with anhydrous Na2SO4, and evaporated to dryness under reduced pressure. The desired product was purified by silica gel column chromatography.
Methyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl) carbonate (4a)
Yield: 90%. 1H NMR (300 MHz, CDCl3) δ 2.20 (dd, J = 5.14, 1.79 Hz, 1H), 2.58 (dd, J = 6.22, 1.67 Hz, 1H), 3.10 (dq, J = 7.47, 5.72 Hz, 1H), 3.17 – 3.31 (m, 1H), 3.56 (dd, J = 14.35, 5.74 Hz, 1H), 3.97 (s, 3H), 7.08 – 7.19 (m, 4H), 7.25 – 7.34 (m, 2H), 7.86 – 7.96 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 24.3, 26.2, 55.7, 62.7, 117.9, 121.4, 123.0, 130.9, 132.4, 148.1, 152.5, 154.3, 162.8. HRMS-ESI (m/z): [M+Na]+, calcd for C17H16NaO6S2, 403.0281: found, 403.0282.
Ethyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl) carbonate (4b)
Yield: 93%. 1H NMR (500 MHz, CDCl3) δ 1.41 (t, J = 7.08 Hz, 3H), 2.16 (dd, J = 5.18, 1.79 Hz, 1H), 2.54 (dd, J = 6.08, 1.50 Hz, 1H), 3.06 (dq, J = 7.68, 5.68 Hz, 1H), 3.19 (dd, J = 14.16, 7.78 Hz, 1H), 3.48 −3.56 (m, 1H), 4.34 (q, J = 7.18 Hz, 2H), 7.06 – 7.14 (m, 4H), 7.19 – 7.29 (m, 2H), 7.83 – 7.91 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 14.4, 24.4, 26.2, 62.8, 65.3, 117.9, 121.5, 123.1, 131.0, 132.4, 148.2, 152.5, 153.8, 162.9. HRMS-ESI (m/z): [M+Na]+, calcd for C18H18NaO6S2, 417.0437: found, 417.0440.
Propyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl) carbonate (4c)
Yield: 88%. 1H NMR (500 MHz, CDCl3) δ 1.02 (t, J = 7.48 Hz, 3H), 1.78 (sxt, J = 7.14 Hz, 2H), 2.15 (dd, J = 5.18, 1.79 Hz, 1H), 2.53 (dd, J = 6.08, 1.69 Hz, 1H), 3.05 (dq, J = 7.35, 5.66 Hz, 1H), 3.19 (dd, J = 14.16, 7.78 Hz, 1H), 3.51 (dd, J = 14.26, 5.68 Hz, 1H), 4.23 (t, J = 6.68 Hz, 2H), 7.05 – 7.14 (m, 4H), 7.20 – 7.27 (m, 2H), 7.82 – 7.90 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 10.3, 22.1, 24.4, 26.2, 62.7, 70.7, 117.9, 121.4, 123.0, 130.9, 132.4, 148.2, 152.4, 153.8, 162.8. HRMS-ESI (m/z): [M+Na]+, calcd for C19H20NaO6S2, 431.0594: found, 431.0565.
Isopropyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl) carbonate (4d)
Yield: 76%. 1H NMR (500 MHz, CDCl3) δ 1.43 (d, J = 6.38 Hz, 6H), 2.20 (dd, J = 4.98, 1.79 Hz, 1H), 2.57 (dd, J = 6.18, 1.79 Hz, 1H), 3.03 – 3.14 (m, 1H), 3.22 (dd, J = 14.36, 7.78 Hz, 1H), 3.55 (dd, J = 14.26, 5.68 Hz, 1H), 5.03 (quin, J = 6.28 Hz, 1H), 7.08 – 7.16 (m, 4H), 7.23 – 7.31 (m, 2H), 7.86 – 7.94 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 21.9, 24.4, 26.3, 62.8, 73.6, 117.9, 121.5, 123.1, 131.0, 132.4, 148.3, 152.4, 153.3, 162.9. HRMS-ESI (m/z): [M+Na]+, calcd for C19H20NaO6S2, 431.0594: found, 431.0586.
Syntheses of the Carbamate Derivatives of 2 (5a–d)
Allyl(4-(4-(methoxymethoxy)phenoxy)phenyl)sulfane (10)
A mixture of compound 8 (6.5 g, 30 mmol), compound 9 (5.0 g, 30 mmol), as previously synthesized,30 Cs2CO3 (15 g, 46 mmol), N,N-dimethylglycine hydrochloride salt (1.3 g, 9.0 mmol) CuI (0.85 g, 4.5 mmol) in dry 1,4-dioxane (100 mL) was heated at 110 °C for 24 h. The mixture was diluted with water and extracted with EtOAc. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography to give compound 10 in 83% yield (7.5 g). 1H NMR (500 MHz, CDCl3) δ 3.48 (d, J = 6.98 Hz, 2 H), 3.51 (s, 3 H), 5.02 – 5.10 (m, 2 H), 5.16 (s, 2 H), 5.81 – 5.93 (m, J = 16.92, 9.99, 6.98, 6.98 Hz, 1 H), 6.87 – 6.92 (m, 2 H), 6.95 – 7.00 (m, 2 H), 7.02 −7.08 (m, 2 H), 7.32 – 7.36 (m, 2 H). 13C NMR (126 MHz, CDCl3) δ 38.8, 56.1, 95.0, 117.6, 117.7, 118.3, 120.8, 128.6, 133.2, 134.0, 151.0, 153.7, 157.6. HRMS-ESI (m/z): [M+H]+, calcd for C17H19O3S, 303.1079: found, 303.1053.
4-(4-(Allylthio)phenoxy)phenol (11)
To a solution of compound 10 (1.1 g, 3.6 mmol) in methanol (20 mL) was added 0.5 mL of concentrated HCl. The mixture was refluxed for 1 h, after which the solvent was evaporated to dryness. The crude product was taken up into water and EtOAc. Layers were separated and the aqueous layer was extracted with EtOAc. The organic extracts were dried with over anhydrous Na2SO4 and concentrated in vacuo. Purification by silica gel chromatography gave the title compound in 99% yield (0.93 g). 1H NMR (500 MHz, CDCl3) δ 3.48 (d, J = 6.78 Hz, 2 H), 4.67 (s, 1 H), 5.01 – 5.09 (m, 2 H), 5.81 – 5.92 (m, J = 16.82, 9.65, 7.20, 7.20 Hz, 1 H), 6.79 – 6.85 (m, 2 H), 6.85 – 6.90 (m, 2 H), 6.90 – 6.97 (m, 2 H), 7.29 – 7.36 (m, 2 H). 13C NMR (126 MHz, CDCl3) δ 39.0, 116.6, 117.7, 118.1, 121.3, 128.4, 133.4, 134.0, 150.0, 152.1, 158.0. HRMS-ESI (m/z): [M+H]+, calcd for C15H15O2S, 259.0787: found, 259.0795.
