

Abstract
A series of gold(III) and palladium(II) heterometallic complexes with new iminophosphorane ligands derived from ferrocenyl-phosphanes [{Cp-P(Ph2)=N-Ph}2Fe] (1), [{Cp-P(Ph2)=N-CH2-2-NC5H4}2Fe] (2) and [{Cp-P(Ph2)=N-CH2-2-NC5H4}Fe(Cp)] (3) have been synthesized and structurally characterized. Ligands 2 and 3 afford stable coordination complexes [AuCl2(3)]ClO4, [{AuCl2}2(2)](ClO4)2, [PdCl2(3)] and [{PdCl2}2(2)]. The complexes have been evaluated for their antripoliferative properties in human ovarian cancer cells sensitive and resistant to cisplatin (A2780S/R), in human breast cancer cells (MCF7) and in a non-tumorigenic human embryonic kidney cell line (HEK-293T). The highly cytotoxic trimetallic derivatives M2Fe (M = Au, Pd) are more cytotoxic to cancer cells than their corresponding monometallic fragments. Moreover, these complexes were significantly more cytotoxic than cisplatin in the resistant A2780R and the MCF7 cell lines. Studies of the interactions of the trimetallic compounds with DNA and the zinc-finger protein PARP-1 indicate that they exert anticancer effects in vitro based on different mechanisms of actions with respect to cisplatin.
Keywords: Gold and palladium complexes, ferrocenyl phosphane-iminophosphorane ligands, cytotoxicity, DNA, zinc-finger proteins
INTRODUCTION
Cisplatin and the follow-on drugs carboplatin (paraplatin™) and oxaliplatin (eloxatin™) are used to treat 40–80% of cancer patients alone or in combination chemotherpay.1 However, their effectiveness is still hindered by clinical problems, including acquired or intrinsic resistance, a limited spectrum of activity, and high toxicity leading to side effects.1,2 Promising anticancer activities of a variety of other metal complexes (including organometallic compounds) have been reported in the past two decades.3–8 Recent progress in this field has brought a better understanding on the mode of action for some of these derivatives.9 In particular, a number of gold compounds have overcome cisplatin resistance to specific cancer cells10 which makes them attractive potential therapeutics. In addition, it has been found that DNA is not the primary target for most gold compounds reinforcing the idea that their mode of action is different with respect to cisplatin.10,11 This has prompted the search for alternative biomolecular targets not only for gold but for other non-platinum complexes10–13 and a revision of the mode of action of anticancer platinum compounds themselves.14 Thus, multiple biological pathways have been proposed for non-platinum derivatives including the inhibition of mitochondrial enzymes and of the proteasome for gold compounds.15–17 Recently, some of us also reported on the potent inhibition of the zinc finger protein poly-(adenosine diphosphate (ADP)-ribose) polymerase 1 (PARP-1) by Au(I) and Au(III) complexes.18,19 PARPs are essential proteins involved in cancer resistance to chemotherapies, and play a key role in DNA repair by detecting DNA strand breaks and catalysing poly(ADP-ribosylation).20,21
Within this frame, there has been a growing interest in heterometallic complexes as potential anticancer agents.22–26 The hypothesis is that the incorporation of two different cytotoxic metals in the same molecule may improve their activity as anti-tumor agents due to interaction of the different metals with multiple biological targets or by the improved chemicophysical properties of the resulting heterometallic compound. The anticancer properties of certain molecules may be increased by the introduction of the organometallic ferrocene motif in the framework due to its low toxicity, high lipophilicity and unique electrochemical behavior.27–29 Thus, several examples of heterometallic complexes incorporating ferrocene and a second cytotoxic metal (Au(I),30–36 Pt(II),30,37–42 Pd(II),40,43–45 Ru(II),46–50 Rh(I)40,51,52 Ir(I),40 and Cu(I)53,54) appeared in the literature. In most cases the antiproliferative properties of the resulting compounds improved with respect of that of the corresponding ferrocene-based ligand, although synergistic effects of the effects of the two different metals were evaluated only in one case.50
