

Abstract
Hepatitis C virus (HCV) is a major global public health problem. While the current standard of care, a direct-acting antiviral (DAA) protease inhibitor taken in combination with pegylated interferon and ribavirin, represents a major advancement in recent years, an unmet medical need still exists for treatment modalities that improve upon both efficacy and tolerability. Towards those ends, much effort has continued to focus on the discovery of new DAAs, with the ultimate goal to provide interferon-free combinations. The RNA-dependent RNA polymerase enzyme NS5B represents one such DAA therapeutic target for inhibition which has attracted much interest over the past decade. Herein, we report the discovery and optimization of a novel series of inhibitors of HCV NS5B, through the use of structure-based design applied to a fragment-derived starting point. Issues of potency, pharmacokinetics and early safety were addressed in order to provide a clinical candidate in fluoropyridone 19.
INTRODUCTION
Hepatitis C virus (HCV) represents a major global public health problem. Over 150 million individuals suffer from chronic infection, with over 350,000 deaths each year attributed to hepatitis C-related liver diseases such as cirrhosis and liver cancer.1 While vaccines exist for some other hepatitis viruses, there are none available for HCV. Up until 2011, standard of care (SOC) treatment consisted of pegylated interferon (Peg-IFN) in combination with the antiviral ribavirin. Numerous shortcomings existed for this treatment regimen, including success rates of only ~ 50% sustained virologic response (SVR) in patients with HCV genotype 1 (GT-1, by far the most common genotype worldwide, accounting for ~ 75% of all cases), and generally poor tolerability.2-4 Much effort has recently focused on the discovery of direct-acting antivirals (DAAs), which can intervene and interfere at specific points within the viral life cycle.5-9 The first two DAA therapeutics to reach the market, NS3/4A protease inhibitors boceprevir and telaprevir, did so in 2011 and are currently being used in combination with the previous SOC, boosting SVR to 70-80% for GT-1.10,11 Unmet needs remain, however, to further increase the SVR in both treatment naïve and treatment experienced patients, as well as in patients comorbid with other infections, such as HIV. Also, elimination of side effects associated with IFN is highly desirable in terms of tolerability and patient compliance, and towards that end much research continues into the discovery of new DAAs that may ultimately be used in combination with each other as part of an IFN-free drug cocktail.12,13
HCV is a virus of the family Flaviviridae, and contains a positive sense, single-stranded RNA genome. The viral life cycle consists of 8 major stages, including entry (endocytosis), uncoating (fusion), polyprotein synthesis from (+)-RNA, cleavage of the polyprotein into individual proteins (structural and non-structural), RNA replication, viral packaging and maturation, release, and re-infection (Figure 1).14 Any of those stages represent opportunities for direct-acting antiviral therapeutic intervention. As already mentioned, inhibitors of the NS3/4A protease enzyme have now reached the market and are currently in use in combination with the previous SOC. The RNA-dependent RNA polymerase enzyme NS5B (enlarged section of Figure 1) represents another DAA therapeutic target for inhibition, and has been the focus of intense research in recent years.15-18 Early efforts focused on identification of nucleoside inhibitors which engage at the catalytic site, ultimately leading to the discovery of some promising therapeutic agents now in late-stage clinical trials.19 More recently, multiple allosteric sites have been identified on NS5B, including the so-called thumb I, thumb II, palm I and palm II sites.16 Non-nucleoside inhibitors have been reported for all of these sites, including several drug candidates that are currently under clinical evaluation.20-24
Figure 1.

HCV life cycle.14
Previously, we reported the discovery of a novel series of benzothiazine-substituted quinolinediones as inhibitors of NS5B, acting at the palm I allosteric site.25 One example from that series is compound 1 (Figure 2). Although we identified highly potent inhibitors, oral exposures in preclinical in vivo pharmacokinetics studies were not sufficient to reach targeted levels, and ultimately we looked for new chemotypes from which to optimize towards a clinical candidate. More recently, we reported the pioneering use of de novo fragment design that enabled us to generate a novel, low molecular weight (MW) lead structure (compound 2, Figure 2).26 Pyridone 2 exhibits potent inhibition of NS5B enzymatic activity (Table 1), especially considering its small size. Given the low MW and high ligand efficiency (LE = 0.46), we felt 2 was a reasonable starting place from which to develop potent inhibitors of NS5B that would also possess desirable physicochemical and ADMET properties required for a safe, orally delivered drug candidate.
Figure 2.

Previously reported Roche inhibitors of NS5B.
Table 1.
Core-to-Sulfonamide Linkages.
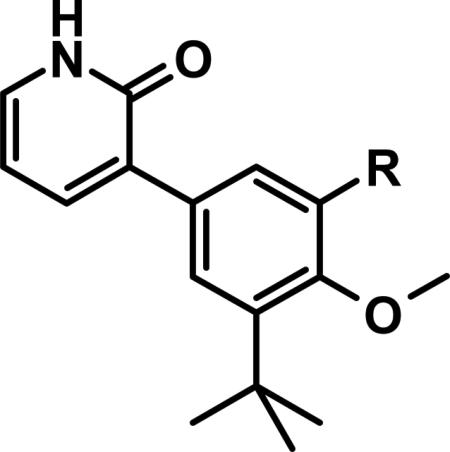
| ||||
|---|---|---|---|---|
| Cmpd | R | NS5B Enzyme IC50 (nM)a | GTla-Replicon EC50 (nM)b | GT1b-Replicon EC50 (nM)b |
| 2 | H | 435 | 8260 | 3860 |
| 3 |
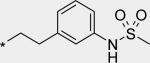
|
45 | -- | 912 |
| 4 |

|
1 | 44 | 30 |
| 5 |

|
2 | 48 | 9 |
| 6 |

|
23 | 1560 | 278 |
| 7 |
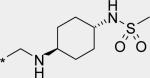
|
40 | 1170 | 218 |
| 8 |
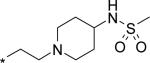
|
40 | 861 | 196 |
| 9 |
|
6 | 61 | 38 |
| 10 |

|
4 | 6 | 1 |
| 11 |
|
15 | 84 | 53 |
| 12 |
|
9 | 28 | 13 |
| 13 |
|
1 | 2 | 1 |
IC50 measured using GT 1b, Conl strain (n ≥ 2)
EC50 measured using GT 1a or GT 1b stable HCV subgenomic replicon (n ≥ 2).
RESULTS AND DISCUSSION
From a structure-based design point of view, we carefully considered the X-ray co-crystal structures of 1 and 2 with NS5B (Figure 3). Both compounds satisfy the large hydrophobic pocket of the palm I allosteric site, with a p-fluorobenzyl moiety for 1, and with tert-butylphenyl for 2. Interestingly for 2, the core benzene ring engages in a classic edge-to-face π-stack with the side chain of Tyr448, while the tert-butyl group completely fills the bottom of the hydrophobic pocket, making close hydrophobic contact with the side chain of Pro197, among other residues. Both ligands engage in H-bonding with the adjacent backbone area, via the quinolinedione oxy-anion of 1 and analogously via the pyridone moiety of 2. As we previously reported, one of the remarkable features of the highly ligand-efficient 2 is the donor-acceptor engagement by the 2(1H)-pyridone moiety with two backbone residues (NH of Tyr448 and C=O of Gln446). In further comparing the co-crystal structures of 1 and 2, superimposing the two structures on each other (Figure 3C), the major obvious difference is the filling of the palm I pocket by 1 in the area adjacent to and partially overlapping with the catalytic pocket. In fact, 1 directly interacts with the side-chain of one of the key catalytic aspartates, Asp318, in a hydrogen bond via the sulfonamide NH. Our design concept for the evolution of 2 towards a lead series was to “grow” from the small template towards the catalytic site of NS5B, adding some of the key interactions found between 1 and the enzyme that are absent from 2, while retaining what we felt was a superior, highly efficient starting point regarding the interactions 2 was already making with the palm I site. Towards such an end, linkers were designed to connect the core benzene ring of 2 together with the highly optimized N-arylmethanesulfonamide fragment present in 1. In the X-ray co-crystal structure of 1, it is apparent that the carbocyclic ring of the benzothiazine moiety makes an edge-to-face π-stack interaction with Phe193 side chain, and all polar atoms of the sulfonamide moiety make productive hydrogen bonds to either polar side chains or a bound water molecule. Molecular modeling using co-crystal structures of 1 and 2 enabled the in silico evaluation of a range of possible linker compositions and positions. The goal was to retain the highly optimized interactions between the N-arylmethanesulfonamide moiety and residues of the palm I pocket, with particular care taken to ensure low strain energy of the final bound conformation. A simple two-atom connection to the position adjacent to the methoxy substituent of the core benzene ring of 2 satisfied these requirements.
Figure 3.