4-(4-(Allylthio)phenoxy)phenyl methylcarbamate (12a)
A solution of compound 11 (0.42 g, 1.6 mmol) in dry CH2Cl2 (4 mL) was cooled in an ice-water bath and treated with TEA (0.46 mL, 3.3 mmol). PNP-chloroformate (0.41 g, 2.0 mmol) in dry THF (3 mL) was added dropwise and the resulting mixture was stirred for 30 min. The mixture was diluted with CH2Cl2, washed with saturated NH4Cl, and concentrated in vacuo. The crude intermediate (4-(4-(allylthio)phenoxy)phenyl (4-nitrophenyl) carbonate) was reconstituted in CH2Cl2 (5 mL), treated with MeNH2 (2.4 mmol) in THF, and stirred for 30 min at room temperature. The mixture was diluted with CH2Cl2 and washed with saturated NaHCO3, followed by water. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. Purification of the crude product by silica gel chromatography gave 0.34 g (67%) of compound 12a. 1H NMR (300 MHz, CDCl3) δ 2.86 (d, J = 5.02 Hz, 3H), 3.43 – 3.53 (m, 2H), 4.98 – 5.12 (m, 2H), 5.19 (q, J = 4.54 Hz, 1H), 5.77 – 5.95 (m, 1H), 6.87 – 7.03 (m, 4H), 7.04 – 7.15 (m, 2H), 7.29 – 7.40 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 27.8, 38.6, 117.7, 119.0, 119.8, 123.0, 129.4, 133.0, 133.8, 146.9, 154.0, 155.5, 156.7. HRMS-ESI (m/z): [M+H]+, calcd for C17H18NO3S, 316.1002: found, 316.1026.
General Procedure for the Syntheses of Compounds 12b–d
To a solution of 11 (0.33 g, 1.3 mmol) in dry toluene (5 mL) were added the alkyl isocyanate (1.4 mmol) and a catalytic amount of TEA (18 µL, 0.13 mmol). The mixture was refluxed for 3 h, after which the solvent was removed under reduced pressure. The desired product was purified by silica gel chromatography.
4-(4-(Allylthio)phenoxy)phenyl ethylcarbamate (12b)
Yield: 92%. 1H NMR (300 MHz, CDCl3) δ 1.17 (t, J = 7.29 Hz, 3H), 3.27 (quin, J = 6.88 Hz, 2H), 3.47 (d, J = 7.18 Hz, 2H), 4.96 – 5.11 (m, 2H), 5.27 (t, J = 5.38 Hz, 1H), 5.76 – 5.94 (m, 1H), 6.86 – 7.03 (m, 4H), 7.03 – 7.14 (m, 2H), 7.27 – 7.38 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 15.1, 36.2, 38.5, 117.6, 118.9, 119.4, 119.8, 123.0, 129.1, 129.3, 132.9, 133.8, 146.8, 156.6. HRMS-ESI (m/z): [M+H]+, calcd for C18H20NO3S, 330.1158: found, 330.1153.
4-(4-(Allylthio)phenoxy)phenyl propylcarbamate (12d)
Yield: 95%. 1H NMR (300 MHz, CDCl3) δ 0.94 (t, J = 7.41 Hz, 3H), 1.56 (sxt, J = 7.27 Hz, 2H), 3.09 – 3.27 (m, 2H), 3.47 (d, J = 6.94 Hz, 2H), 4.97 – 5.12 (m, 2H), 5.30 (t, J = 5.62 Hz, 1H), 5.75 – 5.93 (m, 1H), 6.85 – 7.03 (m, 4H), 7.04 – 7.15 (m, 2H), 7.26 – 7.39 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 11.3, 23.1, 38.5, 43.0, 117.6, 118.9, 119.8, 121.0, 123.0, 129.3, 132.9, 133.8, 146.9, 156.7. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO3S, 344.1315: found, 344.1307.
4-(4-(Allylthio)phenoxy)phenyl isopropylcarbamate (12c)
Yield: 93%. 1H NMR (300 MHz, CDCl3) δ 1.23 (d, 6H), 3.43 – 3.52 (m, 2H), 3.88 (dq, J = 13.84, 6.71 Hz, 1H), 4.96 – 5.14 (m, 3H), 5.76 – 5.94 (m, 1H), 6.85 – 7.03 (m, 4H), 7.04 – 7.14 (m, 2H), 7.27 – 7.38 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 22.9, 38.6, 43.5, 116.3, 117.7, 117.7, 118.9, 119.8, 121.0, 123.0, 133.0, 133.8, 146.8, 156.7. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO3S, 344.1315: found, 344.1313.
General Procedure for the Syntheses of Oxiranes 13a–d
A solution of compound 12 (0.95 mmol) in CH2Cl2 (10 mL) was cooled in an ice-water bath and was treated with m-CPBA (4.8 mmol). The mixture was stirred for 1 h, warmed to room temperature, and stirred for 6 days. The suspension was filtered and the filtrate was washed with saturated Na2S2O3, followed by saturated NaHCO3. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The desired product was purified by silica gel column chromatography.
4-(4-((Oxiran-2-ylmethyl)sulfonyl)phenoxy)phenyl methylcarbamate (13a)
Yield: 53%. 1H NMR (500 MHz, CDCl3) δ 2.45 (dd, J = 4.79, 1.79 Hz, 1H), 2.75 – 2.81 (m, 1H), 2.85 (d, J = 4.98 Hz, 3H), 3.24 – 3.34 (m, 3H), 5.32 (q, J = 4.59 Hz, 1H), 7.00 – 7.10 (m, 4H), 7.11 – 7.17 (m, 2H), 7.80 – 7.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 27.8, 45.9, 45.9, 59.6, 117.5, 121.3, 123.4, 130.6, 132.5, 148.2, 151.6, 155.2, 163.0. HRMS-ESI (m/z): [M+Na]+, calcd for C17H17NNaO6S, 386.0669: found, 386.0682.