Some of us have reported that non-toxic iminophosphorane or iminophosphane (IM) compounds serve as stabilizing (C,N- or N,N) chelating ligands in the preparation of anticancer organometallic and coordination d8 metal complexes.55–58 Organogold(III) complexes containing iminophosphorane (C,N-IM) ligands of the type PPh3=NPh displayed a high cytotoxicity in vitro against human ovarian cancer and leukemia cell lines (in some cases with IC50 values in the nanomolar range)55,56 while being less toxic to normal T-lymphocytes by a non-cisplatin mode of action (involving mitochondrial production of reactive oxygen species).56 More recently, we have reported on the cytotoxicity of coordination and organometallic Pt(II) and Pd(II) complexes with water-soluble iminophosphorane ligands.57,58 Interestingly, we observed that some of these Pt and Pd compounds were more cytotoxic than cisplatin to leukemia cell lines (both T-Jurkat and cisplatin-resistant Jurkat sh-Bak) by a mode of action different to that of cisplatin.57,58
Within this frame, we aimed to explore the biological properties of related IM complexes containing Au(III) and Pd(II) centers incorporating the ferrocene motif, and we report here on the synthesis of new iminophosphorane ligands derived from ferrocenyl-phosphines [{Cp-P(Ph2)=NPh}2Fe] (1), [{Cp-P(Ph2)=N-CH2-2-NC5H4}2Fe] (2) and [{Cp-P(Ph2)=N-CH2-2-NC5H4}Fe(Cp)] (3). While ligand 1 becomes easily protonated while reacted with Au(III) derivatives, ligands 2 and 3 incorporating pyridyl groups serve as an excellent precursors to trimetallic M2Fe and dimetallic MFe derivatives (M = Au, Pd). The antiproliferative properties of the organometallic iron-based ligands and the heterometallic complexes have been evaluated in human ovarian cancer cells sensitive and resistant to cisplatin (A2780S/R), in human breast cancer cells (MCF7) and nontumorigenic human embryonic kidney cell line (HEK-293T).
The compounds were also tested for their possible interactions with plasmid (pBR322) DNA used as a model nucleic acid, and for their reactivity with the transport protein serum albumin (HSA). Moreover, the most promising compounds out of the cytotoxicity screening, the trimetallic M2Fe complexes, were tested for PARP-1 inhibition directly on the purified protein. The obtained results have been discussed in relation to the compounds possible mechanisms of anticancer action.
RESULTS AND DISCUSSION
1. Synthesis and Characterization
Ligands 1 and 2 derived from 1,1′-Bis-diphenylphosphino-ferrocene (DPPF) can be conveniently prepared in moderate yields by reaction of commercial DPPF and phenyl azide (1) or 2-methylpyridine azide (2) by the Staundinger59 method (Scheme 1). Ligand 3 can be prepared in an analogous way in high yields by reacting diphenylphosphino-ferrocene (MPPF)60 and methylpyridine azide. The ligands hydrolyze quickly to amines and the corresponding phosphine oxides at air in solution or as solids due to moisture. They can be conveniently stored in dry glassware under nitrogen for months.
Scheme 1.

Preparation of iminophosphorane ligands 1–3 via the Staudinger59 method.
Ligands 2 and 3 serve as excellent precursors to bimetallic or trimetallic derivatives incorporating Au(III) and Pd(II) centers (Scheme 2). The reaction of ligand 1 to obtain a dinuclear derivative Fe-Au with the Au coordinated to the 2N of the iminophosphorane fragments did not proceed as expected. The reaction of 1 with an in situ solution of [AuCl2(CH3CN)2](ClO4) led to the protonation of the two nitrogen from the iminophosphorane ligand and the obtention of a cationic salt. This compound can indeed be prepared and formulated as [{Cp-P(Ph2)=NH-Ph}2Fe](AuCl4)2 4 by reaction of 1 with 2 equivalents of HAuCl4x3H2O (see experimental). The reaction of ligand 1 with one equivalent of [PdCl2(COD)] resulted in mixtures of coordination and cyclometallated compounds which could not be separated.
Scheme 2.

Preparation of new trimetallic Fe-M2 (M = Au (5), Pd (6)) and bimetallic Fe-M (M = Au (4, 7); Pd (8)). i) 2K[AuCl4]:4AgClO4 in dry CH3CN; ii) 2HAuCl4x3H2O in CH3CN; iii) 2[PdCl2(COD)] in dry CH2Cl2; iv) K[AuCl4]:2AgClO4 in dry CH3CN; v) [PdCl2(COD)] in CH2Cl2. All reactions carried out at RT and under inert atmosphere of N2 or Ar.