(A) X-ray co-crystal structure of compound 1 complexed with NS5B at 2.2 Å, showing the palm I allosteric site;25 (B) compound 2 with NS5B at 2.8 Å; (C) overlay 1 and 2 in binding pocket from (A).
Extension from the core structure of 2 to the catalytic pocket resulted in potent inhibitors of NS5B (Table 1). Comparing 3 and 4, it can be seen that the optimal connection point of a two carbon linker from the core structure 2 to an N-phenylmethanesulfonamide group is at the position para to the substituent nitrogen on the pendant benzene ring. Ethylene-linked 4 not only exhibited potent inhibition of NS5B in the enzymatic assay, but was also efficacious in the cell-based replicon assay, with EC50 < 50 nM for both genotypes 1a and 1b. We further evaluated 4 for its physicochemical and ADME properties (Table 2). Compound 4 demonstrated low solubility (as measured in a high throughput thermal solubility assay) and medium to high permeability (as measured in the Caco2 assay). It was metabolically unstable both in vitro (human and rat liver microsomes) and in vivo (rat), and gave only modest oral bioavailability in rat. The reason for low %F may be attributed to absorption limited by poor solubility, as well as to high first pass metabolism (i.v. clearance > liver blood flow). We also evaluated 4 in some of our cytochrome P450-based early safety in vitro assays (Table 3). It exhibited potent reversible (IC50 = 0.34 μM) as well as time-dependent (kobs 6-fold greater than ethynylestradiol) inhibition of CYP3A4. Inhibition of four other CYP isoforms evaluated (1A4, 2C9, 2C19, 2D6) was not observed (all IC50 > 5 μM, data not shown). Time dependent inhibition (TDI) of CYP3A4 was especially concerning, as it could indicate the formation of reactive metabolites, further compounding the risk for drug-drug interactions (DDIs) from reversible inhibition of CYP3A4, and even potentially leading to autoimmune reactions and severe liver toxicities.27
Table 2.
Physicochemical and ADME comparison for linkers.
| Cmpd | PSAa | clogPb | Caco2 ABc | Caco2 ERd | Sol.e | HLMf | RLMf | i.v. Clg | %Fh |
|---|---|---|---|---|---|---|---|---|---|
| 4 | 75.3 | 4.60 | 1.5 | 3.4 | < 1 | 185 | 1240 | 82 | 11 |
| 5 | 98.1 | 2.47 | 0.3 | 48 | < 1 | 50 | 26 | 41 | 0 |
| 10 | 75.8 | 4.85 | -- | -- | < 1 | 78 | 103 | -- | -- |
| 12 | 93.0 | 3.70 | 0.9 | 19 | < 1 | 40 | 302 | 29 | 0.4 |
| 13 | 96.8 | 3.50 | 0.3 | 15 | -- | 638 | 388 | -- | -- |
polar surface area, calculated in Å2
calculated log P
permeability coefficient, 10−6 cm/s
ratio of BA/AB permeability coefficients
thermal aqueous solubility, pH 6.4 (high throughput assay), μg/mL
human or rat liver microsome intrinsic clearance, μL/min/mg protein
clearance in rat after 0.5 mg/kg i.v. injection, mL/min/kg
bioavailability in rat after 2 mg/kg p.o. administration.
Table 3.
Inhibition of CYP3A4.
| Cmpd | IC50a | % TDIb | kobsc | kobs ratiod |
|---|---|---|---|---|
| 4 | 0.34 | 66 | 0.0921 | 6.01 |
| 5 | > 50 | 4 | 0.0023 | 0.11 |
| 8 | > 50 | 3 | 0.0003 | 0.01 |
| 10 | 1.1 | < 1 | 0.0028 | 0.15 |
| 11 | 0.04 | 36 | 0.0386 | 1.78 |
inhibition of midazolam turnover, μM
normalized percent activity remaining (12 min preincubation with cmpd at 10 μM)
inactivation rate, min−1
ratio of inactivation rates between compound and ethynylestradiol (< 1 considered low TDI potential).
In vitro metabolite ID studies conducted in microsomal preparations revealed three main regions of oxidative metabolism / hydroxylation: the ethylene linker, the pyridone ring, and the tert-butyl group (Figure 4, see also Supporting Information for metabolite ID study of 4). We felt that any of these areas could be responsible, at least in part, for the observed high first pass clearance as well as for the potential production of reactive metabolites (e.g., quinone methide-type structures from oxidation of the benzylic positions of the ethylene linker, pyridinedione from the pyridone, dimethylacetaldehyde from tert-butyl). Our strategy was therefore set to address some of the shortcomings of 4 (poor metabolic stability, DDI / TDI risk) by taking a stepwise approach to modify each of those three key regions with substitutions and changes that we expected to confer resistance to oxidation, either through direct steric blocking or through reduction of relative electron richness, or through a combination. Furthermore, we felt that while compound 4 was quite potent in both biochemical and cell-based assays, improvement in potency, especially in the cell-based replicon system, would ultimately lead to lower targeted exposures in vivo and thus mitigate some of the liabilities a final candidate may have in terms of limited bioavailability or safety margins. Optimization of potency was therefore to be addressed concomitantly with stability and safety.
Figure 4.
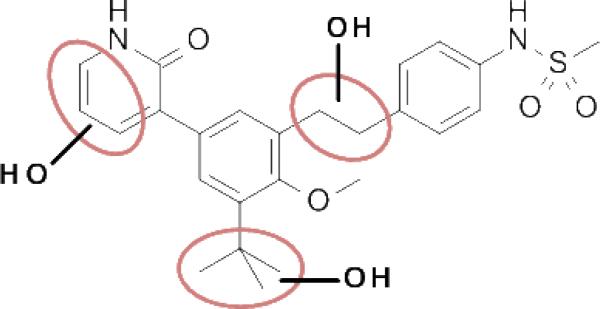
Regions of microsomal oxidation of compound 4, as indicated by the observation of hydroxylated species during metabolite ID studies.
Beginning our optimization of 4 by further investigation into various linker groups (Table 1), we identified carboxamide 5 as an inhibitor of comparable potency to 4 as measured in the enzymatic assay, and with increased efficacy in the genotype 1b replicon cellular assay.28 Compound 5 is significantly stabilized compared to 4 when incubated with liver microsome preparations (Table 2), and exhibits much reduced DDI and TDI liabilities (Table 3). Presumably oxidative metabolism has both been directly blocked at the ethylene linker of 4, as well as indirectly decreased by potential reduction of partitioning into the microsomal fractions because of increased overall polarity of 5 (increased PSA, decreased clogP, Table 2). The increased polarity, however, especially by nature of increased hydrogen bonding donors and acceptors, leads to reduced permeability and increased efflux. Reduced permeability, together with poor solubility, led to essentially no observed bioavailability from p.o. dosing in rat. From i.v. dosing, however, it was noted that in vivo clearance did improve relative to 4 (Table 2).
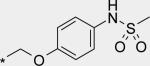
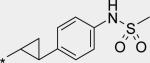
Next, a variety of 2-atom linker analogs of 4 and 5 were designed and synthesized. Compounds 6 – 11 (Table 1) are representative analogs that were predicted via modeling to maintain the critical interactions made by the N-phenylmethanesulfonamide moiety while binding in a low strain energy conformation. In addition, some of these linkers were designed to improve solubility by decreasing planarity (6 – 8) as well as by introducing an ionizeable moiety (7 and 8). While solubility was improved (data not shown), significant loss of potency in both biochemical and cell-based assays was seen as an unacceptable trade-off. We anticipated a requirement for exposures at multiples above efficacious concentrations, especially to enable preclinical toxicological studies. Thus we set aggressive targets for potency, especially potency in a serum-shifted cell-based assay (vida infra), in addition to looking for ideal ADMET properties to allow for safe, oral delivery. Note that piperidine 8 was also devoid of TDI activity (Table 3), demonstrating that the 1,2-diarylethylene moiety was most likely responsible for the observed effect in 4. This hypothesis is also supported by the observed low TDI signal for stilbene analog 10, as well as by the moderate TDI signal observed for the 1,2-trans-cyclopropane-linked analog 11, which has stereoelectronic properties in-between those of the saturated and unsaturated 2 carbon linkers of 4 and 10, respectively. Compound 10 was also remarkable to us for its robust potency in the replicon assay, for both genotypes 1a and 1b. Its improved in vitro (microsomal) stability compared to 4 does not likely owe to a partitioning / polarity difference (similar PSA, clogP, Table 2), and TDI activity is less (Table 3). While we were unable to measure in a Caco-2 assay because of experimental difficulties relating most likely to poor compound solubility, we were hopeful that an expected reasonable permeability of 10 relative to 5 (with fewer H-bonding groups and lower PSA for 10) would make for a better starting point for optimization. Thus considering its advantages in potency and overall ADMET properties, 10 became our lead template upon which we could further refine towards a clinical candidate.
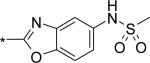
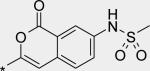
Before we fully committed to the stilbene template of 10, however, we did design and synthesize a number of analogs of 4 and 5 in which the linker was cyclized onto the aryl ring of the N-arylmethanesulfonamide terminus. Design ideas were evaluated and filtered via modeling, where we looked to further restrict the rotational freedom of molecules such as 4 - 11 into what we believed was the most likely active conformation, positioning all of the key interactions as seen in the overlay of crystal structures for 1 and 2 (Figure 3C). Hydrogen bond accepting moieties were included in the fused ring designs to engage with the water molecule network observed in the palm I pocket. Example successful embodiments of this design concept are oxazole 12 and isocoumarin 13.29 We obtained X-ray co-crystal structures of 12 and 13 bound with NS5B (Figure 5), and were pleased to observe them in their predicted binding modes. Of interest to note is that the water molecule hydrogen-bonding with the oxazole nitrogen of 12 is effectively replaced by the isocoumarin carbonyl oxygen of 13. Bicyclic compounds 12 and 13 were both quite potent (Table 1), however both also exhibited deficiencies that ultimately led to their deprioritization. In the case of oxazole 12, poor oral bioavailability was at issue (Table 2), with near-zero exposure from oral dosing most likely owing to a combination of solubility-limiting absorption and efflux-mediated low permeability. In the case of isocoumarin 13, low stability when incubated with human and rat liver microsome preparations, as well as observed efflux which could limit oral exposure (PK experiment not performed), led to its deprioritization. Returning to the stilbene template 10, we next explored modification of the pyridone ring (Table 4). Although change of the saturated to unsaturated linker from 4 to 10 resulted in improved stability to microsomes, as well as reduced TDI potential, we sought to improve stability further so that our aggressive exposure targets could be better met. Recalling that metabolite ID studies had identified the pyridone ring as one of three sites of oxidation (in addition to the linker and tert-butyl group), we sought to stabilize the pyridone either by replacement with more electron deficient heterocycles (14 – 18), or by introduction of small substituents to potentially block oxidation directly (19 – 22). In either case, we viewed retention of the 1,3-donor-acceptor motif of the 2(1H)-pyridone as a requirement, so that the unique engagement with the backbone amides of residues Gln446 and Tyr448 would be maintained. Also, at this mature stage of our lead optimization effort we began to focus more on efficacy in the HCV replicon assay performed in the presence of 40% human serum (HuS). We targeted potency in this in vitro system that would allow for exposures in vivo at 5-fold coverage above the measured EC90. Note that stilbene 10 exhibits a 20-fold shift reduction in efficacy in this assay. However, we still considered it a very potent compound with EC50 and EC90 of 21 and 150 nM, respectively, in the presence of 40% HuS. Note also that enzymatic data is no longer included on Table 4 (and later also not on Table 6), as all of the compounds 14 – 27 exhibited IC50 between 1 and 3 nM (data not shown), even for the least active examples, such as 15. Under our biochemical assay conditions (enzyme and substrate concentrations at 3 nM each), all IC50 < 3 nM are indisinguishable, as the enzyme is effectively titrated with inhibitor at those potencies.
Figure 5.

(A) X-ray co-crystal structure of compound 12 bound in the palm I allosteric site of NS5B at 2.05 Å; (B) compound 13 with NS5B at 2.05 Å.
Table 4.
Pyridone Replacements and Substitutions.
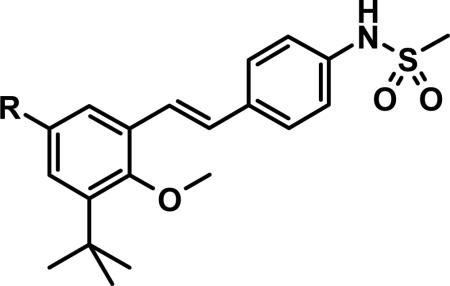
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | R | clogPa | GT1a EC50 (nM)b | GT1b EC50 (nM)b | 40% HuS EC50 (nM)c | 40% HuS EC90 (nM)c |
| 10 |
|
4.85 | 6 | 1 | 21 | 150 |
| 14 |
|
4.16 | 1 | 1 | 5 | 20 |
| 15 |
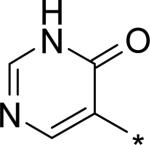
|
4.60 | 30 | 25 | 276 | 1830 |
| 16 |

|
4.38 | 4 | 2 | 8 | 58 |
| 17 |
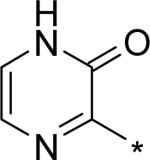
|
4.17 | 9 | 4 | 29 | 134 |
| 18 |
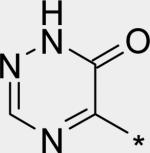
|
2.84 | 8 | 7 | 57 | 311 |
| 19 |
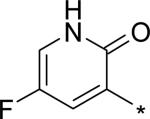
|
5.17 | 8 | 3 | 22 | 129 |
| 20 |
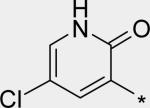
|
5.74 | 39 | 6 | 43 | 211 |
| 21 |
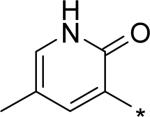
|
5.34 | 63 | 6 | 43 | 193 |
| 22 |

|
5.34 | 9 | 2 | 25 | 145 |
Calculated log P
EC50 measured using GT 1a or GT 1b stable HCV subgenomic replicon (n ≥ 2)
EC50 and EC90 measured using GT 1b stable HCV subgenomic replicon in the presence of 40% human serum (n ≥ 2).
Table 6.
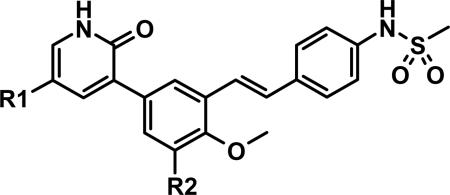
tert-Butyl Modifications and Replacements.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | clogPa | GT1a EC50b | GT1b EC50b | 40% HuS EC50c | 40% HuS EC90c | Caco2 ABa | Caco2 ERf |
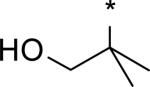
| 23 | H |
|
3.01 | 5 | 3 | 7 | 39 | 0.19 | 77 |
| 24 | H |
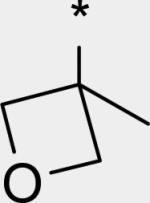
|
2.79 | 16 | 8 | 30 | 195 | 0.36 | 52 |
| 25 | H |
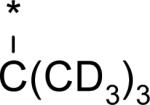
|
4.85 | 2 | -- | 7d | 104d | -- | -- |
| 26 | H |
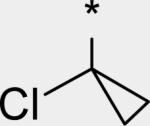
|
4.15 | 10 | 4 | 41 | 286 | 1.56 | 3.6 |
| 27 | F |
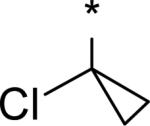
|
4.47 | 30 | 8 | 85 | 566 | -- | -- |
Calculated log P
EC50 measured using GT 1a or GT 1b stable HCV subgenomic replicon, nM (n ≥ 2)
EC50 and EC90 measured using GT 1b (accept where noted) stable HCV subgenomic replicon in the presence of 40% human serum (HuS), nM (n ≥ 2)
GT 1a used
permeability coefficient, 10−6 cm/s
ratio of BA/AB permeability coefficients.
For heteroaromatic replacements of pyridone, examples 14 – 18 are representative of the effort. We were able to obtain our first X-ray co-crystal structure of a stilbene series compound with 14 (Figure 6). A co-crystal structure of the prototype ethylene-linker 4 is also shown for comparison. It can clearly be seen that the unsaturated C=C linker adopts a more coplanar disposition between the two aryl rings than does the saturated ethylene linker, and this bound conformation is very close to the low-energy ground state conformation typical for stilbenes, which would help account for the intrinsic potency increase over diarylethylenes. Note that in the structure for 4 (Figure 6B), it is curious that the –CH3 group of the methoxy is rotated in a different direction than that observed in all of our other co-crystal structures, pushing side chain of Cys366 away from the ligand. The reason for this observed difference is unclear.
Figure 6.

A) X-ray co-crystal structure of compound 14 bound in the palm I allosteric site of NS5B at 1.90 Å; B) compound 4 with NS5B at 2.09 Å.
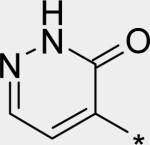
The significant (4- to 7-fold) potency increase for pyridazinone 14 relative to pyridone 10 in the serum-shifted replicon assay caught our attention. While decreased lipophilicity (clogP, Table 4) may be limiting nonspecific binding to serum proteins for 14, it is also possible that there is an intrinsic potency increase due to specific interactions with the binding pocket. We observe what could be an n-to-π* interaction between the ring nitrogen lone pair and the backbone carbonyl carbon of Gly410 (Figure 7), providing an enthalpic improvement to the binding energy, although the vector angle for this interaction does not appear to be ideal. Pyrimidinone 15 is significantly less potent than the other aza-modified pyridines in Table 4. This observation could be explained by increased acidity, whereby an anion form is dominant, and thus cannot provide an H-bond donation to the backbone carbonyl of Tyr448 (we did not measure the pKa of 15 to test this hypothesis). An alternative explanation would be that the lone pair of the additional nitrogen in 15 is not able to make any productive contact with protein residues, and thus the cost of desolvation is significantly detracting from the binding energy.30 Note that uracil analog 16 recovers potency lost in 15, which could be due to reduction in relative acidity, the ability to make more productive contacts with residues in the binding pocket (putatively Gly410 backbone and Asn411 side chain, in addition to Gln446 and Tyr448 backbone), or both. Pyrazinone 17 exhibits a similar efficacy profile to the parent pyridone 10, whereas triazinone 18 is slightly less active.
Figure 7.