4-(4-((Oxiran-2-ylmethyl)sulfonyl)phenoxy)phenyl ethylcarbamate (13b)
Yield: 52%. 1H NMR (500 MHz, CDCl3) δ 1.17 (t, J = 7.28 Hz, 3H), 2.44 (dd, J = 4.88, 2.09 Hz, 1H), 2.69 −2.81 (m, 1H), 3.20 – 3.32 (m, 5H), 5.40 (t, J = 5.58 Hz, 1H), 6.97 – 7.10 (m, 4H), 7.10 – 7.20 (m, 2H), 7.79 – 7.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 15.1, 36.2, 45.9, 45.9, 59.6, 117.5, 121.3, 123.4, 130.5, 132.5, 148.1, 151.5, 154.4, 162.9. HRMS-ESI (m/z): [M+Na]+, calcd for C18H19NNaO6S, 400.0825: found, 400.0849.
4-(4-((Oxiran-2-ylmethyl)sulfonyl)phenoxy)phenyl propylcarbamate (13c)
Yield: 64%. 1H NMR (500 MHz, CDCl3) δ 0.93 (t, J = 7.38 Hz, 3H), 1.56 (sxt, J = 7.30 Hz, 2H), 2.44 (dd, J = 4.79, 1.99 Hz, 1H), 2.74 – 2.82 (m, 1H), 3.14 – 3.23 (m, 2H), 3.23 – 3.32 (m, 3H), 5.40 (t, J = 5.78 Hz, 1H), 6.98 – 7.09 (m, 4H), 7.10 – 7.20 (m, 2H), 7.78 – 7.88 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 11.3, 23.0, 43.0, 45.9, 45.9, 59.6, 117.5, 121.3, 123.4, 130.6, 132.5, 148.2, 151.5, 154.6, 162.9. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO6S, 392.1162: found, 392.1153.
4-(4-((Oxiran-2-ylmethyl)sulfonyl)phenoxy)phenyl isopropylcarbamate (13d)
Yield: 50%. 1H NMR (500 MHz, CDCl3) δ 1.21 (d, J = 6.38 Hz, 6H), 2.45 (dd, J = 4.98, 1.60 Hz, 1H), 2.74 −2.82 (m, 1H), 3.22 – 3.34 (m, 3H), 3.86 (dq, J = 13.86, 6.68 Hz, 1H), 5.16 (d, J = 7.78 Hz, 1H), 6.98 – 7.10 (m, 4H), 7.15 (d, J = 8.77 Hz, 2H), 7.81 – 7.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 22.9, 43.6, 45.9, 45.9, 59.6, 117.5, 121.3, 123.5, 130.6, 132.5, 148.2, 151.6, 153.7, 163.0. HRMS-ESI (m/z): [M+Na]+, calcd for C19H21NNaO6S, 414.0982: found, 414.1006.
General Procedure for the Syntheses of Thiiranes 5a–d
To a solution of oxirane 13 (0.41 mmol) in CH2Cl2 (2 mL) was added a mixture of thiourea (0.82 mmol) in methanol (3 mL). The resulting mixture was stirred for 24 h at room temperature, after which the solvent was removed under reduced pressure. The residue was partitioned between CH2Cl2 and water. The organic layer was dried over anhydrous Na2SO4, and the suspension was filtered. The solution was concentrated in vacuo to afford the crude product, which was subsequently purified by silica gel column chromatography.
4-(4-((Thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl methylcarbamate (5a)
Yield: 90%. 1H NMR (500 MHz, CDCl3) δ 2.13 (dd, J = 5.18, 1.79 Hz, 1H), 2.51 (dd, J = 6.18, 1.79 Hz, 1H), 2.86 (d, J = 4.98 Hz, 3H), 3.03 (dq, J = 7.65, 5.69 Hz, 1H), 3.18 (dd, J = 14.16, 7.78 Hz, 1H), 3.50 (dd, J = 14.16, 5.78 Hz, 1H), 5.30 (q, J = 4.59 Hz, 1H), 7.00 – 7.11 (m, 4H), 7.11 – 7.18 (m, 2H), 7.81 – 7.87 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 24.3, 26.2, 27.8, 62.6, 117.6, 121.3, 123.5, 130.8, 131.9, 148.2, 151.6, 155.2, 163.0. HRMS-ESI (m/z): [M+Na]+, calcd for C17H17NNaO5S2, 402.0440: found, 402.0448.
4-(4-((Thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl ethylcarbamate (5b)
Yield: 80%. 1H NMR (500 MHz, CDCl3) δ 1.20 (t, J = 7.18 Hz, 3H), 2.14 (dd, J = 5.18, 1.79 Hz, 1H), 2.52 (dd, J = 6.18, 1.40 Hz, 1H), 3.04 (dq, J = 7.45, 5.76 Hz, 1H), 3.17 (dd, J = 14.16, 7.78 Hz, 1H), 3.24 −3.36 (m, 2H), 3.51 (dd, J = 14.26, 5.68 Hz, 1H), 5.25 (t, J = 5.28 Hz, 1H), 7.08 (d, J = 8.77 Hz, 2H), 7.05 (d, J = 8.77 Hz, 2H), 7.17 (d, J = 8.97 Hz, 2H), 7.85 (d, J = 8.97 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 15.2, 24.3, 26.2, 36.3, 62.7, 117.7, 121.3, 123.5, 130.8, 132.0, 148.2, 151.7, 154.4, 163.1. HRMS-ESI (m/z): [M+H]+, calcd for C18H20NO5S2, 394.0777: found, 394.0798.
4-(4-((Thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl propylcarbamate (5c)
Yield: 76%. 1H NMR (300 MHz, CDCl3) δ 0.96 (t, J = 7.41 Hz, 3H), 1.60 (sxt, J = 7.32 Hz, 2H), 2.15 (dd, J = 5.14, 1.79 Hz, 1H), 2.52 (dd, J = 5.86, 1.55 Hz, 1H), 3.00 – 3.10 (m, 1H), 3.15 – 3.29 (m, 3H), 3.52 (dd, J = 14.23, 5.62 Hz, 1H), 5.24 (t, J = 5.86 Hz, 1H), 7.00 – 7.13 (m, 4H), 7.13 – 7.22 (m, 2H), 7.80 – 7.89 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 11.4, 23.2, 24.4, 26.2, 43.1, 62.7, 76.8, 77.2, 77.7, 117.7, 121.3, 123.5, 130.9, 132.0, 148.2, 151.7, 154.6, 163.1. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO5S2, 408.0934: found, 408.0945.