The incorporation of one or two pyridyl group in the iminophosphorane ligands 2 and 3 allows for the preparation of coordination dimetallic (7, 8) and trimetallic (5, 6) Au(III) and Pd(II) complexes. While ligands 1–3 are air sensitive the new heterometallic complexes are obtained as air stable solids. The bimetallic complex Fe-Au 7 (Scheme 2) is a cationic derivative while the trimetallic compound Fe-Au2 (5) and the protonated salt 4 are a dicationic species (3 electrolytes) as demonstrated by the conductivity measurements (see Experimental). Both the trimetallic and bimetallic complexes containing Pd(II) centers are neutral species. The presence of only one signal in the 31P{1H} NMR spectra of 5 and 7 suggests a symmetric structure (further confirmed by DFT calculations).
In order to make comparisons about the antiproliferative properties of the new ligands and heterometallic complexes and that of Au(III) and Pd(II) monometallic analogues we prepared previously described ligand Ph3P=N-CH2-2-NC5H4 (IM-PPh3) 961 and monometallic complexes [(Ph3P=N-CH2-2-NC5H4)AuCl2]ClO4 1061 and [(Ph3P=N-CH2-2-NC5H4)PdCl2] 11 (figure 1).
Figure 1.
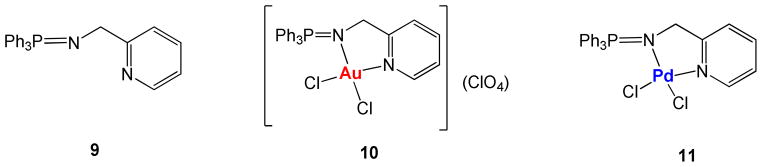
Previously reported IM-PPh3 ligand 9,61 gold(III) derivative 1061 and new Pd(II) complex 11 evaluated for their antiproliferative properties (Table 3) for comparison purposes with the new IM ligands containing ferrocenyl-phosphanes (1–3) and the new IM heterometallic complexes (4–8).
The stability of the compounds in solution can be easily ascertained by 31P{1H} NMR spectroscopy in deuterated solvents (see supporting information). Most compounds are very stable in CD3CN solution (weeks). The stability in d6-DMSO solution was also evaluated. While ligands 2 and 3 (1 is insoluble in d6-DMSO) have half lives of minutes (giving phosphane oxide immediately) the metallic complexes 4–11 have half-lives of weeks (4, 5) or days (6, 7, 11) or several hours (8, 10). The most stable complexes are the protonated ligand (4), the trimetallic complexes M2Fe (M = Au 5, Pd 6) and gold dimetallic (7) species. All the palladium complexes and the gold (10) with the Ph3P=N-CH2-2-NC5H4 (IM-PPh3) ligand (9) decompose to cyclometalled species (in the case of ligand 9 the metallation occurs at the aryl group of the PPh3 phosphane while in the case of the ferrocenyl-phosphane derivatives the metalation occurs both at the aryl ring of the PPh2 fragment and at the cyclopentadienyl of the ferrocene fragment). The stability of 10 in DMSO-H2O is very poor, and, therefore, the incorporation of ferrocene fragments to the iminophosphorane ligand was advantageous to improve the stability of the related gold complexes. In all cases the decomposition products contain the ligand still bound to the metallic fragments. Of note, all complexes are soluble in mixtures DMSO:H2O (1:99 ratio) in micromolar concentrations (relevant for subsequent biological studies).
It is clear that there is an advantage to use the new heterometallic complexes in biological studies as opposed to use the iminophosphorane ligands derived from ferrocenyl phosphanes (1–3) in combination with metallic salts (K[AuCl4] and [PdCl2(COD)]). The new metallic complexes, particularly the trinuclear derivatives 5 and 6, have a much higher stability in DMSO and DMSO:H2O solution than the parent ligands 1–3.
In order to further characterize the stability of the compounds, in physiological-type conditions, we recorded the UV-visible spectra of the metal complexes in PBS solution over time. Representative spectra of compound 5 and 10 are reported in Figure S22 in the supporting information. As it can be seen in the case of 5, the compound’s absorption spectrum is almost unaltered even after 12 h incubation at room temperature, whereas the absorption rapidly decreases with time in the case of the mononuclear gold compound 10.
The structure of compounds 8 and 11 have also been determined by X-ray diffraction analysis (Figure 2). Selected bond lengths and angles for both compounds are collected in Table 1.
Figure 2.