Enlargement from co-crystal structure of 14 with NS5B (Figure 6), showing potential n-to-π* interaction between pyridazinone ring nitrogen and Gly410 backbone carbonyl
For single substituents, a sampling of our effort is summarized by pyridones 19 – 22. Placing the substituents at the 5-position (19 – 21) was intended to directly block the putative site of metabolic oxidation, a strategy that was at least partially successful (vida infra). In terms of potency, it was interesting to see that no loss was observed with the 5-fluoro substitution in 19. The slightly larger substituents chloro and methyl in 20 and 21, respectively, affected modest declines in potency relative to the unsubstituted pyridone 10, with a trend towards a greater decline in the genotype 1a replicon assay. We were unable to obtain co-crystal structures of our molecules complexed with genotype 1a NS5B, and so structure-based explanations of this trend can only be speculative. Several residues in the palm I region near the pyridone ring binding site are changed in genotype 1a relative to 1b, including Y415F, D444N and Q446E, which could play a role in the observed potency differences. Note that when the substituent is moved from the 5- to the 6-position (methyl group on 22), potency is restored to the levels observed for 10 and 19, for both subtypes 1a and 1b.
As a primary motivation behind the design of pyridone analogs 14 – 22 was to improve metabolic stability, we evaluated them under both in vitro and in vivo conditions (Table 5). In general, pyridone substitutions (19 – 22) had a larger impact on reducing clearance than did replacement with heterocyclic analogs containing additional nitrogens (14 – 18). Considering stability category as defined in Table 5 (high, medium, low), data from human liver microsomes and hepatocytes were in higher concordance with each other than were the corresponding data from rat. For all of the compounds reported on Table 5, human in vitro stability either fell within or very close to the medium range, which represents a significant improvement relative to the in vitro stability measured for 10 (Table 2). In vitro stability data from rat was not highly predictive of in vivo, with no clear advantage to using either microsomal or hepatocytic systems (both predicting the in vivo stability category with only ~ 50% accuracy). For the substituted pyridones evaluated, two exhibited high stability in vivo (19 and 22), with one more in the medium-to-high range (20).
Table 5.
Pyridone replacement in vitro and in vivo stability.a
| Cmpd | HLMb | HHc | RLMd | RHe | i.v. Clf |
|---|---|---|---|---|---|
| 14 | 24 (M) | 21 (L) | 202 (L) | 29 (M) | 53 (L) |
| 16 | 30 (M) | 4 (M) | 36 (M) | 20 (M) | 56 (L) |
| 17 | -- | 20 (L) | -- | 65 (L) | 75 (L) |
| 18 | 20 (M) | 7 (M) | 78 (M) | 42 (L) | 31 (M) |
| 19 | 17 (M) | 10 (M) | 48 (M) | 16 (M) | 17 (H) |
| 20 | 5 (H) | 9 (M) | 10 (H) | 9 (M) | 23 (M) |
| 21 | 29 (M) | 10 (M) | 92 (L) | 18 (M) | 44 (L) |
| 22 | 15 (M) | 6 (M) | 25 (H) | 5 (H) | 10 (H) |
Stability category H/M/L = high/medium/low
human liver microsome intrinsic clearance, μL/min/mg protein (high stability < 6.5, low stability > 35)
human hepatocyte intrinsic clearance, μL/min/106 cells (high stability < 2.8, low stability > 15)
rat liver microsome intrinsic clearance, μL/min/mg protein (high stability < 15, low stability > 90)
rat hepatocyte intrinsic clearance, μL/min/106 cells (high stability < 5.4, low stability > 29)
clearance in rat after 0.5 mg/kg i.v. injection, mL/min/kg (high stability < 20, low stability > 40).
While several candidates had been identified from the exploration of pyridone group modifications which exhibited medium to high stability (18, 19, 20 and 22) in vitro and in vivo, we decided to extend the effort to block or retard metabolic oxidation to modifications on the tert-butyl group. Representative examples of this effort are shown in Table 6,31 and will be discussed first in terms of efficacy and cell permeability, before presenting the effects of these changes on clearance. Hydroxy-tert-butyl analog 23 is very potent, especially in the serum-shifted assay. Oxetane 24 is less potent than 23, but similar to 10 and other potent stilbene series compounds. Comparing the polarities (clogP) of 10, 23 and 24, it is not clear that increased polarity is driving the increased replicon activities of alcohol 23, as clogP for 23 and 24 are very close. Unfortunately, it is more clear for both compounds that increasing polarity leads to reduced intrinsic permeability, as well as increased efflux, with 24 fairing slightly better in this regard than 23. We generally targeted permeability as measured by the Caco2 assay to be > 10−6 cm/s, with efflux ratio < 10, to advance molecules with the best chance for efficient intestinal absorption, so data for both 23 and 24 fell outside of those targets. Deuterated tert-butyl analog 25 was designed to test the impact of a primary kinetic isotope effect on metabolic oxidation of tert-butyl CH. Replacement of hydrogens with deuteria does not decrease potency (data only available for genotype 1a, which typically gives lowered measured potencies for this series). We also explored the 1-chloro-1-cyclopropyl group as a replacement for tert-butyl that would be a good size mimetic, without presenting any terminal methyl groups for easy oxidation. 1-Chloro-1-cyclopropyl appeared to give an advantage in permeability, with 26 looking much improved relative to oxo-substituted tert-butyl analogs 23 and 24. Potency drops only 2-fold (compare 26 to 10, Table 4), showing that the 1-chloro-1-cyclopropyl moiety is well tolerated in the place of tert-butyl. The loss of potency is slightly greater when combined with a pyridone substitution in 27 (3- to 4-fold, compare to data for 19, Table 4), which was designed to see if blocking metabolism at both sites simultaneously would produce additive or synergistic effects on stability.
We evaluated promising tert-butyl replacements for stability (Table 7). Oxetane 24 did show improvement relative to 10 in vitro, with all measurements at either medium or high stability. In vivo stability observed in rat, however, fell in the low range. 1-Chloro-1-cyclopropyl analog 26 showed a very similar in vitro stabilization when incubated with liver microsome preparations from both human and rat. Combination of the tert-butyl stabilizing group 1-chloro-1-cyclopropyl with a pyridone stabilizing substitution (5-fluoro), however, did not result in an additive or synergistic effect (compare hepatocyte stability for 27 vs. 19 from Table 5). Interestingly, one of the most effective substitution we found for stabilizing the template in vitro through tert-butyl modification was deuterium, as in the perdeutero analog 25, demonstrating that an isotope effect can significantly impact rate of metabolic clearance on this template. Although retarding oxidative metabolism by replacing or substituting at the tert-butyl group had some effect on overall rate of clearance (at least in vitro), it appeared that the effect achieved by stabilizing through pyridone modification alone was sufficient to reach the desired target of medium to high stability.
Table 7.
tert-Butyl substitution effect on in vitro and in vivo stability.a
| Cmpd | HLM | HH | RLM | RH | i.v. Cl |
|---|---|---|---|---|---|
| 24 | 22 (M) | 2 (H) | 74 (M) | 21 (M) | 50 (L) |
| 25 | 19 (M) | 2 (H) | 28 (M) | 13 (M) | -- |
| 26 | 17 (M) | -- | 77 (M) | -- | -- |
| 27 | -- | 9 (M) | -- | 27 (M) | -- |
refer to Table 5 for explanation of abbreviations.
We decided to focus on fluoropyridone 19 for further preclinical development based on its balance of high potency with medium to high metabolic stability. For a summary of in vitro data for 19 not already presented, see Table 8. Intrinsic permeability was measured at slightly below the desired target of 10−6 cm/s, however significant efflux was not observed and the molecule was still advanced. CYP3A4-mediated DDI and TDI liabilities were low. Inhibition of four other CYP isoforms (1A4, 2C9, 2C19, 2D6) was not observed (all IC50 > 50 μM, data not shown).
Table 8.
Additional in vitro data for 19.
permeability coefficient, 10−6 cm/s
ratio of BA/AB permeability coefficients
inhibition of midazolam turnover, μM
normalized percent activity remaining (12 min pre-incubation with cmpd at 10 μM)
inactivation rate, min−1
ratio of inactivation rates between compound and ethynylestradiol (< 1 considered low TDI potential).
Further evaluation of 19 included in vivo pharmacokinetic (PK) studies, as well as in vitro potency studies against a panel of genotype 1 HCV NS5B clinical isolates. With regards to in vivo PK, we felt that in order to enable preclinical toxicology studies, sustained exposures in preclinical species should exceed by at least several multiples the exposures expected to be useful in human efficacy studies (i.e., where plasma trough levels, Cmin, exceed the 40% human serum-shifted replicon EC90). Thus, a target Cmin of 303.5 ng/mL, equating to 5-fold above the serum-shifted EC90 for GT-1b, was considered a minimum requirement following oral dosing. Furthermore, those levels were targeted to be maintained for > 12 hours following a single dose, so that b.i.d. or q.d. dosing regimens could be implemented. Studies in rat are summarized in Table 9 and on Figure 8. Upon dosing compound 19 as a suspension in our typical vehicle (hypromellose 2910, polysorbate 80, benzyl alcohol and water), modest oral bioavailability (7.2%) was observed at 2 mg/kg, with a non-dose proportional drop-off to 2.1% observed at the higher 20 mg/kg dose. Importantly, exposures did not cover the targeted plasma level of 5-fold over serum-shifted EC90, even at Cmax. Moving into a “nanosuspension”32 and dosing up to 50 mg/kg showed slightly increased bioavailability (now 2.9%), however targeted plasma concentrations were again not reached. Considering low solubility for this series (for 19, aqueous solubility in pH 6.5 phosphate buffer = 0.1 μg/mL), we viewed it as a strong possibility that oral absorption was being limited by slow dissolution. We therefore investigated solution dosing using a polyethylene glycol / caprylocaproyl macrogol-8 glyceride (Labrasol®) vehicle,33 in which compound 19 was completely dissolved and not merely in suspension. Indeed, increases in bioavailability and exposures above targeted levels in plasma were observed at the two doses tested. The increased exposures observed during these studies from solution formulation support the hypothesis that absorption was limited because of solubility / dissolution factors in the previous studies using suspensions. At the high solution dose (50 mg/kg), plasma concentrations remained above 303.5 ng/mL for over 8 hours (with extrapolation suggesting up to 12 hours, Figure 8), sufficient coverage to enable toxicology studies in rat. Similar experiments in dog using PEG 400 / Labrasol® solution dosing also demonstrated good kinetics and oral bioavailability (Table 10).
Table 9.
In vivo pharmacokinetics for 19 in rat.
| Dose / Route | Clb | Vdssc | t 1/2 d | C max e | AUCf | %F |
|---|---|---|---|---|---|---|
| 0.5 mg/kg i.v. | 17.4 | 5.12 | 5.3 | 281 | 470 | -- |
| 2 mg/kg p.o., suspension | -- | -- | -- | 12.7 | 138 | 7.2 |
| 20 mg/kg p.o., suspension | -- | -- | -- | 32.4 | 407 | 2.2 |
| 50 mg/kg p.o., nanosuspension | -- | -- | -- | 104 | 1370 | 2.9 |
| 14 mg/kg p.o., solutiona | -- | -- | -- | 398 | 4460 | 33 |
| 50 mg/kg p.o., solutiona | -- | -- | -- | 817 | 10100 | 21 |
1:1 PEG 400 / Labrasol
clearance, mL/min/kg
volume of distribution at steady state, L/kg
half-life, hr
peak concentration, ng/mL
area under curve, ng·h/mL.
Figure 8.