4-(4-((Thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl isopropylcarbamate (5d)
Yield: 87%. 1H NMR (500 MHz, CDCl3) δ 1.21 (d, J = 6.58 Hz, 6H), 2.12 (dd, J = 5.18, 1.79 Hz, 1H), 2.50 (dd, J = 6.08, 1.50 Hz, 1H), 3.02 (dq, J = 7.38, 5.78 Hz, 1H), 3.17 (dd, J = 14.16, 7.78 Hz, 1H), 3.49 (dd, J = 14.26, 5.68 Hz, 1H), 3.86 (dq, J = 13.81, 6.70 Hz, 1H), 5.20 (d, J = 7.78 Hz, 1H), 7.06 (d, J = 8.77 Hz, 2H), 7.03 (d, J = 8.97 Hz, 2H), 7.15 (d, J = 8.97 Hz, 2H), 7.78 – 7.87 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 22.8, 24.3, 26.1, 43.5, 62.6, 117.6, 121.2, 123.5, 130.7, 131.9, 148.1, 151.5, 153.6, 163.0. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO5S2, 408.0934: found, 408.0957.
Syntheses of the Urea Derivatives of 3 (6a–d)
1-Methyl-3-(4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)urea (6a)
PNP-chloroformate (36 mg, 0.17 mmol) was dissolved in dry CH2Cl2 (2 mL) and cooled in an ice-water bath. Thiirane 3 (46 mg, 0.14 mmol) in dry CH2Cl2 (2 mL) was added dropwise and the resulting mixture was stirred for 30 min in an ice-water bath. It was diluted with CH2Cl2, washed with saturated NH4Cl, and concentrated in vacuo. The crude intermediate (4-nitrophenyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)carbamate) was reconstituted in THF (3 mL), treated with MeNH2 (0.28 mmol) in THF, and stirred for 15 min at room temperature. The mixture was diluted with CH2Cl2 and washed with saturated NaHCO3, followed by water. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. Purification of the crude product by silica gel chromatography gave 29 mg (53%) of compound 6a. 1H NMR (500 MHz, CDCl3/CD3OD) δ 1.94 (dd, J = 5.08, 1.69 Hz, 1 H), 2.32 (dd, J = 6.18, 1.79 Hz, 1 H), 2.59 (s, 3 H), 2.77 – 2.86 (m, J = 7.40, 6.12, 6.12, 5.28 Hz, 1 H), 3.06 (dd, J = 14.36, 7.38 Hz, 1 H), 3.30 (dd, J = 14.36, 5.98 Hz, 1 H), 6.75 – 6.83 (m, 2 H), 6.83 – 6.90 (m, 2 H), 7.18 – 7.25 (m, 2 H), 7.59 – 7.66 (m, 2 H). 13C NMR (126 MHz, CDCl3/CD3OD) δ 23.6, 25.7, 26.0, 62.3, 117.0, 120.4, 120.9, 130.5, 130.9, 137.0, 149.0, 157.3, 163.6. HRMS-ESI (m/z): [M+H]+, calcd for C17H19N2O4S2, 379.0781: found, 379.0767.
General Procedure for the Syntheses of Thiiranes 6b–d
A suspension of thiirane 3 as the HCl salt (80 mg, 0.17 mmol) in THF (3 mL) was mixed with N-methylmorpholine (22 µL, 0.20 mmol). The alkyl isocyanate (0.34 mmol) was added dropwise and the reaction was stirred for 24 h at room temperature. The mixture was concentrated in vacuo and the desired urea derivative was purified by silica gel column chromatography.
1-Ethyl-3-(4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)urea (6b)
Yield: 61%. 1H NMR (500 MHz, CDCl3/CD3OD) δ 0.92 (t, J = 7.18 Hz, 3H), 1.90 (dd, J = 5.08, 1.69 Hz, 1H), 2.23 – 2.32 (m, 1H), 2.73 – 2.83 (m, 1H), 2.94 – 3.12 (m, 3H), 3.26 (dd, J = 14.36, 5.98 Hz, 1H), 6.71 – 6.79 (m, 2H), 6.79 – 6.87 (m, 2H), 7.10 – 7.22 (m, 2H), 7.54 – 7.64 (m, 2H). 13C NMR (126 MHz, CDCl3/CD3OD) δ 14.7, 23.5, 25.6, 34.3, 62.2, 116.8, 117.0, 120.3, 120.8, 130.3, 130.5, 130.8, 136.9, 148.9, 156.5, 163.5. HRMS-ESI (m/z): [M+H]+, calcd for C18H21N2O4S2, 393.0937: found, 393.0923.
1-Propyl-3-(4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)urea (6c)
Yield: 58%. 1H NMR (500 MHz, CDCl3/CD3OD): δ 0.67 (t, J = 7.5 Hz, 3H), 1.27 (sxt, J = 7.3 Hz, 2H), 1.86 (dd, J = 5.1, 1.7 Hz, 1H), 2.23 (dd, J = 6 .2, 1.6 Hz, 1H), 2.74 (dt, J = 12.5, 6.1Hz, 1H), 2.89 (t, J = 7.1Hz, 2H), 3.01 (dd, J = 14.3, 7.5 Hz, 1H), 3.22 (dd, J = 14.4, 6.0 Hz, 1H), 6.64 – 6.75 (m, 2H), 6.76 – 6.83 (m, 2H), 7.11 – 7.18 (m, 2H), 7.52 – 7.58 (m, 2H). 13C NMR (126 MHz, CDCl3/CD3OD) δ 10.7, 22.9, 23.3, 25.5, 41.2, 62.1, 116.8, 116.9, 120.2, 120.7, 130.3, 130.4, 130.8, 136.9, 148.8, 156.5, 163.4. HRMS-ESI (m/z): [M+H]+, calcd for C19H23N2O4S2, 407.1094: found, 407.1098.