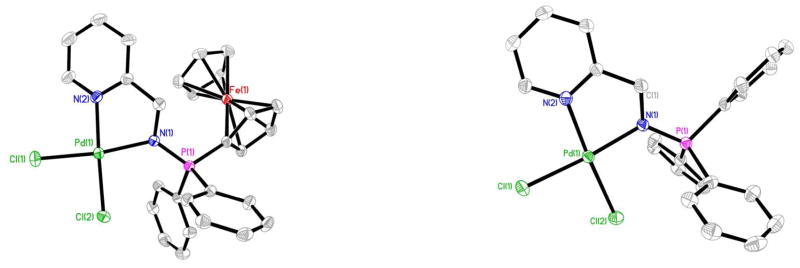
Molecular structure of the compounds 8 and 11 with the atomic numbering scheme.
Table 1.
Selected Structural Parameters of complexes 8 and 11 obtained from X-ray single crystal diffraction studies. Bond lengths in Å and angles in °.
| 8 | 11 | ||
|---|---|---|---|
| P(1)-N(1) | 1.608(4) | P(1)-N(1) | 1.610(3) |
| N(1)-C(1) | 1.469(7) | N(1)-C(1) | 1.463(5) |
| Pd(1)-N(1) | 2.041(4) | Pd(1)-N(1) | 2.038(3) |
| Pd(1)-N(2) | 2.025(5) | Pd(1)-N(2) | 2.029(3) |
| Pd(1)-Cl(1) | 2.2938(15) | Pd(1)-Cl(1) | 2.3081(11) |
| Pd(1)-Cl(2) | 2.2845(16) | Pd(1)-Cl(2) | 2.2895(11) |
| N(2)-Pd(1)-N(1) | 79.51(17) | N(2)-Pd(1)-N(1) | 80.87(12) |
| Cl(2)-Pd(1)-Cl(1) | 90.08(7) | Cl(2)-Pd(1)-Cl(1) | 90.13(4) |
| N(1)-Pd(1)-Cl(1) | 172.28(13) | N(1)-Pd(1)-Cl(1) | 173.45(9) |
| N(2)-Pd(1)-Cl(2) | 174.06(14) | N(2)-Pd(1)-Cl(2) | 175.94(9) |
| N(1)-Pd(1)-Cl(2) | 97.63(13) | N(1)-Pd(1)-Cl(2) | 95.61(9) |
| N(2)-Pd(1)-Cl(1) | 92.84(13) | N(2)-Pd(1)-Cl(1) | 93.50(9) |
| Fe(1)-C | 2.035(6) – 2.051(7) | ||
The geometry about the Pd(II) centers is pseudo-square planar with the N(2)-Pd(1)-N(1) angle of 79.51(17)° (8) and 80.87(12)° (11) suggesting a rigid ‘bite’ angle of the chelating ligand. The Pd centers are on an almost ideal plane with negligible deviations from the least-squares plane. The main distances Pd-N(1) or Pd-N(iminic) and Pd-N(2) found for 8 and 11 are similar to that in related iminophosphorane complexes like [PdCl2(TPA=N-C(O)-2-NC5H4)],57 [Pd{k2-C,N-C6H4(PPh2=NC6H4Me-4′)-2}{μ-OAc)}]2,62 [Pd{k2-C,N-C6H4(PPh2=NC6H4Me-4′)-2}(tmeda)] ClO4,62 [Pd(C6H4CH2NMe2)(Ph3PNC(O)-2-NC5H4)]ClO4,63 and [PtCl(PMe2Ph){(N(p-Tol)=PPh2)2CHCH3}]Cl.64 The distances P=N are shorter than in compounds which incorporate a carbonyl group bonded to the iminic nitrogen (due to a smaller delocalized charge density as compared to the CO group).57,58
Because of the difficulties of obtaining experimental crystal structures for the gold and palladium complexes 5–7, we used computational DFT methods to verify if the suggested molecular structures would be favourable. The initial geometry for the models was taken either from the crystal structure of 8 and 11 or modelled using resembling structures obtained from the Cambridge Crystallographic Data Base. Figure 3 shows the energetically most favourable structures for the complexes, the freely optimized ligand structures can be found in the supplementary material (Figure S1).
Figure 3.

Optimized structures for FeM and FeM2 compounds 5–8 and monometallic analogues 10 and 11. Hydrogens have been omitted for clarity.