Exposure curves for compound 19 following oral dosing in rat, with 5X EC90 line marking 303.5 ng/mL.
Table 10.
In vivo pharmacokinetics for compound 19 in dog.
| Dose / Route | Clb | Vdssc | t 1/2 d | C max e | AUCf | %F |
|---|---|---|---|---|---|---|
| 0.5 mg/kg i.v. | 3.28 | 1.11 | 4.9 | 666 | 2410 | -- |
| 2 mg/kg p.o., solutiona | -- | -- | -- | 301 | 4260 | 45 |
1:1 PEG 400 / Labrasol
clearance, mL/min/kg
volume of distribution at steady state, L/kg
half-life, hr
peak concentration, ng/mL
area under curve, ng·h/mL.
In addition to profiling 19 for its PK properties, we studied its ability to inhibit various HCV strains by measuring its activity against replicons containing NS5B sequences derived from clinical isolates from treatment naïve genotype 1 HCV patients. Preexisting amino acid polymorphisms may confer reduced sensitivity to direct-acting antivirals, and drug pressure-induced selection may increase the prevalence of those strains within a patient's quasispecies, leading to increased potential for resistance.34 Using a transient replicon assay, we measured EC50 against a number patient-derived NS5B isolates from both genotypes 1a and 1b (Table 11 and Figure 9).35 The H77 and Con1 strains were included as references for genotypes 1a and 1b, respectively. Also for comparison, we evaluated another non-nucleoside inhibitor of NS5B, PF-00868554, which binds to a different allosteric site, the “thumb II” site. Pyridone 19 is very potent against all isolates tested from both genotypes, with EC50 ranging from 0.4 – 6.8 nM. Activity for the thumb II inhibitor PF-00868554 is also consistent across all isolates tested, with EC50 measured in the range of 18.7 – 95.8 nM and in agreement with previously published data.36 Interestingly, the largest shift observed for 19 relative to a reference strain was for the GT-1a isolate RO-38 (~ 7-fold), which contains the mutation S556G, a known resistance mutation to palm I non-nucleoside inhibitors.35 Serine 556 is present in the palm I binding site (see Figs. 3, 5 and 6). Even considering this shift, 19 is still an extremely potent inhibitor with an EC50 of 6.8 nM.
Table 11.
Antiviral activity of 19 and PF-00868554 against GT 1a and GT 1b NS5B clinical isolates.
| GT | Isolate | Cmpd 19 EC50 (nM)a | PF-00868554 EC50 (nM)a |
|---|---|---|---|
| 1a | H77 | 1.0±0.3 | 95.8±9.7 |
| RO-18 | 1.3±0.1 | 47.0±6.2 | |
| RO-22 | 4.1±1.0 | 42.8±7.6 | |
| RO-24 | 3.1±0.1 | 37.0±1.2 | |
| RO-28 | 1.8±0.1 | 38.8±2.3 | |
| RO-32 | 2.3±0.1 | 26.0±1.2 | |
| RO-33 | 1.1±0.1 | 34.4±3.4 | |
| RO-34 | 2.2±0.1 | 25.0±0.8 | |
| RO-35 | 0.4±0.1 | 22.0±4.9 | |
| RO-38 | 6.8±0.5 | 56.0±6.0 | |
| RO-41 | 1.5±0.2 | 48.0±6.8 | |
| (mean)b | 1.8 | 39.5 | |
| 1b | Con1 | 1.4±0.3 | 51.1±11.2 |
| RO-1 | 0.5±0.1 | 39.3±3.0 | |
| RO-2 | 0.7±0.1 | 21.3±4.9 | |
| RO-3 | 1.7±0.4 | 28.5±7.2 | |
| RO-4 | 0.4±0.1 | 25.5±7.4 | |
| RO-7 | 1.8±0.7 | 20.8±5.4 | |
| RO-8 | 2.0±0.2 | 18.7±4.8 | |
| RO-9 | 3.0±0.1 | 23.5±6.9 | |
| RO-10 | 2.8±0.1 | 39.7±7.2 | |
| (mean)b | 1.3 | 28.2 |
EC50 measured using transient HCV subgenomic replicon assay (n ≥ 2), nM±SEM (Standard Error)
calculated geometric mean.
Figure 9.

Antiviral activity of 19 and PF-00868554 against replicons from genotype 1 NS5B clinical isolates; reference strains shown as solid shapes, geometric means shown as dotted lines.
CONCLUSION
In summary, a novel, highly potent series of non-nucleoside inhibitors of HCV NS5B has been identified from a fragment-derived starting point, and optimized to a clinical candidate in fluoropyridone 19. A structure-based approach was applied to the design of various linkers connecting the hydrophobic core to the arylsulfonamide that engages the catalytic residues adjacent to the palm I allosteric site. We identified conformationally restricted linkers such as in carboxamide 5 and trans-stilbene 10, predisposed to their active, target-bound conformations in the ground state, giving them robust intrinsic potency. Evaluation of linkers based on early safety criteria such as time-dependent inhibition of CYP3A4, pharmacokinetic criteria such as in vitro and in vivo stability towards clearance, and physicochemical criteria such as membrane permeability led to the selection of the trans-stilbene connector as the preferred template upon which to further optimize. The results of metabolite ID studies focused our attention on the pyridone and tert-butyl moieties as potential regions for tuning properties, especially towards further improving stability. Substituted pyridones provided the largest effect in this direction, and fluoropyridone 19 was ultimately selected as the clinical candidate. While rat in vivo pharmacokinetics from i.v. dosing for 19 fell into a desirable range, low solubility limited oral absorption. In order to achieve exposures needed to support preclinical toxicology evaluation at multiples above the human serum-adjusted replicon EC90, we solved the problem of poor oral absorption by moving to a lipid-based solution formulation. Finally, we showed that 19 maintains its robust, low nanomolar potency across 17 different human HCV NS5B clinical isolates from both genotypes 1a and 1b. Outcomes from further development studies of 19 will be reported in due course.
CHEMISTRY
Syntheses of selected representatives from compounds 3 – 27 are depicted in Schemes 1 – 5. Full synthetic details for all compounds 3 – 27 that are not outlined here may be found in the Supporting Information. In Schemes 1 and 2 are depicted the syntheses of a few different linkers from Table 1: 1,2-diarylethylene 4 and trans-stilbene 10 (Scheme 1); and carboxamide 5 (Scheme 2). The sequence towards 4 and 10 began with 3-tert-butyl-2-hydroxybenzaldehyde 28, which was brominated and O-methylated in high yield to produce 5-bromo-3-tert-butyl-2-hydroxy-benzaldehyde 29. Suzuki cross-coupling with (2-methoxy-3-pyridyl)boronic acid provided biaryl intermediate 30, which was then reacted with a 4-nitrobenzyl Wittig reagent to give stilbene 31 as the trans isomer. Tandem hydrogenation of both the nitro and stilbene groups provided aniline 32 with a saturated ethylene linker. Compound 32 was N-mesylated and selectively O-dealkylated to afford the final pyridone 4. Alternatively, intermediate 31 could have its nitro group selectively reduced using elemental iron, resulting in aniline 33. Mesylation and deprotection as before yielded stilbene 10. In Scheme 2 is depicted the synthesis of carboxamide 5. First, 5-bromo-1-tert-butyl-2-methoxy-3-nitrobenzene 34 was cross-coupled under Suzuki conditions to give biaryl 35. Hydrogenation afforded aniline 36, which was then treated with 4-nitrobenzoyl chloride in the presence of triethylamine to provide carboxamide 37 in high yield. Nitro group reduction was achieved under hydrogenation conditions with catalytic palladium hydroxide to give 38. Finally, N-mesylation followed by O-demethylation afforded pyridone 5.
Scheme 1.

Scheme 5.

Scheme 2.

In Schemes 3 and 4 are shown two different routes used to prepare pyridone replacement analogs pyridazinone 14 and pyrimidinone 15, respectively. Beginning with Scheme 3, 4-bromo-2-tert-butylanisole is converted to an organomagnesium reagent under Grignard conditions, and then reacted in excess (2.5 equiv) with 4,5-dichloro-1H-pyridazin-6-one in refluxing THF. Chlorine substitution occurred at the more reactive 4-position, with good selectivity and high yield. The remaining chloro substituent was removed by hydrogenolysis in water / DMF in the presence of palladium(0) on carbon and potassium hydroxide to afford pyridazinone 40.37 Selective bromination at the benzene ring position ortho to the methoxy group was achieved by treatment with N-bromosuccinimide in DMF at 50 °C for 18 hours to give 41. The concise, 4-step synthesis of 14 was completed by a final step cross coupling with N [4 [(E) 2 (4,4,6-trimethyl-1,3,2-dioxaborinan-2-yl)vinyl]phenyl]methanesulfonamide under Suzuki conditions and microwave heating to 125 °C in a sealed tube. Forcing conditions were required to achieve the modest 18% yield of the final product, as the bromo group of 41 is quite sterically hindered (bulky tert-butyl group forces methoxy methyl to rotate away from it, thus hindering access to the bromine). In Scheme 4, we began with 5-bromo-3-tert-butyl-2 hydroxy-benzaldehyde 29 (from Scheme 1) and stereoselectively installed the trans-stilbene via Horner-Wadsworth-Emmons reaction with the benzaldehyde, followed by reduction of the terminal nitro group with elemental iron to afford aniline 42 in high yield. N-mesylation provided 43, which was then converted to boronate 44, providing a versatile intermediate which could be used for cross-coupling reactions with heteroaryl bromides to make pyridones and pyridone replacements. In this example, boronate 44 was reacted with 4-benzyloxy-5-bromopyrimidine to give a corresponding biaryl intermediate in 34% yield. Removal of the O-benzyl group via treatment with HBr in acetic acid at room temperature afforded the final product pyrimidinone 15 in 71% yield.
Scheme 3.

Scheme 4.