1-Isopropyl-3-(4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)urea (6d)
Yield: 55%. 1H NMR (500 MHz, CDCl3/CD3OD) δ 0.82 (d, J = 6.38 Hz, 6H), 1.78 (dd, J = 5.08, 1.69 Hz, 1H), 2.16 (dd, J = 6.18, 1.60 Hz, 1H), 2.62 – 2.69 (m, 1H), 2.91 – 3.01 (m, 2H), 3.15 (dd, J = 14.36, 6.18 Hz, 1H), 3.55 (dt, J = 13.11, 6.50 Hz, 1H), 6.61 – 6.66 (m, 2H), 6.69 – 6.75 (m, 2H), 7.02 – 7.09 (m, 2H), 7.43 – 7.52 (m, 2H). 13C NMR (126 MHz, CDCl3/CD3OD) δ 22.2, 23.1, 25.3, 41.2, 61.9, 116.6, 116.7, 120.1, 120.6, 130.2, 130.3, 130.8, 136.8, 148.8, 155.7, 163.4. HRMS-ESI (m/z): [M+H]+, calcd for C19H23N2O4S2, 407.1094: found, 407.1114.
Syntheses of the Carbamate Derivatives of 3 (7a–d)
General Procedure for the Syntheses of Thiiranes 7a–d
A suspension of thiirane 3 as the HCl salt (60 mg, 0.17 mmol) in THF (3 mL) was mixed with N-methylmorpholine (22 µL, 0.20 mmol) in an ice-water bath. The alkyl chloroformate (0.34 mmol) was added dropwise and the reaction was stirred for 20 min. It was concentrated in vacuo and reconstituted in ethyl acetate. The resulting mixture was washed with water, dried with anhydrous Na2SO4, and evaporated to dryness under reduced pressure. The desired product was purified by silica gel column chromatography.
Methyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)carbamate (7a)
Yield: 80%. 1H NMR (500 MHz, CDCl3) δ 2.16 (dd, J = 5.18, 1.79 Hz, 1H), 2.54 (dd, J = 6.18, 1.60 Hz, 1H), 3.06 (dq, J = 7.63, 5.70 Hz, 1H), 3.18 (dd, J = 14.26, 7.88 Hz, 1H), 3.52 (dd, J = 14.26, 5.48 Hz, 1H), 3.79 (s, 3H), 6.89 (s, 1H), 7.00 – 7.10 (m, 4H), 7.46 (d, J = 8.37 Hz, 2H), 7.82 – 7.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 24.4, 26.3, 52.7, 62.8, 117.5, 120.7, 121.4, 130.9, 131.8, 135.5, 150.3, 154.3, 163.4. HRMS-ESI (m/z): [M+Na]+, calcd for C17H17NNaO5S2, 402.0440: found, 402.0447.
Ethyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)carbamate (7b)
Yield: 80%. 1H NMR (500 MHz, CDCl3) δ 1.31 (t, J = 7.08 Hz, 3H), 2.15 (dd, J = 4.98, 1.79 Hz, 1H), 2.53 (dd, J = 5.98, 1.40 Hz, 1H), 3.00 – 3.09 (m, 1H), 3.18 (dd, J = 14.26, 7.88 Hz, 1H), 3.52 (dd, J = 14.26, 5.68 Hz, 1H), 4.23 (q, J = 7.18 Hz, 2H), 6.89 (s, 1H), 6.98 – 7.09 (m, 4H), 7.46 (d, J = 8.37 Hz, 2H), 7.80 – 7.88 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 14.7, 24.4, 26.2, 61.5, 62.8, 117.5, 120.7, 121.4, 130.9, 131.8, 135.6, 150.2, 153.9, 163.4. HRMS-ESI (m/z): [M+Na]+, calcd for C18H19NNaO5S2, 416.0597: found, 416.0607.
Propyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)carbamate (7c)
Yield: 76%. 1H NMR (500 MHz, CDCl3) δ 0.98 (t, J = 7.38 Hz, 3H), 1.71 (sxt, J = 7.14 Hz, 2H), 2.16 (dd, J = 4.98, 1.79 Hz, 1H), 2.53 (dd, J = 6.18, 1.20 Hz, 1H), 3.06 (dq, J = 7.68, 5.75 Hz, 1H), 3.17 (dd, J = 14.16, 7.78 Hz, 1H), 3.53 (dd, J = 14.06, 5.48 Hz, 1H), 4.14 (t, J = 6.68 Hz, 2H), 6.84 (s, 1H), 6.99 – 7.09 (m, 4H), 7.46 (d, J = 8.57 Hz, 2H), 7.81 – 7.88 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 10.5, 22.4, 24.4, 26.3, 62.8, 67.2, 117.5, 120.6, 121.4, 130.9, 131.8, 135.6, 150.2, 154.0, 163.5. HRMS-ESI (m/z): [M+Na]+, calcd for C19H21NNaO5S2, 430.0753: found, 430.0756.
Isopropyl (4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)phenyl)carbamate (7d)
Yield: 49%. 1H NMR (500 MHz, CDCl3) δ 1.31 (d, J = 6.38 Hz, 6H), 2.16 (dd, J = 5.08, 1.69 Hz, 1H), 2.54 (dd, J = 6.08, 1.50 Hz, 1H), 3.06 (dq, J = 7.70, 5.67 Hz, 1H), 3.17 (dd, J = 14.16, 7.78 Hz, 1H), 3.53 (dd, J = 14.16, 5.58 Hz, 1H), 4.94 – 5.10 (m, 1H), 6.72 (s, 1H), 6.98 – 7.12 (m, 4H), 7.46 (d, J = 8.57 Hz, 2H), 7.79 – 7.90 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 21.6, 22.3, 24.4, 26.3, 62.8, 117.5, 120.6, 121.4, 130.9, 131.8, 135.7, 148.3, 150.2, 163.5. HRMS-ESI (m/z): [M+H]+, calcd for C19H22NO5S2, 408.0934: found, 408.0940.
Enzyme Inhibition Studies
Human recombinant active MMP-2 and MMP-7 and catalytic domains of MMP-3 and MMP-14 were purchased from EMD Biosciences (La Jolla, CA, USA). The catalytic domains of human recombinant MMP-1, MMP-8 and MMP-9 were from Biomol International (Plymouth Meeting, PA, USA). Fluorogenic substrate MOCAcPLGLA2pr(Dnp)AR-NH2 was purchased from Peptides International (Louisville, KY, USA); MOCAcRPKPVE(Nva)WRK(Dnp)-NH2, (Dnp)P(Cha)GC(Me)HAK(NMa)NH2 and McaKPLGL(Dpa)AR-NH2 were obtained from R&D Systems (Minneapolis, MN, USA). The Km values used for the reaction of MMP-2, MMP-9cat and MMP-14cat with the fluorogenic substrate MOCAcPLGLA2pr(Dnp)AR-NH2 were 18.9 ± 1.0, 5.0 ± 0.1, 5.6 ± 0.4 µM, respectively. The Km value used for the reaction of MMP-8cat with the fluorogenic substrate McaKPLGL(Dpa)AR-NH2 was 13.3 ± 1.89 µM. Inhibitor stock solutions (10 mM) were prepared in DMSO. The methodology for enzyme inhibition and assays was the same as reported previously.10 Substrate hydrolysis was measured with a Cary Eclipse fluorescence spectrophotometer (Varian, Walnut Creek, CA, USA). Compounds 4–7 were stable in the buffers that were used in the kinetic assays.