In all complexes, the coordination of the metal atoms (Au, Pd) was slightly twisted square planar. The most important difference between the freely optimized ligand structures and corresponding complexes was the torsional behaviour of the nitrogen atoms and also of the ferrocene rings. In separately optimized ligands, the minimum energy conformation of the N-C-C-N(py) torsion was near 180 degrees, while obviously the complexation of the nitrogens resulted in cis-confomation. Also, for the doubly substituted free ligand 2 the ferrocene rings optimized in staggered conformation instead of the effectively eclipsed one obtained for the metallic complexes Fe-Au2 5 and Fe-Pd2 6 (see Figures 3 and S1). For these compounds the energetically most stable structures were obtained when the substituents were located at the 1 and 3′ carbon atoms of the ferrocene rings.
It should be noted, that we tested the effect of the initial substitution sites (1,2′) also for compounds 5 and 6. In the case of the cationic Fe-Au2 complex 5 the ferrocene rings turned into a staggered conformation resulting effectively again into 1,3′ substitution. In the case of the Fe-Pd2 complex 6, the optimization resulted in a more “closed” structure, which had only a slightly higher total energy. It could be concluded therefore, that both structures are feasible for the Fe-Pd2 compound 6 (see Figure S2 in the supplementary material).
Table 2 lists the selected bond distances and electronic properties for the compounds. The calculated IR spectra are also compared for selected stretching modes in Table 2. The trend in the shifts for the ν(M-Cl) values is clear: the Au complexes 5, 7 and 10 exhibited larger wavenumbers for the asymmetric and symmetric M-Cl stretch than the corresponding palladium complexes 6, 8 and 11 (see more details in the supporting information section Figures S3–S8).
Table 2.
Selected geometrical and electronic properties of the FeM and FeM2 compounds 5–8, 10 and 11. Gibbs free energies refer to formation reactions, where the corresponding metal ions react with chloride ions and the corresponding free ligands. GAP is the energy difference of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).
| 5 | 6 | 7 | 8 | 10 | 11 | |
|---|---|---|---|---|---|---|
| d(P=N) | 1.650 | 1.616 | 1.659 | 1.623 | 1.654 | 1.618 |
| d(M-Cl) | 2.256 | 2.285 | 2.258 | 2.283 | 2.258 | 2.283 |
| ΔrG [kJmol−1] | −1797b | −701b | −1824 | −711 | −1811 | −708 |
| GAP [eV] | 3.34 | 4.55 | 2.91 | 4.35 | 4.08 | 4.58 |
| ν(P=N) [cm−1]a | 948b (940) | 953b (923) | 948 (940) | 957 (929) | 942 (930) | 951 (930) |
| ν(M-Cl) [cm−1]a | 374;385 (382 br) | 345;357 (331;349) | 370;384 (384 br) | 344;357 (331;347) | 371;384 (359 br) | 343;358 (331;351) |
IR spectra were simulated in aqueous solvent; (experimental values obtained from nujol mulls between polyethylene sheets).
For the doubly substituted complexes, the reaction energies were scaled per metal atom.
This correlates very well in the optimized M-Cl distances, since the Au-Cl distances were about 0.03 Å shorter than the Pd-Cl distances. The ν(P=N) values in the complexes were found to be very similar. The same trends are seen also in the experimental frequencies, even though the computational absolute values are slightly overestimated, which is typical for the applied DFT method.65
The formation energies of the compounds were also investigated (see Table 2 and supporting information for details) and it was found that all the studied complexes are readily formed. The larger HOMO-LUMO gap, on the other hand, indicates that palladium complexes 6 and 8 are more stable and thus less reactive than the gold analogues 5 and 7. This is in accordance with our previous observation for gold(III) coordination compounds containing iminophosphorane ligands.56 Interestingly, the gold complex 10 does not show similar reduction in the HOMOLUMO gap, indicating that the ferrocene motif can increase the reactivity of the compounds and that the ferrocene moiety has a larger effect on gold complexes that on palladium ones. Experimentally 10 is the most unstable compound of the series most likely due to a cyclometallation process in DMSO. However, this cyclometallation process was not in the scope of our computational studies, and therefore the energetics could not reveal this particular detail of the relative stability.