In Scheme 5 is depicted the synthesis of compound 24, containing one of the tert-butyl replacement groups (3-methyloxetan-3-yl) from Table 6. In the first step, 4,6-dibromo-2-iodoanisole 45 is coupled to diethyl malonate in the presence of cesium carbonate and catalytic amounts of copper(I) iodide and 2-phenylphenol following Buchwald's procedure,38 generating α-arylmalonate 46 via selective reaction at aryliodide position. Methylation of 46 at the α-malonate position, followed by reduction of the esters to alcohols with lithium aluminum hydride at low temperature afforded diol 47. Ring closure of the 1,3-diol to the oxetane was affected by subjection to Mitsunobu-type conditions (DIAD, triphenylphosphine, toluene) in a sealed tube at 140 °C (microwave), yielding intermediate 48. Taking advantage of the steric hindrance differential between the two bromines of 48, a Suzuki cross coupling with (2-oxo-1H-pyridin-3-yl)boronic acid occurred with high regioselectivity at the less hindered aryl bromide to give the biaryl product 49. Also, we found by this point that the pyridone group did not need protection to carry through many of the chemistries that we used to prepare compounds in this series, thus sparing deprotection steps at the ends of syntheses. The final step here was cross-coupling with N-[4-[(E)-2-(4,4,6-trimethyl-1,3,2-dioxaborinan-2-yl)vinyl]phenyl]methanesulfonamide under Suzuki conditions with microwave heating to 120 °C in a sealed tube, affording trans-stilbene 24 in 64% yield.
EXPERIMENTAL
General information
All reactions were performed in round-bottom flasks unless otherwise noted. The flasks were fitted with rubber septa and reactions were conducted under inert atmosphere. Stainless steel syringes were used to transfer air- and moisture-sensitive liquids. Most commercial reagents were purchased from Sigma Aldrich, Alfa Aesar, Strem, Lancaster, AstaTech, or TCI, and used as received. Solvents were purchased from Mallinckrodt and anhydrous dimethylformamide (DMF) and tetrahydrofuran (THF) from Sigma Aldrich in a Sure/Seal system. Flash chromatography was performed using AnaLogix IntelliFlashTM 280 or Teledyne Isco CombiFlash® Companion® purification systems using silica gel columns RediSep Rf (particle size 40-63 powder). Deuterated solvents were purchased from Sigma-Aldrich. Proton nuclear magnetic resonance (1H NMR) spectra was recorded on Bruker Avance 300 MHz, 400 MHz, or 500 MHz spectrometers at 25 °C. Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3 δ 7.27, DMSO δ 2.50). Data is represented as follows: chemical shift, multiplicity (br. = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hertz (Hz), and integration. Liquid chromatography mass spectrometry (LC/MS) analyses were carried out using an Agilent 6140 single quadrupole in ES/APCI dual mode, and Agilent LC with a Zorbax SB-C18 2.1×30mm, 3.5 m column using a 0.02% TFA/water-acetonitrile solvent system. Microwave reactions were carried out in a Biotage Initiator microwave synthesizer. Purity of tested compounds was ≥95% as confirmed by HPLC/MS.
5-Bromo-3-tert-butyl-2-methoxy-benzaldehyde (29)
To a solution of 3-tert-butyl-2-hydroxybenzaldehyde (28) (5.0 g, 28.0 mmol) in CH2Cl2 (20 mL) at 0 °C was added dropwise a solution of Br2 (1.45 mL, 28.0 mmol) in CH2Cl2 (15 mL) over a period of 30 min. After the addition was complete, the reaction was stirred for 1 h before the organic volatiles were removed under reduced pressure to afford 5-bromo-3-tert-butyl-2-hydroxy-benzaldehyde (7.23 g, 100%) as a light yellowish solid. 1H NMR (300 MHz, CDCl3) δ 11.76 (s, 1 H), 9.84 (s, 1 H), 7.67-7.48 (m, 2 H), 1.44 (s, 9 H) ppm.
A mixture of 5-bromo-3-tert-butyl-2-hydroxy-benzaldehyde (3.83 g, 15.0 mmol), MeI (2.32 mL, 37.0 mmol) and K2CO3 (6.18 g, 45.0 mmol ) in DMF (50 mL) was heated at 50 °C for 1 h then cooled to rt and diluted with Et2O and water. The organic layer was thrice washed with water then brine, dried (MgSO4) and concentrated to afford 29 (3.99 g, 99%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 10.30 (s, 1 H), 7.86 (d, J = 2.3 Hz, 1 H), 7.67 (d, J = 2.6 Hz, 1 H), 3.97 (s, 3 H), 1.43 (s, 9 H) ppm.
3-tert-Butyl-2-methoxy-5-(2-methoxy-3-pyridyl)benzaldehyde (30)
A sealed tube containing 29 (2.0 g, 7.38 mmol), (2-methoxy-3-pyridyl)boronic acid (1.68 g, 10.98 mmol), Pd(PPh3)4 (0.66 g, 0.57 mmol), Na2CO3 (1.96 g, 18.49 mmol) in a mixture of MeOH (11 mL) and CH2Cl2 (3 mL) was heated with a microwave reactor at 115 °C for 30 min. The reaction mixture was concentrated, diluted with EtOAc, washed with brine, and dried over Na2SO4. Chromatography on SiO2 (EtOAc/hexane, 0 to 10%) gave 30 (2.123 g, 96%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.60-10.15 (m, 1 H), 8.25 8.14 (m, 1 H), 7.97-7.88 (m, 1 H), 7.87-7.75 (m, 1 H), 7.70-7.54 (m, 1 H), 7.12-6.91 (m, 1 H), 4.08-3.98(m, 6 H), 1.48 (s, 9 H) ppm.
3-[3-tert-Butyl-4-methoxy-5-[(E)-2-(4-nitrophenyl)vinyl]phenyl]-2-methoxy-pyridine (31)
To a slurry of NaH (0.17 g, 4.159 mmol, 60% in mineral oil) in THF (5 mL) containing 0.1 equivalent of 15-crown-5 cooled to 0 °C was added a solution of diethyl (4-nitro-phenyl)-phosphonate (1.14 g, 4.16 mmol) in THF (5 mL). When gas evolution ceased, a slurry of 30 (1.13 g, 3.78 mmol) was added and stirred at rt for 18 h. An additional equivalent of NaH was added to the partially reacted mixture and stirring continued for 1 h. The reaction mixture was partitioned between water and Et2O and the organic extract was sequentially washed with water and brine, dried (Na2SO4), filtered, and evaporated. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 50%) to afford 31 as a sole geometric isomer. 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.6 Hz, 2 H), 8.17 (d, J = 4.5 Hz, 1 H), 7.73-7.61 (m, 4 H), 7.57 (d, J = 16.2 Hz, 1 H), 7.53-7.47 (m, 1 H), 7.13 (d, J = 16.7 Hz, 1 H), 7.03-6.95 (m, 1 H), 4.01 (s, 3 H), 3.84 (s, 3 H), 1.45 (s, 9 H) ppm.
4-[2-[3-tert-Butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]ethyl]aniline (32)
To a solution of 31 (351 mg, 0.840 mmol) in EtOAc (15 mL) and MeOH (15 mL), was added Pd(OH)2 (20% on carbon, 166 mg, 0.237 mmol). H2 gas was bubbled into the solution for 15 min. The reaction was stirred under an atmosphere of H2 for 18 h. The reaction was filtered, concentrated, and purified over SiO2 by chromatography (EtOAc/hexane, 5% to 20%) to give 32 (106 mg, 32%) as a colorless oil. LC/MS (ES/APCI): 391 (M + H)+.
N-[4-[2-[3-tert-Butyl-2-methoxy-5-(2-oxo-1H-pyridin-3yl)phenyl]ethyl]phenyl]methanesulfonamide (4)
To a solution of 32 (106 mg, 0.272 mmol) in pyridine (5 mL) at 0 °C was added methanesulfonyl chloride (0.035 mL, 0.450 mmol). The reaction was gradually warmed to rt and stirred for 1.5 h. The solution was diluted with EtOAc, washed with saturated CuSO4 solution, 1N HCl solution, and dried over Na2SO4. Purification over SiO2 by chromatography (EtOAc/hexane, 10% to 25%) gave N-[4-[2-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]ethyl]phenyl]methanesulfonamide (104 mg, 82%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.13 (dd, J = 4.9, 1.5 Hz, 1 H), 7.57 (dd, J = 7.3, 1.7 Hz, 1 H), 7.43-7.27 (m, 2 H), 7.25-7.10 (m, 4 H), 6.96 (dd, J = 7.2, 5.3 Hz, 1 H), 6.39 (s, 1 H), 3.98 (s, 3 H), 3.83 (s, 3 H) 2.99 (s, 4 H), 2.97 (s, 3 H), 1.43 (s, 9 H) ppm.
A solution of N-[4-[2-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]ethyl]phenyl]methanesulfonamide (104 mg, 0.222 mmol), 48% HBr (0.1 mL, 0.87 mmol) in AcOH (3 mL) was heated at 65 °C for 18 h in a sealed tube. The reaction mixture was carefully poured into saturated aqueous NaHCO3 ice solution, extracted with EtOAc, and dried over Na2SO4. Purification by SiO2 preparative TLC (EtOAc/hexane, 1:1 followed by EtOAc/hexane, 2:1) gave 4 (51 mg, 51%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.68 (br s, 1 H), 9.59 (s, 1 H) 7.54 (dd, J = 6.9, 2.1 Hz, 1 H), 7.51 (d, J = 2.2 Hz, 1 H), 7.46 (d, J = 2.2 Hz, 1 H), 7.34 (dd, J = 6.4, 2.0 Hz, 1 H), 7.25 (d, J = 8.5 Hz, 2 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.26 (t, J = 6.8 Hz, 1 H), 3.76 (s, 3 H), 2.93 (s, 3 H), 2.88 (s, 4 H), 1.36 (s, 9 H) ppm. LC/MS (ES/APCI): 455 (M + H)+.
4-[(E)-2-[3-tert-Butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]vinyl]aniline (33)
To a suspension of 31 (0.29 g, 0.693 mmol) in MeOH (5 mL) and water (5 mL) was added NH4Cl (0.37 g, 6.93 mmol) followed by Fe powder (0.19 g, 3.464 mmol). The solution was heated at reflux for 1 h, cooled and filtered. The filtrate was concentrated in vacuo. The filtrate was taken up in EtOAc, washed with brine, dried (Na2SO4), filtered and evaporated. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 30%) to afford 33 (0.26 g, 100%) as a yellow foam. 1H NMR (300 MHz, CDCl3) δ 8.15 (dd, J = 4.9, 1.9 Hz, 1 H), 7.63 (d, J = 2.3 Hz, 2 H), 7.44 7.33 (m, 3 H), 7.17 (d, J = 15.8 Hz, 1 H), 7.02 6.93 (m, 2 H), 6.69 (d, J = 8.3 Hz, 2 H), 4.00 (s, 3 H), 3.83 (s, 3 H), 3.81 3.69 (m, 2 H), 1.44 (s, 9 H) ppm.
N-[4-[(E)-2-[3-tert-Butyl-2-methoxy-5-(2-oxo-1H-pyridin-3-yl)phenyl]vinyl]phenyl] methanesulfonamide (10)
To a solution of 33 (0.153 g, 0.394 mmol) in pyridine (4 mL) at 0 °C was added methanesulfonyl chloride (0.037 mL, 0.055 g, 0.473 mmol). Stirring was continued at 0 °C for 2 h, then at rt for an additional 2 h. The reaction mixture was diluted with EtOAc, washed sequentially with aqueous CuSO4 and 2N HCl, dried (Na2SO4), filtered, and concentrated. The crude residue was purified over SiO2 by chromatography (EtOAc/hexane, 40%) to afford N-[4-[(E)-2-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]vinyl]phenyl] methanesulfonamide (0.178 g, 96%) as a yellow foam. 1H NMR (400 MHz, CDCl3) δ 8.16 (dd, J = 4.5, 1.5 Hz, 1 H), 7.64 (d, J = 2.5 Hz, 2 H), 7.54 (d, J = 8.6 Hz, 2 H), 7.46 (d, J = 2.0 Hz, 1 H), 7.36 (d, J = 16.7 Hz, 1 H), 7.23 (d, J = 8.1 Hz, 2 H), 7.05 (s, 1 H), 7.02-6.95 (m, 1 H), 6.42 (s, 1 H), 4.00 (s, 3 H), 3.83 (s, 3 H), 3.04 (s, 3 H), 1.45 (s, 9 H) ppm.
A solution of N-[4-[(E)-2-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]vinyl]phenyl]methanesulfonamide (0.14 g, 0.3 mmol), AcOH (3 mL) and 48% HBr (0.105 mL) was heated at 60 °C for 18 h. The reaction mixture was cooled to rt, diluted with EtOAc and water to give a precipitate, which was filtered and washed with water and ether to give 10 (115 mg, 67%) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, J = 1.5 Hz, 1 H), 7.67 (dd, J = 6.9, 2.0 Hz, 1 H), 7.58-7.51 (m, 3 H), 7.37 (dd, J = 6.3, 1.8 Hz, 1 H), 7.17 (d, J = 16.4 Hz, 1 H) 7.17-7.07 (m, 3 H), 6.29 (t, J = 6.6 Hz, 1 H), 3.75 (s, 3 H), 2.91 (s, 1 H), 1.39 (s, 9 H) ppm. LC/MS (ES/APCI): 453 (M + H)+.
3-(3-tert-Butyl-4-methoxy-5-nitro-phenyl)-2-methoxy-pyridine (35)
A mixture of 5-bromo-1-tert-butyl-2-methoxy-3-nitrobenzene (34) (0.48 g, 1.7 mmol), (2-methoxy-3-pyridyl)boronic acid (0.28 g, 1.8 mmol), Pd(PPh3)4 (0.19 g, 0.17 mmol), and Na2CO3 (0.53 g, 5.0 mmol) in CH2Cl2/MeOH (3:1, 8 mL) was subjected to microwave heating to 115 °C for 35 min. The reaction mixture was filtered and the filtrate concentrated. The crude residue was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 20%) to afford 35 (0.44 g, 82%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.19 (dd, J = 4.9, 1.5 Hz, 1 H), 7.87 (d, J = 2.3 Hz, 1 H), 7.72 (d, J = 1.9 Hz, 1 H), 7.61 (dd, J = 7.5, 1.9 Hz, 1 H), 7.00 (dd, J = 7.3, 5.1 Hz, 1 H), 3.99 (s, 3 H), 3.85 (s, 3 H), 1.44 (s, 9 H) ppm.
3-tert-Butyl-2-methoxy-5-(2-methoxy-3-pyridyl)aniline (36)
To a solution of 35 (0.98 g, 3.1 mmol) in EtOAc/MeOH (2:1, 30 mL) was added 10% Pd/C (0.10 g). A stream of H2 was bubbled through the solution for 20 min then the solution was stirred at rt for 16 h. The reaction mixture was filtered and concentrated to afford 36. 1H NMR (300 MHz, CDCl3) δ 8.12 (dd, J = 4.9, 1.9 Hz, 1 H), 7.59 (dd, J = 7.36, 1.7 Hz, 1 H), 7.02-6.84 (m, 3 H), 3.97 (s, 3 H), 3.84 (s, 3 H), 3.70 (br s, 2 H), 1.41 (s, 9 H) ppm.