Ultra Performance Liquid Chromatography (UPLC)
The chromatographic system consisted of a Waters Acquity UPLC System (Waters Corporation, Milford, MA, USA) equipped with a binary solvent manager, an autosampler, a column heater and a photodiode array detector. Samples were analyzed on an Acclaim RSLC 120 C18 column (2.2 µm, 2.1 mm i.d. × 100 mm, Dionex, Sunnyvale, CA, USA). The mobile phase consisted of elution at 0.5 mL/min with 85% A/15% B for 1 min, followed by a 6-min linear gradient to 10% A/90% B and then 10% A/90% B for 2 min (A = water; B = acetonitrile). Effluent was monitored by UV detection at 245 nm.
Multiple-Reaction Monitoring (MRM)-Mass spectrometry
All MRM-mass spectrometric experiments were performed with a Waters TQD tandem quadrupole detector (Milford, MA, USA) monitored with MassLynx MS software. Mass spectrometry acquisition was performed in the positive or negative electrospray ionization mode with MRM. The capillary voltage, cone voltage, extractor voltage, and RF lens voltage were set at 2.8 kV, 35 V, 3 V, and 0.3 V, respectively. Desolvation gas flow rate was 650 L/h (nitrogen). The cone gas flow rate was set at 20 L/h (nitrogen) for (+)-ESI-MRM, and at 50 L/h (nitrogen) for (−)-ESI-MRM. The source temperature and desolvation temperature were held at 150 °C and 350 °C, respectively. Samples were analyzed on an Acclaim RSLC 120 C18 column (2.2 µm, 2.1 mm i.d. × 100 mm, Dionex, Sunnyvale, CA, USA). The mobile phase consisted of elution at 0.5 mL/min with 85% A/15% B for 1 min, followed by a 6-min linear gradient to 10% A/90% B and then 10% A/90% B for 2 min (for UPLC/(+)-ESI: A = 1% formic acid in water; B = 1% formic acid in acetonitrile; for UPLC/(−)-ESI: A = water; B = acetonitrile).
Liquid Chromatography/ MicroTOF-Q II Mass Spectrometry
The LC system consisted of a Dionex Ultimate 3000 RSLC (Dionex, Sunnyvale, CA, USA) equipped with Dionex Ultimate 3000 autosampler and Dionex Ultimate 3000 diode array detector. Mass spectrometric experiments were performed with a Bruker MicroTOF-Q II (Billerica, MA, USA) monitored with Hystar 3.2 software. Mass spectrometry acquisition was performed using electrospray ionization with the following parameters: end plate offset voltage = −0.5 kV, capillary voltage = 4.5 kV, nitrogen as both nebulizer and dry gas (8.0 L/min flow rate, 200 °C dry temperature, 4.0 bar). Samples were analyzed on Acclaim RSLC 120 C18 column (2.2 µm, 2.1 mm i.d. × 100 mm, Dionex, Sunnyvale, CA, USA). The mobile phase consisted of elution at 0.5 mL/min with 85% A/15% B for 3 min, followed by a 9-min linear gradient to 10% A/90% B, then 10% A/90% B for 2 min (A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile).
Microsomal Stability
Incubations consisted of male rat liver S9 (1.0 mg, BD Biosciences, Woburn, MA, USA), NADPH (0.5 mM, final concentration), and 10 µM compound (final concentration) in potassium phosphate buffer (50 mM, pH 7.4) at 37 °C in a total volume of 1 mL. Aliquots were drawn at different time points and the reactions were terminated by addition of one volume of acetonitrile containing 1 µM of internal standard 14 or 15, synthesized as described previously.10 The precipitated protein was centrifuged (10 min, 10,000 g) and the supernatant was analyzed by reversed-phase UPLC/ESI-MRM. Compounds 2, 4b, 6b, 7a, 7b, and internal standard 14 were analyzed by (−)-ESI-MRM of the transitions 321→184 for 2, 393→321 for 4b, 391→256 for 6b, 378→274 for 7a, 392→274 for 7b, and 398→261 for 14. Compounds 3, 5b, and internal standard 15 were analyzed by (+)-ESI-MRM of the transitions 322→185, 394→185, and 300→93, respectively. Peak area ratios relative to the internal standard were measured and the half-lives of the compounds in male rat liver S9 were calculated according to the first-order rate law.
Metabolite Identification
Compounds (50 µM of 5b and 7a) were incubated with rat liver S9 (1.0 mg) and 0.5 mM NADPH at 37°C for 30 min in 50 mM potassium phosphate buffer at pH 7.4. The incubations were terminated by addition of one volume of acetonitrile. The precipitated protein was centrifuged (10 min, 13,000 g) and the supernatant was analyzed by reversed-phase LC/microTOF-Q II mass spectrometry.
Computational Method
Coordinates of the thiirane analogs were built in Maestro v 9.3 environment and energy minimized using LigPrep v2.52. (Schrödinger LLC). MMP-2 protein structure coordinate, which was obtained from a previous study,31 was prepared for docking using the Protein Preparation Wizard protocol in Schrödinger Suite 2012. A constrained energy minimization using the OPLS2005 forcefield was carried out with a 0.30 Å RMSD convergence threshold. The docking grid was calculated for a cubic box that would allow ligands up to 20 Å length to dock at the catalytic site centered on the ligand. A core-constraint for the thiirane ring for 1 Å was enforced in the Glide SP v5.8 docking and top scoring poses were retained for further inspection.32
Animals
Mice (female C57Bl/6J, 6–8 weeks old, 16–19 g body weight, specific pathogen free, n = 3 per time point) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were provided with Teklad 2019 Extruded Rodent Diet (Harlan, Madison, WI, USA) and water ad libitum. Animals were maintained in polycarbonate shoebox cages with hardwood bedding in a room under a 12:12 h light:dark cycle and at 72 ± 2 °F. All procedures were performed in accordance with the University of Notre Dame Institutional Animal Care and Use Committee.