2. Biological Activity
2.1. Antiproliferative Studies
The antiproliferative properties of the new ligands and heterometallic complexes and of ligand Ph3P=N-CH2-2-NC5H4 (IM-PPh3) 961 and monometallic complexes [(Ph3P=N-CH2-2-NC5H4)AuCl2]ClO4 1061 and [(Ph3P=N-CH2-2-NC5H4)PdCl2] 11 (figure 1) were assayed by monitoring their ability to inhibit cell growth using the MTT assay (see Experimental Section). Cytotoxic activity of the compounds was determined as described in the Experimental Section in the human ovarian cancer A2780 cell line, and its cisplatin resistant variant (A2780cisR), as well as in the human breast cancer cell line MCF7, in comparison to cisplatin. The results are summarized in Table 3. In general the ferrocene-based ligands are poorly cytotoxic in all tested cell lines and this may be caused by their poor solubility in aqueous solution. Interestingly, ligands 1–3 incorporating the ferrocenyl-phosphanes are considerably more cytotoxic than the purely organic ligand 9 containing triphenylphosphane. This is in agreement with the enhanced antripoliferative properties for a vast majority of derivatives in which an aryl group is substituted by the ferrocene motif.20,21 Concerning the dimetallic complexes derived from ligand 3 (based on MPPF), the Pd(II) derivative 8 is markedly more cytotoxic than 3 and of the Au(III) derivative 7. Moreover, such dimetallic complexes are less active than their monometallic IM-PPh3 analogues 10 (Au) and 11 (Pd) on the ovarian cancer cell lines A2780S/R. It must be noted, that the reduced effect of the gold compound 10 may also be due to its reduced stability in aqueous medium as explained in the previous section. Concerning the trimetallic M2Fe (M = Au 5, Pd 6) compounds with ligand 2 (based on DPPF), these complexex exhibit important cytotoxic effects in the low micromolar range in all the studied cell lines. Notably, they are also significantly more cytotoxic than cisplatin towards the resistant A2780R and MCF7 cell lines. The trimetallic compounds also resulted to be significantly more cytotoxic than ligands 1 and 2 and more effective than the monometallic Au and Pd derivatives 10 and 11. In order to assess the compounds selectivity for cancerous cells with respect to normal cell lines, the most cytotoxic heterometallic compounds were also screened for their antiproliferative effects on the non-tumorigenic human embryonic kidney cells HEK293T. In most cases the cytotoxicity is comparable for the cancerous and normal cell lines.
Table 3.
IC50 (UM) of ligands 1–3 and 9, DPPF oxide and metal complexes 4–8, 10–11 and cisplatin in human cell lines.a All compounds were dissolved in 1% of DMSO and diluted with water before addition to cell culture medium for a 74 h incubation period. Cisplatin was dissolved in H2O.
| A2780 | A2780cisR | MCF7 | HEK-293T | |
|---|---|---|---|---|
| 1 | 40.3 ±5.2 | 32.7 ±6.0 | - | 45 ±5.0 |
| 2 | 5.3±0.8 | 13.1±4.0 | 15.0±6.0 | 10.5±4.0 |
| 3 | 36.7±6.0 | 30.3 ±1.6 | - | >50 |
| 4 | 1.4 ±0.8 | 1.3 ±1.0 | 3.0±0.8 | 1.2±0.6 |
| 5 | 2.7±0.8 | 2.4±0.5 | 3.5±1.5 | 4.0±0.8 |
| 6 | 2.0±0.4 | 1.8±0.7 | 3.0±0.6 | 5.6±0.3 |
| 7 | 30.1±5.0 | 25.2±4.3 | 40.0±7.0 | 32.2±5.0 |
| 8 | 17.4±4 | 11.9±2.5 | 10.2±3.7 | 15.0±3.2 |
| 9 | >100 | >50 | >100 | >100 |
| 10 | 19.0±1.1 | 8.0±2.4 | 30.0±3.8 | 30.5±1.5 |
| 11 | 7.1±1.7 | 6.2±0.3 | 25.0±4.3 | 45.0±5.0 |
| DDPF-oxide | 113±72 | 85±38 | >50 | >50 |
| cisplatinb | 1.9±0.6 | 35±7.0 | 20.0±3.0 | 11.0±2.9 |
Data are expressed as mean ± SD (n =4).
Ref. 18.
Only the in the case of monometallic Pd compound 11 is markedly (ca. 7.5-fold) more toxic on the cancerous cell lines (IC50 6–7 μM) than on the non-cancerous HEK293T cells (IC50 ca. 45 μM). Finally, compound 4 was among the most cytotoxic in all the tested cell lines including the HEK293T cells, and this may be attributed to the presence of the AuCl4− anion in the compound with a highly oxidative character leading to marked and unselective cell death. Such type of gold-based ionic compound being highly toxic also in the non-tumorigenic cell line will not be certainly selected for further biological screening.