N-[3-tert-Butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]-4-nitro-benzamide (37)
To a solution of 36 (0.24 g, 0.85 mmol) and Et3N (0.18 mL, 1.3 mmol) in CH2Cl2 (10 mL) at 0 °C was added 4-nitrobenzoyl chloride (0.19 g, 1.0 mmol). The resulting mixture was stirred at 0 °C for 3 h, and then warmed to rt. The mixture was then diluted with additional CH2Cl2, washed sequentially with water and brine, dried (Na2SO4), filtered, and concentrated. The crude residue was purified over SiO2 by chromatography to afford 37 (0.36 g, 97%) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.42-8.35 (m, 3 H), 8.27 (br s, 1 H), 8.16 (dd, J = 5.1, 1.5 Hz, 1 H), 8.10 (d, J = 8.56 Hz, 2 H), 7.66 (dd, J = 7.6, 1.5 Hz, 1 H), 7.40 (d, J = 2.0 Hz, 1 H), 6.98 (dd, J = 7.1, 5.0 Hz, 1 H), 4.00 (s, 3 H), 3.84 (s, 3 H), 1.45 (s, 9 H) ppm.
4-Amino-N-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]benzamide (38)
To a solution of 37 (0.36 g, 0.82 mmol) in EtOAc/MeOH (2:1, 15 mL) was added 10% Pd/C (0.035 g). A stream of H2 gas was bubbled through the solution for 20 min, and the white solid precipitate that formed was recovered by filtration to afford 38 (quantitative) which was carried onto the subsequent step without further purification. 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 2.3 Hz, 1 H), 8.13 (d, J = 5.0 1.9 Hz, 1 H), 8.08 (br s, 1 H), 7.77 (d, J = 8.7 Hz, 2 H), 7.66 (dd, J = 7.3, 1.7 Hz, 1 H), 7.34 (d, J = 1.9 Hz, 1 H), 6.95 (dd, J = 7.2, 4.9 Hz, 1 H), 6.73 (d, J = 8.3 Hz, 2 H), 4.04 (br s, 2 H), 3.99 (s, 3 H), 3.83 (s, 3 H), 1.44 (s, 9 H) ppm.
N-[3-tert-Butyl-2-methoxy-5-(2-oxo-1H-pyridin-3-yl)phenyl]-4-(methanesulfonamido) benzamide (5)
To a solution of 38 (0.10 g, 0.25 mmol) in pyridine (2 mL) at 0 °C was added methanesulfonyl chloride (0.034 g, 0.30 mmol). Stirring was continued at 0 °C for 2 h, then at rt for an additional 2 h. The reaction mixture was diluted with EtOAc, washed sequentially with aqueous CuSO4 and 2N HCl, dried (Na2SO4), filtered, and concentrated. The crude residue was purified over SiO2 by chromatography (EtOAc/hexane, 50% to 100%) to afford N-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]-4-(methanesulfonamido)benzamide (0.12 g, 99%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 1.9 Hz, 1 H), 8.23-8.10 (m, 2 H), 7.94 (d, J = 8.7 Hz, 2 H), 7.65 (dd, J = 7.2, 1.9 Hz, 1 H), 7.40-7.30 (m, 3 H), 6.95 (dd, J = 7.3, 5.0 Hz, 1 H), 6.90 (br s, 1 H), 3.99 (s, 3 H), 3.84 (s, 3 H), 3.10 (s, 3 H), 1.45 (s, 9 H) ppm.
A solution of N-[3-tert-butyl-2-methoxy-5-(2-methoxy-3-pyridyl)phenyl]-4-(methanesulfonamido)benzamide (0.12 g, 0.25 mmol), 48% HBr (40 μL) and AcOH (3 mL) was heated in a sealed tube at 60 °C for 15 h. The reaction mixture was cooled to rt, diluted with EtOAc, and washed with water. Upon addition of saturated aqueous NaHCO3, a solid precipitated, which was collected and triturated with Et2O to afford 5 (70 mg, 60%) as a white solid. mp 274.0-276.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.74 (br s, 1 H), 10.20 (s, 1 H), 9.87 (s, 1 H), 8.04 (d, J = 8.6 Hz, 2 H), 7.71 (d, J = 2.0 Hz, 1 H), 7.61 (dd, J = 6.8, 1.8 Hz, 1 H), 7.53 (d, J = 2.0 Hz, 1 H), 7.36 (dd, J = 6.1, 1.5 Hz, 1 H), 7.30 (d, J = 8.6 Hz, 2 H), 6.29 (t, J = 6.6 Hz, 1 H), 3.72 (s, 3 H), 3.10 (s, 3 H), 1.39 (s, 9 H) ppm. LC/MS (ES/APCI): 470 (M + H)+.
5-(3-tert-Butyl-4-methoxy-phenyl)-1H-pyridazin-6-one (40)
A dry round-bottom flask was charged with 4-bromo-2-tert-butylanisole (39) (2.933 g, 12 mmol), THF (15 mL) and magnesium turnings (0.29 g, 12 mmol) were added. The reaction mixture was heated to reflux and stirred for 45 min then cooled to rt. The resulting solution was added dropwise at rt to a stirred solution of 4,5-dichloro-1H-pyridazin-6-one (0.796 g, 4.8 mmol), THF (10 mL) and Et2O (20 mL). The reaction mixture was then heated at reflux for 18 h. The reaction was cooled to 0 °C and quenched with saturated aqueous NH4Cl and extracted with EtOAc (150 mL). The aqueous phase was withdrawn and re-extracted with EtOAc (150 mL). Each extract was washed sequentially with water and brine. The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo. The residue was triturated with EtOAc/hexane (1:1) to afford 5-(3-tert-butyl-4-methoxy-phenyl)-4-chloro-1H-pyridazin-6-one (1.087 g, 77%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 11.25 (br s, 1 H), 7.85 (s, 1 H) 7.53-7.35 (m, 2 H), 6.96 (d, J = 8.3 Hz, 1 H), 3.89 (s, 3 H), 1.39 (s, 9 H) ppm.
A Parr Shaker bottle was charged with 5-(3-tert-butyl-4-methoxy-phenyl)-4-chloro-1H-pyridazin-6-one (1.08 g), a solution of KOH (0.52 g in 11 mL of H2O), and DMF (1.3 mL). To this mixture was added 10% Pd/C and the bottle was connected to a Parr shaker and flushed three times with H2 then shaken for 18 h at rt under an atmosphere of ca. 50 psi of H2. To the resulting solution was added 5M KOH to dissolve the precipitate then the solution was filtered through a glass microfiber filter and rinsed with 5M KOH and H2O. The filtrate was acidified with concentrated HCl and the resulting mixture extracted with CH2Cl2 (100 mL). The aqueous layer was withdrawn and re-extracted with EtOAc. The combined extracts were dried (Na2SO4), filtered, and evaporated to afford 40 (0.791 g, 83%) as a white powder. 1H NMR (400 MHz, CDCl3) δ 10.78 (br s, 1 H), 7.83 (d, J = 4.0 Hz, 2 H), 7.72 (d, J = 2.0 Hz, 1 H), 7.30 (d, J = 4.0 Hz, 1 H), 6.95 (d, J = 8.6 Hz, 1 H), 3.89 (s, 3 H), 1.40 (s, 9 H) ppm.
5-(3-Bromo-5-tert-butyl-4-methoxy-phenyl)-1H-pyridazin-6-one (41)
To a solution of 40 (0.1 g, 0.39 mmol) and DMF (2 mL) was added NBS (0.069 g, 0.39 mmol) and the resulting solution stirred at 50 °C for 18 h. The reaction was concentrated in vacuo and the residue partitioned between Et2O and water. The aqueous layer was withdrawn and re-extracted with Et2O. The organic layers were twice washed with water (5 mL) and once with brine (5 mL). The organic layers were combined, dried (Na2SO4), filtered, and evaporated. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 30%) to afford 41 (0.068 g, 52%) as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 2.3 Hz, 1 H), 7.91-7.86 (m, 1 H), 7.77-7.73 (m, 1 H), 7.32 (d, J = 4.1 Hz, 1 H), 3.97 (s, 3 H), 1.42 (s, 9 H) ppm.
N-[4-[(E)-2-[3-tert-Butyl-2-methoxy-5-(6-oxo-1H-pyridazin-5-yl)phenyl]vinyl]phenyl] methanesulfonamide (14)
A microwave tube was charged with 41 (0.068 g), N-[4-[(E)-2-(4,4,6-trimethyl-1,3,2-dioxaborinan-2-yl)vinyl]phenyl]methanesulfonamide (0.078 g), Na2CO3 (0.064 g), Pd(PPh3)4 (0.023 g), MeOH (1.8 mL), and CH2Cl2 (0.6 mL). The tube was flushed with Ar, sealed and irradiated in a microwave synthesizer at 125 °C for 40 min. The reaction mixture was cooled and concentrated in vacuo. The residue was partitioned between CH2Cl2 (25 mL) and water (5 mL). The organic layer was washed with brine (5 mL). The aqueous phase was twice extracted with CH2Cl2 (25 mL). The organic layers were combined, dried (Na2SO4), filtered, and evaporated. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 60%) to give 14 (0.175 g, 18%) as a yellow powder. mp: 240.0-242.0 °C. 1H NMR (400 MHz, CDCl3) δ 11.19 (s, 1 H), 8.50 (s, 1 H), 8.25 (d, J = 2.2 Hz, 1 H), 7.93 (d, J = 4.2 Hz, 1 H), 7.56-7.50 (m, 3 H), 7.41 (d, J = 4.2 Hz, 1 H), 7.38-7.30 (m, 3 H), 7.07 (d, J = 16.4 Hz, 1 H), 3.84 (s, 3 H), 3.03 (s, 3 H), 1.45 (s, 9 H) ppm. LC/MS (ES/APCI): 454 (M + H)+.
4-[(E)-2-(5-Bromo-3-tert-butyl-2-methoxy-phenyl)vinyl]aniline (42)
To a suspension of NaH (0.35 g, 8.73 mmol, 60% mineral oil dispersion) and dry THF (9 mL) was added 15-crown-5 (0.27 mL, 1.36 mmol) and the resulting mixture cooled to 0 °C. A solution of diethyl (4-nitro-benzyl)-phosphonate (2.33 g, 8.53 mmol) in dry THF (10.5 mL) was added slowly while maintaining the temperature at 0 °C. The reaction was stirred for 15 min then a solution of 5-bromo-3-tert-butyl-2-methoxy-benzaldehyde (29) (1.93 g, 7.12 mmol) and dry THF (21 mL) was added. The reaction mixture was stirred at 0 °C for 2 h then allowed to warm to rt and stirred for 18 h. The reaction mixture was quenched with water (50 mL) and the resulting solution thrice extracted with Et2O (3 × 60 mL). The combined extracts were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 5%) to afford 5-bromo-1-tert-butyl-2-methoxy-3-[(E)-2-(4-nitrophenyl)vinyl]benzene (2.70 g, 98%). 1H NMR (300 MHz, CDCl3) δ 8.24 (d, J = 8.7 Hz, 2 H), 7.66 (d, J = 8.7 Hz, 2 H), 7.60 (d, J = 2.3 Hz, 1 H), 7.51-7.34 (m, 2 H), 7.09 (d, J = 16.6 Hz, 1 H), 3.78 (s, 3 H), 1.40 (s, 9 H) ppm.
A mixture of 5-bromo-1-tert-butyl-2-methoxy-3-[(E)-2-(4-nitrophenyl)vinyl]benzene (0.788 g, 2.02 mmol), iron (0.471 g, 8.44 mmol), NH4Cl (0.867 g, 16.2 mmol), MeOH (25 mL), and water (25 mL) was heated at reflux for 4 h. The reaction mixture was cooled to rt and filtered. The filtrate was thrice extracted with EtOAc and the combined extracts washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford 42 (0.71 g, 95%) as a yellow solid which was used without further purification.
N-[4-[(E)-2-(5-Bromo-3-tert-butyl-2-methoxy-phenyl)vinyl]phenyl]methanesulfonamide (43)
To a solution of 42 (0.161 g, 0.45 mmol) and pyridine (5 mL) cooled to 0 °C was added methanesulfonyl chloride (38 μL, 0.493 mmol). After stirring for 45 min at 0 °C, the reaction mixture was washed with 1N HCl and extracted three times with EtOAc. The organic extracts were washed with saturated CuSO4 solution three times and once with brine. The extracts were dried over Na2SO4, filtered, and concentrated to give 43 (193 mg, 99%) as a brown oil. 1H NMR (300 MHz, CDCl3) δ 7.57 (d, J = 2.3 Hz, 1 H), 7.53 (d, J = 8.3 Hz, 2 H), 7.34 (d, J = 2.3 Hz, 1 H), 7.29-7.17 (m, 3 H), 6.99 (d, J = 16.4 Hz, 1 H), 6.50 (s, 1 H), 3.77 (s, 3 H), 3.05 (s, 3 H), 1.39 (s, 9 H) ppm.
N-[4-[(E)-2-[3-tert-Butyl-2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]vinyl]phenyl]methanesulfonamide (44)
A mixture of 43 (0.476 g, 1.086 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (0.303 g, 1.19 mmol), PdCl2(PPh3)2 (0.177 g, 0.217 mmol), and KOAc (0.32 g, 3.26 mmol) under an Ar atmosphere was dissolved in DMSO (25 mL). The reaction mixture was then heated to 90 °C for 18 h. The reaction was cooled to rt and partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc and the combined extracts were washed five times with water and once with brine. The organic layers were dried, filtered, and evaporated. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 30%) to give 44 (0.311g, 53%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.92 (s, 1 H) 7.78-7.67 (m, 1 H) 7.54 (d, J = 8.3 Hz, 2 H), 7.38-7.06 (m, 4 H), 6.37 (s, 1 H), 3.80 (s, 3 H), 3.04 (s, 3 H), 1.47-1.41 (m, 9 H), 1.38-1.33 (m, 12 H) ppm.
N-[4-[(E)-2-[3-tert-Butyl-2-methoxy-5-(6-oxo-1H-pyrimidin-5-yl)phenyl]vinyl]phenyl]methanesulfonamide (15)
A tube was charged with 44 (0.130 g, 0.144 mmol), 4-benzyloxy-5-bromopyrimidine (0.070 g, 0.264 mmol), Pd(PPh3)4 (0.028 g, 0.024 mmol), Na2CO3 (0.077 g, 0.726 mmol), MeOH (3 mL), and CH2Cl2 (1 mL). The tube and solution were sparged with Ar, sealed and heated at 120 °C for 40 min. The solution was cooled, filtered through CELITE®, and the filtrate concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 50%) to afford N-[4-[(E)-2-[5-(4-benzyloxypyrimidin-5-yl)-3-tert-butyl-2-methoxy-phenyl]vinyl]phenyl]methanesulfonamide (45 mg, 34%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1 H), 8.58 (s, 1 H), 7.67 (d, J = 2.0 Hz, 1 H), 7.56-7.43 (m, 5 H), 7.34 (d, J = 7.6 Hz, 4 H), 7.22 (d, J = 8.6 Hz, 2 H), 6.94 (d, J = 16.7 Hz, 1 H), 6.34 (s, 1 H), 5.53 (s, 2 H), 3.83 (s, 3 H), 3.05 (s, 3 H), 1.39 (s, 9 H) ppm.