Animal Dosing and Sample Collection
Compound 5b was formulated as a solution at a concentration of 5.0 mg/mL in 10% DMSO/25% Tween 80/65% water. Compound 6b was dissolved in 80% propylene glycol/20% DMSO. The solutions were sterilized by filtration through an Acrodisc syringe filter (Pall Life Sciences, 0.2 µm, 13 mm diameter, PTFE membrane). Mice (n = 3 per time point) were administered a single 100 µL subcutaneous (sc) dose of compound 5b or 6b (equivalent to 25 mg/kg). Terminal blood samples (n=3 per time point) were collected in heparin through the posterior vena cava at specific time points. Blood samples were stored on ice and centrifuged to obtain plasma. Whole brain samples were harvested after transcardiac perfusion with saline, weighed, and immediately flash-frozen in liquid nitrogen and stored at −80 °C until analysis.
Sample Analysis
A 50-µL aliquot of plasma was mixed with 100-µL of internal standard 14 or 15 in acetonitrile to a final concentration of 5 µM. The sample was centrifuged at 20 000g for 15 min. Brain samples were weighed and homogenized for 5 min in one volume equivalent of cold acetonitrile containing internal standard 14 or 15 using a bullet blender (Next Advance, Inc., Averill Park, NY, USA) at 4 °C. The brain homogenates were centrifuged at 20 000g for 20 min at 4 °C. The plasma and brain supernatants were analyzed by reversed-phase UPLC/ESI-MRM of the transitions 394→320 for 5b (positive ion), 391→256 for 6b (negative ion), 321→184 for 2 (negative ion), 322→185 for 3 (positive ion), 398→261 (negative ion) for internal standard 14, and 300→93 for internal standard 15. Calibration curves of compounds 2, 3, 5b, and 6b were prepared by fortification of blank mouse plasma and blank mouse brain with compounds 2, 3, 5b, and 6b at concentrations up to 50 µM. Quantification was performed by using peak area ratios relative to the internal standard and linear regression parameters calculated from the calibration curve standards. The assay was linear from 0.05 to 5 µM, with a coefficient of determination, R2, of 0.994 for 2 and 0.991 for 6b.
Mass spectrometry acquisition of compound 2 was performed in the negative ESI mode with MRM. The capillary voltage, cone voltage, extractor voltage, and RF lens voltage were set at 3.0 kV, 30 V, 3 V, and 0.1 V, respectively. Desolvation gas flow rate was 450 L/h (nitrogen) and the cone gas flow rate was 50 L/h (nitrogen). The source temperature and desolvation temperature were held at 150 °C and 250 °C, respectively. Samples were analyzed on an Acquity BEH Shield RP 18 column (1.7 µm, 2.1 mm i.d. × 100 mm, Waters). The mobile phase consisted of elution at 0.5 mL/min with a 2-min linear gradient from 40% water/60% acetonitrile to 20% water/80% acetonitrile, followed by a 6-min linear gradient to 100% acetonitrile.
Pharmacokinetic Parameters
The area under the mean concentration-time curve up to the last quantifiable sampling time (AUC0-last) was calculated by the trapezoidal rule using PK Solutions software version 2.0 (Summit Research Services, Montrose, CO, USA). AUC0-∞ was calculated as AUC0-last + (Clast/k), where Clast is the concentration at the last quantifiable sampling time and k is the elimination rate constant. Half-lives (t½α and t½β) were estimated from the linear portion of the concentration-time data by linear regression, where the slope of the line is the rate constant k and t½ = ln2/k.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant CA122417 from the National Institutes of Health (to M.C. and S.M.). M.G. is a Fellow of the Chemistry-Biochemistry-Biology Interface (CBBI) Program at the University of Notre Dame, supported by training grant T32 GM075762.
ABBREVIATIONS USED
- AUC
area under the concentration-time curve
- BBB
blood-brain barrier
- m-CBPA
meta-chloroperbenzoic acid
- CNS
central nervous system
- DMSO
dimethyl sulfoxide
- Dnp
2,4-dinitrophenyl
- ESI
electrospray ionization
- MMP
matrix metalloproteinase
- MOCAc
(7-methoxycoumarin-4-yl)acetyl
- MOM
methoxymethyl
- MRM
multiple reaction monitoring
- MS
mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate reduced
- sc
subcutaneous
- TEA
triethylamine
- UPLC
ultra-performance liquid chromatography
Footnotes
ASSOCIATED CONTENT
Supporting Information
Table S1 showing the kinetic parameters for the slow-binding inhibition of MMPs; Tables S2 and S3 reporting the concentrations of compounds 2 and 6b in plasma and brain. This material is available free of charge via the Internet at http://pubs.acs.org.
ANCILLARY INFORMATION
PDB ID for the TOC graphic: 3MA2 (MMP-14 coordinate) and PDB ID: 1CK7 (MMP-2 coordinate).