2.2. Reactivity with biomolecules
Interactions with plasmid DNA
Since DNA replication is a key event for cell division, it is among critically important targets in cancer chemotherapy. Most cytotoxic platinum drugs form strong covalent bonds with the DNA bases.66 However, a variety of platinum compounds act as DNA intercalators upon coordination to the appropriate ancillary ligands.67 There are also reports on palladium derivatives interacting with DNA in covalent68,69 and non-covalent fashions.70,71 Conversely, most gold-based compounds do not display a strong interaction with DNA.10,11
Thus, we performed agarose gel electrophoresis studies to unravel the effects of the new heterometallic compounds 5–8 and 10, 11 on plasmid (pBR322) DNA (Fig. 4). This plasmid has two main forms: OC (open circular or relaxed form, Form II) and CCC (covalently closed or supercoiled form, Form I). Changes in electrophoretic mobility of both forms are usually taken as evidence of metal-DNA binding. Generally, the larger the retardation of supercoiled DNA (CCC, Form I), the greater the DNA unwinding produced by the drug.72 Binding of cisplatin to plasmid DNA, for instance results in a decrease in mobility of the CCC form and an increase in mobility of the OC form (see lanes a–d for cisplatin in Fig. 4).
Fig. 4.

Electrophoresis mobility shift assays for cisplatin and compounds 5–8 and 10, 11 (see Experimental for details). DNA refers to untreated plasmid pBR322. a, b, c and d correspond to metal/DNAbp ratios of 0.25, 0.5, 1.0 and 2.0 respectively.
Treatment with increasing amounts of Au(III) compounds with IM ligands containing ferrocene 5, and 7 and the Au(III) compound with an IM-PPh3 ligand 10 do not affect the mobility of the faster-running supercoiled form (Form I) even at the highest molar ratios (d). This is also in accordance with previously reported results on organogold(III) compounds containing iminophosphorane ligands did not interact with DNA.55
Conversely, the Pd(II) compounds with IM ligands containing ferrocene 8 or triphenylphosphine 11 have a similar behaviour to that of a coordination iminophosphorane palladium complex described by us [PdCl2(TPA=N-C(O)-2-NC5H4)].56 In fact, these compounds significantly modify the electrophoretic mobility and the retardation of the faster-running supercoiled form (Form I or CCC) and an increased electrophoretic mobility of form II (OC) was clearly observed for 6, 8 and 11 at lower ratios than those for cisplatin. Moreover, the Pd complexes 8 and 11 are able to induce a coalescence of the two plasmid forms, an effect that has already been described for cisplatin, but at higher drug/DNAbp ratios.73 The presence of a coalescence point indicates a strong unwinding of the pBR322 plasmid DNA for compounds 8 and 11. The trimetallic Fe-Pd2 compound 6 (with ligand 2 derived from DPPF) also shows a coalescence point at the lowest metal/DNA ratio (0.25, line a), most likely followed by a change of conformation of plasmid DNA from negative supercoil to positive supercoil at the highest metal compound/DNAbp ratios. Overall, our results indicates an interaction of the new iminophosphorane palladium complexes mono and heterometallic (Fe-Pd) with DNA stronger than that observed for cisplatin.
Inhibition of PARP-1
As previously mentioned the zinc finger protein PARP-1 has been recently identified as a possible taget for Au(III) complexes in vitro.18,19 Information on the reactivity of the gold complexes with the PARP-1 zinc-finger domain was also obtained by high-resolution mass spectrometry, and an excellent correlation between PARP-1 inhibition in protein extracts and the ability of the complexes to bind to the zinc finger motif (in competition with zinc) was established.18 Thus, in order to further investigate the mechanisms of action of the new heteronuclear complexes, we tested the mononuclear compounds 5 and 6 for their PARP-1 inhibition properties in vitro as reported in the Experimental section. The obtained preliminary results showed that the trinuclear Au-Fe compound 5 is a potent PARP-1 inhibitor with an IC50 = 1± 0.5 μM, while the trinuclear Pd-Fe analogue 6 is much less effective with an IC50 = 17± 0.8 μM.