To a solution of N-[4-[(E)-2-[5-(4-benzyloxypyrimidin-5-yl)-3-tert-butyl-2-methoxy-phenyl]vinyl]phenyl]methanesulfonamide (0.044 g, 0.081 mmol) and AcOH (1.5 mL) at rt was added 48% HBr (30 μL). The reaction was stirred at rt for 18 h, diluted with EtOAc and poured into saturated NaHCO3. The aqueous layer was extracted with EtOAc and the combined extracts dried, filtered, and evaporated. The crude product was triturated with EtOAc/Et2O to give 15 (26 mg, 71%) as an off-white solid. mp: 253.0 255.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.73 (br s, 1 H), 9.83 (s, 1 H), 8.19 (d, J = 11.6 Hz, 2 H), 7.84 (s, 1 H), 7.63 (d, J = 8.1 Hz, 2 H), 7.57 (s, 1 H), 7.34-7.15 (m, 4 H), 3.77 (s, 3 H), 3.01 (s, 3 H), 1.39 (s, 9 H) ppm. LC/MS (ES/APCI): 454 (M + H)+.
N-[4-[(E)-2-[3-tert-Butyl-5-(5-fluoro-2-oxo-1H-pyridin-3-yl)-2-methoxy-phenyl]vinyl] phenyl]methanesulfonamide (19)
A microwave vial was charged with 43 (0.193 g, 0.442 mmol), (5-fluoro-2-methoxy-3-pyridyl)boronic acid (0.0771 g, 0.451 mmol), Na2CO3 (0.123 g, 1.16 mmol), Pd(PPh3)4 (0.027 g, 0.023 mmol), MeOH (1.2 mL), and CH2Cl2 (0.3 mL), sealed and irradiated in a microwave synthesizer at 115 °C for 35 min. The vial was cooled to rt, filtered, and the solid washed with EtOAc. The filtrate was washed sequentially with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 20 to 50%) to afford N-[4-[(E)-2-[3-tert-butyl-5-(5-fluoro-2-methoxy-3-pyridyl)-2-methoxy-phenyl]vinyl]phenyl]methanesulfonamide (0.127 g, 59%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.99 (d, J = 3.0 Hz, 1 H), 7.74-7.28 (m, 9 H) 7.03 (d, J = 16.2 Hz, 1 H), 3.98 (s, 3 H), 3.83 (s, 3 H), 3.04 (s, 3 H), 1.45 (s, 9 H) ppm.
A vial was charged with N-[4-[(E)-2-[3-tert-butyl-5-(5-fluoro-2-methoxy-3-pyridyl)-2-methoxy-phenyl]vinyl]phenyl]methanesulfonamide (0.087 g, 0.18 mmol), AcOH (2 mL) and 48% HBr (38 μL), sealed and heated at 60 °C for 18 h. The reaction was cooled to rt and added to a mixture of ice and saturated aqueous NaHCO3. The resulting mixture was thrice extracted with EtOAc and the combined extracts washed sequentially with saturated aqueous NaHCO3 and brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford 19 (0.071 g, 84%) as a solid. 1H NMR (400 MHz, DMSO d6) δ 9.82 (s, 1 H), 7.88 (d, J = 2.0 Hz, 1 H), 7.83 (dd, J = 8.8, 3.3 Hz, 1 H), 7.68-7.52 (m, 4 H), 7.35-7.13 (m, 4 H), 3.77 (s, 3 H), 3.01 (s, 3 H), 1.39 (s, 9 H) ppm. LC/MS (ES/APCI): 471 (M + H)+.
Diethyl 2-(3,5-dibromo-2-methoxy-phenyl)propanedioate (46)
A mixture of 1,5-dibromo-3-iodo-2-methoxy-benzene (45) (5.4 g, 14 mmol), diethyl malonate (4.0 g, 25 mmol), CuI (0.13 g, 0.69 mmol), 2-phenylphenol (0.24 g, 1.4 mmol) and Cs2CO3 (6.8 g, 21 mmol) in THF (14 mL) was stirred at 70 °C for 15 h. The resulting mixture was cooled to rt and partitioned between EtOAc and saturated aqueous NH4Cl. The organic extract was then washed sequentially with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 10%) to afford 46 (4.1 g, 55%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 2.0 Hz, 1 H), 7.28 (d, J = 2.0 Hz, 1 H), 4.24 (dd, J = 7.1, 4.5 Hz, 4 H), 4.12 (q, J = 7.1 Hz, 1 H), 3.85 (s, 3 H), 1.33-1.17 (m, 6 H) ppm.
2-(3,5-Dibromo-2-methoxy-phenyl)-2-methyl-propane-1,3-diol (47)
To a suspension of NaH (0.86 g, 22 mmol, 60% dispersion in mineral oil) in THF (50 mL) cooled to 0 °C was added dropwise a solution of 46 (4.1 g, 7.7 mmol) in THF (10 mL). The resulting mixture was stirred at 0 °C for 20 min then iodomethane (3.6 g, 26 mmol) was added dropwise and the reaction warmed to rt and stirred for 48 h. The reaction mixture was partitioned between Et2O and saturated aqueous NH4Cl. The organic extract was washed sequentially with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 10%) to afford diethyl 2-(3,5-dibromo-2-methoxy-phenyl)-2-methyl-propanedioate (2.9 g, 84%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1 H), 7.28 (s, 1 H), 4.30-4.15 (m, 4 H), 3.85 (s, 1 H), 1.81 (s, 3 H), 1.27 (t, J = 7.1 Hz, 6 H) ppm.
To a solution of diethyl 2-(3,5-dibromo-2-methoxy-phenyl)-2-methyl-propanedioate (0.919 g, 2.1 mmol) in THF (4 mL) cooled to 0 °C was added dropwise LiAlH4 (2.40 mL, 2.4 mmol, 1.0 M solution in THF). The reaction mixture was stirred at 0 °C for 1 h, and then saturated aqueous potassium sodium tartrate was added and the resulting mixture was stirred at rt for 2 h. The mixture was extracted with EtOAc, and the organic extract washed sequentially with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was purified over SiO2 by chromatography (EtOAc/hexane, 0 to 50%) to afford 47 (0.369 g, 52%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 2.5 Hz, 1 H), 7.56 (d, J = 2.0 Hz, 1 H), 4.15-4.06 (m, 2 H), 3.94 (s, 3 H), 3.90 (dd, J = 11.1, 5.6 Hz, 2 H), 2.46 (t, J = 5.6 Hz, 2 H), 2.05 (s, 3 H).
3-(3,5-Dibromo-2-methoxy-phenyl)-3-methyl-oxetane (48)
A tube was charged with 47 (0.34 g, 0.97 mmol) and PPh3 (0.51 g, 1.93 mmol), flushed with Ar and sealed. A solution of diisopropyl azodicarboxylate (0.39 g, 1.93 mmol) and toluene (6 mL) was added via syringe and the resulting mixture was irradiated in a microwave reactor at 140 °C for 30 min. The reaction mixture was concentrated in vacuo and the crude product purified over SiO2 by chromatography (EtOAc/hexane, 0 to 10%) to afford 48 (0.138 g, 43%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 2.5 Hz, 1 H), 6.91 (d, J = 2.5 Hz, 1 H), 4.98 (d, J = 6.1 Hz, 2 H), 4.50 (d, J = 5.6 Hz, 2 H), 3.87 (s, 3 H), 1.76 (s, 3 H) ppm.
3-[3-Bromo-4-methoxy-5-(3-methyloxetan-3-yl)phenyl]-1H-pyridin-2-one (49)
A microwave vial was charged with 48 (0.138 g, 0.411 mmol), (2-oxo-1H-pyridin-3-yl)boronic acid (0.063 g, 0.453 mmol), Na2CO3 (0.131 g, 1.24 mmol), Pd(PPh3)4 (0.047 g, 0.041 mmol), MeOH (3 mL), and CH2Cl2 (1 mL), sealed and irradiated in a microwave synthesizer at 115 °C for 30 min. The vial was cooled to rt, filtered, and concentrated in vacuo. The crude residue was partitioned between CH2Cl2 and water. The organic extract was washed with brine. The aqueous layers were extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 by chromatography (EtOAc/hexanes, 0 to 100%) to afford 49 (0.095 g, 66%) as an off-white powder. 1H NMR (400 MHz, CDCl3) δ 11.79 (br s, 1 H), 7.80 (d, J = 2.0 Hz, 1 H), 7.54 (dd, J =7.1, 2.0 Hz, 1 H), 7.35 (dd, J = 6.6, 2.0 Hz, 1 H), 7.15 (d, J = 2.0 Hz, 1 H), 6.37 (t, J = 6.8 Hz, 1 H), 5.07 (d, J = 5.6 Hz, 2 H), 4.55 (d, J = 5.6 Hz, 2 H), 3.92 (s, 3 H), 1.80 (s, 3 H) ppm.
N-[4-[(E)-2-[2-Methoxy-3-(3-methyloxetan-3-yl)-5-(2-oxo-1H-pyridin-3-yl)phenyl]vinyl]phenyl]methanesulfonamide (24)
A microwave vial was charged with 49 (0.095 g, 0.271 mmol), N-[4-[(E)-2-(4,4,6-trimethyl-1,3,2-dioxaborinan-2-yl)vinyl]phenyl]methanesulfonamide (0.132 g, 0.408 mmol), Na2CO3 (0.086 g, 0.811 mmol), Pd(PPh3)4 (0.031 g, 0.027 mmol), MeOH (3 mL), and CH2Cl2 (1.5 mL), sealed and irradiated in a microwave synthesizer at 120 °C for 1 h. The vial was cooled to rt, filtered, and concentrated in vacuo. The crude residue was partitioned between CH2Cl2 and water. The organic extract was washed with brine. The aqueous layers were extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 by chromatography (MeOH/CH2Cl2, 0 to 10%) to afford 24 (0.081 g, 64%) as a white powder. mp: 279.0 281.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.76 (br s, 1 H), 9.85 (s, 1 H), 7.89 (d, J = 2.1 Hz, 1 H), 7.70 (dd, J = 7.0, 2.1 Hz, 1 H), 7.62 (d, J = 8.7 Hz, 2 H), 7.39 (d, J = 5.3 Hz, 1 H), 7.30-7.12 (m, 5 H), 6.30 (t, J = 6.8 Hz, 1 H), 4.91 (d, J = 5.7 Hz, 2 H), 4.45 (d, J = 5.7 Hz, 2 H), 3.74 (s, 3 H), 3.01 (s, 3 H), 1.72 (s, 3 H) ppm. LC/MS (ES/APCI): 467 (M + H)+.
Computational chemistry
Molecular modeling was performed using the molecular operating environment (MOE).39 Figures were generated using Pymol 1.5.0.4.40
HCV NS5B polymerase biochemical assay protocol
The enzymatic activity of HCV polymerase (NS5B570n-Con1) was measured as described previously26 with only two changes as follows: concentration of RNA template was 3 nM and concentration of NS5B570n-Con1 enzyme was 3 nM.
Stable HCV replicon assay
The stable GT 1b Con1 and GT 1a H77 subgenomic replicon cell lines expressing the renilla luciferase as a reporter gene (cell lines 2209-23 and pRLucH771b75S/I, respectively) were used as described by Le Pogam et al.41
Transient HCV replicon assay using a panel of GT 1 NS5B clinical isolates
The GT 1b Con1 and GT 1a H77 transient replicons in which the wild type NS5B gene has been replaced by NS5B isolates obtained from untreated patients were used as described by Le Pogam et al.35
Time Dependent CYP3A4 Inhibition
Stock solutions of test compound, midazolam, and 17 α-ethynylestradiol were prepared in 5% DMSO in acetonitrile. Midazolam, 17 α-ethylnylestradiol, and α-nicotinamide adenine dinucleotide phosphate, reduced form (0-NADPH), were obtained from Sigma Chemical Co. Human liver microsomes (HLM, lot no. 0510222) were purchased from XenoTech, LLC (Lenexa, KS), prepared from a mixed-gender pool of 50 human livers. The assay was performed with a two stage robotic procedure in a 96-well format using Biomek 2000 (Beckman) for liquid handling. Inactivation mixtures (180 μL/well) containing 1 mg/mL HLM (Xenotech), 3 mM MgC12, 1 mM EDTA, and concentration 10 μM of test compound in 100 mM potassium phosphate buffer (pH 7.4) were incubated at 37 °C in a 96-well plate. After 3 minutes of pre incubation, the reactions were initiated by adding 20 μL of 10 mM NADPH and allowed to proceed up to 20 minutes. Verapamil and Ethynylestradiol at 10 μM were included as positive controls for time dependent CYP3A4 inactivation.42-43 At 0, 2, 6, 12, and 20 minutes after the start of the pre incubation, 10 μL of the preincubation mixtures were transferred to activity assay mixtures (90 μL/well, pre-warmed at 37 °C) containing 3 mM MgC12, 1 mM EDTA, 1 mM NADPH, and 10 μM of midazolam in 100 mM potassium phosphate buffer (pH 7.4). The final mixtures were incubated for 5 minutes and quenched with 100 μL of 0.1% formic acid in acetonitrile containing 2 Pg/mL 7-hydroxycoumarin as the internal standard. The quenched mixtures were transferred and further diluted in a deep well plate. After centrifugation, the supernatant was analyzed by HPLC-MS/MS.
Metabolite ID
Human liver microsomal proteins were purchased from BD GENTEST, Woburn, MA. The incubation mixture contained 50 mM potassium phosphate pH 7.4, 5 mM magnesium chloride, 2mM NADPH, test compound (20 μM) and microsomal protein (0.5 mg/mL). The total volume of each incubation mixture was 0.5 mL. All reagents except NADPH were combined in a 13 mL round bottom glass test tube on ice. The samples were pre incubated for approximately 2 minutes at 37 °C in a thermostatically controlled shaking water bath. The reaction was started by the addition of NADPH. The reaction was stopped at 30 minutes by quenching with 0.5 mL of methanol. For each microsomal incubation, one control sample (30 minutes) without NADPH was incubated together with the samples. The quenched samples were kept on ice (10-15 minutes) then at room temperature, vortexed briefly, and centrifuged. The supernatant was transferred to test tubes, evaporated to partial dryness (~ 0.2 mL), then transferred to an autosampler vial. Samples analyzed by RP-HPLC/UV/MS. Low resolution mass spectrometric analysis was carried out on a ThermoFinnigan iontrap mass spectrometer (model LTQ) equipped with an electrospray ionization source. The heated capillary temperature in the source was held at 250 °C. In the standard screening mode, the trap collected positive m/z 150 to m/z 800 scans. Collision-induced dissociation tandem MS was conducted with 40% relative collision energy and an isolation width of 3 amu. Product ion analyses were performed following collision induced dissociation of selected precursor ions to obtain substructural fragment ion information. MS3 experiments were also performed.
Pharmacokinetics