REFERENCES
- 1.Fokas E, Steinbach JP, Rodel C. Biology of brain metastases and novel targeted therapies: Time to translate the research. Biochim Biophys Acta. 2013;1835:61–75. doi: 10.1016/j.bbcan.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22:2865–2872. doi: 10.1200/JCO.2004.12.149. [DOI] [PubMed] [Google Scholar]
- 3.Xie TX, Huang FJ, Aldape KD, Kang SH, Liu M, Gershenwald JE, Xie K, Sawaya R, Huang S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188–3196. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 4.Preusser M, Capper D, Ilhan-Mutlu A, Berghoff AS, Birner P, Bartsch R, Marosi C, Zielinski C, Mehta MP, Winkler F, Wick W, von Deimling A. Brain metastases: pathobiology and emerging targeted therapies. Acta Neuropathol. 2012;123:205–222. doi: 10.1007/s00401-011-0933-9. [DOI] [PubMed] [Google Scholar]
- 5.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 7.Taskar KS, Rudraraju V, Mittapalli RK, Samala R, Thorsheim HR, Lockman J, Gril B, Hua E, Palmieri D, Polli JW, Castellino S, Rubin SD, Lockman PR, Steeg PS, Smith QR. Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharm Res. 2012;29:770–781. doi: 10.1007/s11095-011-0601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, Wilner KD. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29:e443–e445. doi: 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 9.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 12.Devy L, Dransfield DT. New strategies for the next generation of matrix-metalloproteinase inhibitors: Selectively targeting membrane-anchored MMPs with therapeutic antibodies. Biochem Res Int. 2011;2011:191670. doi: 10.1155/2011/191670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 14.Talvensaari-Mattila A, Paakko P, Hoyhtya M, Blanco-Sequeiros G, Turpeenniemi-Hujanen T. Matrix metalloproteinase-2 immunoreactive protein: a marker of aggressiveness in breast carcinoma. Cancer. 1998;83:1153–1162. doi: 10.1002/(sici)1097-0142(19980915)83:6<1153::aid-cncr14>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Still K, Robson CN, Autzen P, Robinson MC, Hamdy FC. Localization and quantification of mRNA for matrix metalloproteinase-2 (MMP-2) and tissue inhibitor of matrix metalloproteinase-2 (TIMP-2) in human benign and malignant prostatic tissue. Prostate. 2000;42:18–25. doi: 10.1002/(sici)1097-0045(20000101)42:1<18::aid-pros3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Koshiba T, Hosotani R, Wada M, Miyamoto Y, Fujimoto K, Lee JU, Doi R, Arii S, Imamura M. Involvement of matrix metalloproteinase-2 activity in invasion and metastasis of pancreatic carcinoma. Cancer. 1998;82:642–650. doi: 10.1002/(sici)1097-0142(19980215)82:4<642::aid-cncr5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 17.Nomura H, Fujimoto N, Seiki M, Mai M, Okada Y. Enhanced production of matrix metalloproteinases and activation of matrix metalloproteinase 2 (gelatinase A) in human gastric carcinomas. Int J Cancer. 1996;69:9–16. doi: 10.1002/(SICI)1097-0215(19960220)69:1<9::AID-IJC3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Rojiani MV, Alidina J, Esposito N, Rojiani AM. Expression of MMP-2 correlates with increased angiogenesis in CNS metastasis of lung carcinoma. Int J Clin Exp Pathol. 2010;3:775–781. [PMC free article] [PubMed] [Google Scholar]
- 19.Brown S, Bernardo MM, Li ZH, Kotra LP, Tanaka Y, Fridman R, Mobashery S. Potent and selective mechanism-based inhibition of gelatinases. J Am Chem Soc. 2000;122:6799–6800. [Google Scholar]
- 20.Forbes C, Shi Q, Fisher JF, Lee M, Hesek D, Llarrull LI, Toth M, Gossing M, Fridman R, Mobashery S. Active site ring-opening of a thiirane moiety and picomolar inhibition of gelatinases. Chem Biol Drug Des. 2009;74:527–534. doi: 10.1111/j.1747-0285.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee M, Villegas-Estrada A, Celenza G, Boggess B, Toth M, Kreitinger G, Forbes C, Fridman R, Mobashery S, Chang M. Metabolism of a highly selective gelatinase inhibitor generates active metabolite. Chem Biol Drug Des. 2007;70:371–382. doi: 10.1111/j.1747-0285.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 22.Gooyit M, Lee M, Schroeder VA, Ikejiri M, Suckow MA, Mobashery S, Chang M. Selective water-soluble gelatinase inhibitor prodrugs. J Med Chem. 2011;54:6676–6690. doi: 10.1021/jm200566e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonfil RD, Dong Z, Trindade Filho JC, Sabbota A, Osenkowski P, Nabha S, Yamamoto H, Chinni SR, Zhao H, Mobashery S, Vessella RL, Fridman R, Cher ML. Prostate cancer-associated membrane type 1-matrix metalloproteinase: a pivotal role in bone response and intraosseous tumor growth. Am J Pathol. 2007;170:2100–2111. doi: 10.2353/ajpath.2007.060720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M, Ikejiri M, Klimpel D, Toth M, Espahbodi M, Hesek D, Forbes C, Kumarasiri M, Noll BC, Chang M, Mobashery S. Structure-activity relationship for thiirane-based gelatinase inhibitors. ACS Med Chem Lett. 2012;3:490–495. doi: 10.1021/ml300050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minkkila A, Myllymaki MJ, Saario SM, Castillo-Melendez JA, Koskinen AM, Fowler CJ, Leppanen J, Nevalainen T. The synthesis and biological evaluation of para-substituted phenolic N-alkyl carbamates as endocannabinoid hydrolyzing enzyme inhibitors. Eur J Med Chem. 2009;44:2994–3008. doi: 10.1016/j.ejmech.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Mayer P, Brunel P, Chaplain C, Piedecoq C, Calmel F, Schambel P, Chopin P, Wurch T, Pauwels PJ, Marien M, Vidaluc JL, Imbert T. New substituted 1-(2,3-dihydrobenzo[1, 4]dioxin-2-ylmethyl)piperidin-4-yl derivatives with alpha(2)-adrenoceptor antagonist activity. J Med Chem. 2000;43:3653–3664. doi: 10.1021/jm991121g. [DOI] [PubMed] [Google Scholar]
- 27.Pochetti G, Montanari R, Gege C, Chevrier C, Taveras AG, Mazza F. Extra binding region induced by non-zinc chelating inhibitors into the S1' subsite of matrix metalloproteinase 8 (MMP-8) J Med Chem. 2009;52:1040–1049. doi: 10.1021/jm801166j. [DOI] [PubMed] [Google Scholar]
- 28.Gooyit M, Lee M, Hesek D, Boggess B, Oliver AG, Fridman R, Mobashery S, Chang M. Synthesis, kinetic characterization and metabolism of diastereomeric 2-(1-(4-phenoxyphenylsulfonyl)ethyl)thiiranes as potent gelatinase and MT1-MMP inhibitors. Chem Biol Drug Des. 2009;74:535–546. doi: 10.1111/j.1747-0285.2009.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gooyit M, Suckow MA, Schroeder KL, Wolter WR, Mobashery S, Chang M. Selective gelatinase inhibitor neuroprotective agents cross the blood-brain barrier. ACS Chem Neurosci. 2012;3:730–736. doi: 10.1021/cn300062w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. Potent mechanism-based inhibitors for matrix metalloproteinases. J Biol Chem. 2005;280:33992–34002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 31.Tao P, Fisher JF, Shi Q, Mobashery S, Schlegel HB. Matrix metalloproteinase 2 (MMP2) inhibition: DFT and QM/MM studies of the deprotonation-initialized ring-opening reaction of the sulfoxide analogue of SB-3CT. J Phys Chem B. 2009;114:1030–1037. doi: 10.1021/jp909327y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.