Male Wistar/Han (CRL:WI) rats (Charles River Laboratories, Hollister, CA or Raleigh, NC) used in these studies weighed between 180 and 300 g and were surgically implanted with cannulas in the jugular and/or femoral veins. All studies were conducted under IACUC approved protocols, and animals were allowed free access to food and water. Single intravenous bolus doses were administered into the tail vein or into the jugular cannula of dual cannulated animals using dosing formulations consisting of DMSO (< 10%) / PEG 400 / water; the remaining cannulas were used for blood collection. Single oral doses were administered by gavage using a dosing formulation consisting of hypromellose 2910 (0.5%), polysorbate 80 (0.4%) and benzyl alcohol (0.9%) in water, unless otherwise specified. Blood was collected into tubes containing EDTA at typically 0.08 (iv only), 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. Plasma concentrations were determined by a liquid chromatography/tandem mass spectrometry method (LC/MS/MS) following protein precipitation with acetonitrile.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge all members of the Analytical Department in Roche Palo Alto and the Physical Chemistry Department at Hoffmann-La Roche, Nutley for characterization of the compounds. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
ABBREVIATIONS
- HCV
hepatitis C virus
- NS5B
non-structural 5B
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Synthetic procedures and spectroscopic data, metabolite ID study of compound 4 (MS/MS/MS), HCV NS5B polymerase protein crystallography, references. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Atom coordinates and structure factors for HCV NS5B/ligand complexes have been deposited at the Protein Data Bank under the codes 4MK7 (2), 4MK8 (4), 4MK9 (12), 4MKA (13), 4MKB (14).
REFERENCES
- 1.WHO Fact sheet No164. 2013 Jul; ( http://www.who.int/mediacentre/factsheets/fs164/en/)
- 2.Dienstag JL, McHutchison JG. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology. 2006;130:231–264. doi: 10.1053/j.gastro.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut. 2006;55:1350–1359. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.European Association for the Study of the Liver EASL Clinical Practice Guidelines: Management of Hepatitis C Virus Infection. J. Hepatol. 2011;55:245–264. doi: 10.1016/j.jhep.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 5.Shah N, Pierce T, Kowdley KV. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs. 2013 Jun 4; doi: 10.1517/13543784.2013.806482. Epub ahead of print (doi:10.1517/13543784.2013.806482) [DOI] [PubMed] [Google Scholar]
- 6.Kiser JJ, Flexner C. Direct acting antiviral agents for hepatitis C virus infection. Annu. Rev. Pharmacol. Toxicol. 2013;53:427–449. doi: 10.1146/annurev-pharmtox-011112-140254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013;11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 8.Asselah T, Marcellin P. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 2012:88–102. doi: 10.1111/j.1478-3231.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- 9.Ilyas JA, Vierling JM. An overview of emerging therapies for the treatment of chronic hepatitis C. Clin. Liver Dis. 2011;15:515–536. doi: 10.1016/j.cld.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Poordad F, McCone J, Jr., Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. SPRINT-2 Investigators. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 12.Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int. 2013:93–104. doi: 10.1111/liv.12076. [DOI] [PubMed] [Google Scholar]
- 13.Shi N, Hiraga N, Imamura M, Hayes CN, Zhang Y, Kosaka K, Okazaki A, Murakami E, Tsuge M, Abe H, Aikata H, Takahashi S, Ochi H, Tateno-Mukaidani C, Yoshizato K, Matsui H, Kanai A, Inaba T, McPhee F, Gao M, Chayama K. Combination therapies with NS5A, NS3 and NS5B inhibitors on different genotypes of hepatitis C virus in human hepatocyte chimeric mice. Gut. 2013;62:1055–1061. doi: 10.1136/gutjnl-2012-302600. [DOI] [PubMed] [Google Scholar]
- 14.Figure 1 reproduced from reference (9), with permission from Elsevier and http://mattsmark.com/ (original artist website)
- 15.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA Polymerase. J. Med. Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 16.Mayhoub AS. Hepatitis C RNA-dependent RNA polymerase inhibitors: A review of structure–activity and resistance relationships; different scaffolds and mutations. Bioorg. Med. Chem. 2012;20:3150–3161. doi: 10.1016/j.bmc.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 17.Legrand-Abravanel F, Nicot F, Izopet J. New NS5B polymerase inhibitors for hepatitis C. Expert Opin. Investig. Drugs. 2010;19:963–975. doi: 10.1517/13543784.2010.500285. [DOI] [PubMed] [Google Scholar]
- 18.Powdrill MH, Bernatchez JA, Götte M. Inhibitors of the Hepatitis C Virus RNA-Dependent RNA Polymerase NS5B. Viruses. 2010;2:2169–2195. doi: 10.3390/v2102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013;368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 20.Zeuzem S, Asselah T, Angus P, Zarski JP, Larrey D, Müllhaupt B, Gane E, Schuchmann M, Lohse A, Pol S, Bronowicki JP, Roberts S, Arasteh K, Zoulim F, Heim M, Stern JO, Nehmiz, Kukolj G, Böcher WO, Mensa FJ. Faldaprevir (BI 201335), BI 207127 and ribavirin oral therapy for treatment-naive HCV genotype 1: SOUND-C1 final results. Antivir. Ther. 2013 Apr 4; doi: 10.3851/IMP2567. Epub ahead of print (doi: 10.3851/IMP2567) [DOI] [PubMed] [Google Scholar]
- 21.Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, Heckaman M, Larsen L, Menon R, Koev G, Tripathi R, Pilot-Matias T, Bernstein B. Exploratory study of oral combination antiviral therapy for hepatitis C. N. Engl. J. Med. 2013;368:45–53. doi: 10.1056/NEJMoa1208809. [DOI] [PubMed] [Google Scholar]
- 22.Lawitz E, Poordad F, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, Larsen L, Menon R, Koev G, Tripathi R, Pilot-Matias T, Bernstein B. A phase 2a trial of 12-week interferon-free therapy with two direct-acting antivirals (ABT-450/r, ABT-072) and ribavirin in IL28B C/C patients with chronic hepatitis C genotype 1. J. Hepatol. 2013;59:18–23. doi: 10.1016/j.jhep.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Kapelusznik L, Heil EL, Temesgen Z, Talwani R. Setrobuvir: RNA-directed RNA polymerase inhibitor treatment of hepatitis C virus infection. Drugs of the Future. 2012;37:725–733. [Google Scholar]
- 24.de Bruijne J, van de Wetering de Rooij J, van Vliet AA, Zhou XJ, Temam MF, Molles J, Chen J, Pietropaolo K, Sullivan-Bólyai JZ, Mayers D, Reesink HW. First-in-human study of the pharmacokinetics and antiviral activity of IDX375, a novel nonnucleoside hepatitis C virus polymerase inhibitor. Antimicrob. Agents Chemother. 2012;56:4525–4528. doi: 10.1128/AAC.00451-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Vicente J, Hendricks RT, Smith DB, Fell JB, Fischer J, Spencer SR, Stengel PJ, Mohr P, Robinson JE, Blake JF, Hilgenkamp RK, Yee C, Adjabeng G, Elworthy TR, Tracy J, Chin E, Li J, Wang B, Bamberg JT, Stephenson R, Oshiro C, Harris SF, Ghate M, Leveque V, Najera I, Le Pogam S, Rajyaguru S, Ao-Ieong G, Alexandrova L, Larrabee S, Brandl M, Briggs A, Sukhtankar S, Farrell R, Xu B. Non-Nucleoside Inhibitors of HCV Polymerase NS5B. Part 2: Synthesis and Structure–Activity Relationships of Benzothiazine-Substituted Quinolinediones. Bioorg. Med. Chem. Lett. 2009;19:3642–3646. doi: 10.1016/j.bmcl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Talamas FX, Ao-Ieong G, Brameld KA, Chin E, de Vicente J, Dunn JP, Ghate M, Giannetti AM, Harris SF, Labadie SS, Leveque V, Li J, Lui AS-T, McCaleb KL, Nájera I, Schoenfeld RC, Wang B, Wong A. De Novo Fragment Design: A Medicinal Chemistry Approach to Fragment Based Lead Generation. J. Med. Chem. 2013;56:3115–3119. doi: 10.1021/jm4002605. [DOI] [PubMed] [Google Scholar]
- 27.Riley RJ, Grime K, Weaver R. Time dependent CYP inhibition. Expert Opin. Drug Metab. Toxicol. 2007;3:51–66. doi: 10.1517/17425255.3.1.51. [DOI] [PubMed] [Google Scholar]
- 28.For a recent report of HCV NS5B inhibitors similar to 5: Donner P, Randolph JT, Huang P, Wagner R, Maring C, Lim BH, Colletti L, Liu Y, Mondal R, Beyer J, Koev G, Marsh K, Beno D, Longenecker K, Pilot-Matias T, Kati W, Molla A, Kempf D. High potency improvements to weak aryl uracil HCV polymerase inhibitor leads. Bioorg. Med. Chem. Lett. 2013;23:4367–4369. doi: 10.1016/j.bmcl.2013.05.078.
- 29.For a recent report of HCV NS5B inhibitors employing a template cyclization strategy similar to that used to design 12 and 13: Krueger AC, Randolph JT, DeGoey DA, Donner PL, Flentge CA, Hutchinson DK, Liu D, Motter CE, Rockway TW, Wagner R, Beno DW, Koev G, Lim HB, Beyer JM, Mondal R, Liu Y, Kati WM, Longenecker KL, Molla A, Stewart KD, Maring CJ. Aryl uracil inhibitors of hepatitis C virus NS5B polymerase: synthesis and characterization of analogs with a fused 5,6-bicyclic ring motif. Bioorg. Med. Chem. Lett. 2013;23:3487–3490. doi: 10.1016/j.bmcl.2013.04.057.
- 30.We ruled out undesired tautomers as an explanation based on reported observations that the 4(3H)-pyrimidinone lactam tautomer is dominant over either the 4(1H)-pyrimidinone lactam or 4-hydroxypyrimidine lactim tautomers in aqueous solution, see: Inoue Y, Furutachi N, Nakanishi K. Tautomerism of 4-hydroxy and 4,6-dihydroxypyrimidine. J. Org. Chem. 1966;31:175–178.
- 31.For a recent publication of similar efforts with respect to tert-butyl: Barnes-Seeman D, Jain M, Bell L, Ferreira S, Cohen S, Chen X-H, Amin J, Snodgrass B, Hatsis P. Metabolically Stable tert-Butyl Replacement. ACS Med. Chem. Lett. 2013;4:514–516. doi: 10.1021/ml400045j.
- 32.Patel VR, Agrawal YK. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011;2:81–87. doi: 10.4103/2231-4040.82950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004;21:201–230. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 34.Farci P. New insights into the HCV quasispecies and compartmentalization. Semin. Liver Dis. 2011;31:356–374. doi: 10.1055/s-0031-1297925. [DOI] [PubMed] [Google Scholar]
- 35.Le Pogam S, Seshaadri A, Kosaka A, Chiu S, Kang H, Hu S, Rajyaguru S, Symons J, Cammack N, Najera I. Existence of Hepatitis C Virus NS5B Variants Naturally Resistant to Non-nucleoside, but not to Nucleoside, Polymerase Inhibitors Among Untreated Patients. J. Antimicrob. Chemother. 2008;61:1205–1216. doi: 10.1093/jac/dkn085. [DOI] [PubMed] [Google Scholar]
- 36.Troke PJ, Lewis M, Simpson P, Gore K, Hammond J, Craig C, Westby M. Characterization of resistance to the nonnucleoside NS5B inhibitor filibuvir in hepatitis C virus-infected patients. Antimicrob. Agents Chemother. 2012;56:1331–1341. doi: 10.1128/AAC.05611-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Procedures adapted from: Ward, Robert William; Witherington, Jason: WO 2005/075438 (Preparation 33 and Preparation 34) [Google Scholar]
- 38.Hennessy EJ, Buchwald SL. A General and Mild Copper-Catalyzed Arylation of Diethyl Malonate. Org. Lett. 2002;4:269–272. doi: 10.1021/ol017038g. [DOI] [PubMed] [Google Scholar]
- 39.Molecular Operating Environment (MOE), version 2008.10. Chemical Computing Group, Inc.; Montreal, Canada: 2010. http://www.chemcomp.com. [Google Scholar]
- 40.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA: http://www.pymol.org. [Google Scholar]
- 41.Le Pogam S, Jiang WR, Leveque V, Rajyaguru S, Ma H, Kang H, Jiang S, Singer M, Ali S, Klumpp K, Smith D, Symons J, Cammack N, Nájera I. In Vitro Selected Con1 Subgenomic Replicons Resistant to 2'-C-Methyl-Cytidine or to R1479 Show Lack of Cross Resistance. Virology. 2006;351:349–359. doi: 10.1016/j.virol.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 42.Yeo KR, Yeo WW. Inhibitory effects of verapamil and diltiazem on simvastatin metabolism in human liver microsomes. Br. J. Clin. Pharmacol. 2001;51:461–470. doi: 10.1046/j.1365-2125.2001.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guengerich FP. Oxidation of 17 alpha-ethynylestradiol by human liver cytochrome P-450. Mol. Pharmacol. 1988;33:500–508. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.