Interactions with HSA
Human serum albumin (HSA) is the most abundant carrier protein in plasma and is able to bind a variety of substrates including metal cations, hormones and most therapeutic drugs. It has been demonstrated that the distribution, the free concentration and the metabolism of various drugs can be significantly altered as a result of their binding to the protein.74 HSA possesses three fluorophores, these being tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe) residues, with Trp214 being the major contributor to the intrinsic fluorescence of HSA. This Trp fluorescence is sensitive to the environment and binding of substrates, as well as changes in conformation that can result in quenching (either dynamic or static). Thus, the fluorescence spectra of HSA in the presence of increasing amounts of the representative compounds 5, 7 and 8 and cisplatin were recorded in the range of 300–450 nm upon excitation of the tryptophan residue at 295 nm (Fig. 5). The compounds caused a concentration dependent quenching of fluorescence without changing the emission maximum or shape of the peaks (5, 7, 8), as seen in Fig. 5 (A) for compound 7. All this data indicates an interaction of the heterometallic compounds with HSA. The fluorescence data was analyzed by the Stern-Volmer equation. While a linear Stern-Volmer plot is indicative of a single quenching mechanism, either dynamic or static, the positive deviation observed in the plots of F0/F versus [Q] of our compounds (Fig. 5) suggests the presence of different binding sites in the protein.75 Of note, a similar behavior was observed in the case of coordination iminophosphorane complexes of d8 metals for which we also reported a concentration dependent fluorescence quenching.57,58 In this graph higher quenching by the iminophosphorane complexes was observed compared to that of cisplatin under the chosen conditions, most likely due to the faster reactivity of our compounds with HSA compared to cisplatin.
Figure 5.

(a) Fluorescence titration curve of HSA with compound 7. Arrow indicates the increase of quencher concentration (10–100 μM). (b) Stern-Volmer plot for HSA fluorescence quenching observed with compounds 5, 7, 8 and cisplatin.
In the case of [MCl2(TPA=N-C(O)-2-NC5H4)] (M = Pd, Pt) isothermal titration calorimetry (ITC)57 showed two different binding interactions which explained the lack of linearity observed in the fluorescence quenching studies, as the Stern-Volmer method assumes all binding sites to be equivalent. We believe that a similar reactivity takes place for the iminophosphorane compounds described here.
CONCLUSIONS
In conclusion we have prepared a series of coordination iminophosphorane complexes of gold(III) and palladium(II) derived from ferrocenyl phosphanes. The new compounds are markedly more stable than the ferrocenyl phosphane ligands in DMSO or aqueous solution, and more stable than their monometallic analogues derived from PPh3 [(AuCl2(Ph3P=N-CH2-2-NC5H4)]ClO4 10 and [PdCl2{N-CH2-2-NC5H4}] 11. The most stable trimetallic M2Fe (M = Au, Pd) derivatives (5 and 6) exhibit important cytotoxic effects in the low micromolar range in all the cancer cells studied and are more cytotoxic than their corresponding monometallic fragments (ligand 2 containing ferrocene and monometallic analogues 10 and 11) also indicating a possible synergistic antiproliferative effect of the two different metals. Importantly, these complexes were significantly more cytotoxic than cisplatin in the resistant A2780R and MCF7 cell lines supporting the idea of different mechanisms of action than cisplatin.
It is worth mentioning that heterometallic trinuclear complexes have already been described by some of us to be more stable than their mononuclear and dinuclear precursors, including goldtitanium complexes.24,25 Moreover, improved biological properties of multinuclear complexes with respect to mononuclear compounds have been reported,26 which could be related to favorable modulation of the stability, solubility, and/or lipophilicity of the organometallic scaffolds. Therefore, the results described herein support the idea that there is an advantage in synthesizing heteronuclear complexes as anticancer agents.
When the reactivity of the new heteronuclear complexes was tested with plasmid DNA the Pd(II) derivatives showed strong interactions with nucleic acids, while the Au(III) analogues were practically ineffective. Instead, the Au(III) complexes resulted to be good inhibitors of the zinc-finger protein PARP-1, a possible target enzyme for anticancer metal compounds, while the Pd(II) complexes were markedly less potent. In general, although further studies are necessary to validate the mechanisms of biological action of this new series of heteronuclear metal compounds, our results support the idea of different reactivity of the complexes with the investigated biomolecules depending on the metal ion and not only on the ligand set.
EXPERIMENTAL
All manipulations involving air-free syntheses were performed using standard Schlenk-line techniques under a nitrogen atmosphere or in a glove-box MBraun MOD System. Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc. The substrates [(Cp-PPh2)2Fe] 1,1′-Bis-diphenylphosphino-ferrocene (DPPF), K[AuCl4] and [PdCl2(COD)] were purchased from Strem chemicals and used without further purification. [(Cp-PPh2)Fe(Cp)] diphenylphosphino-ferrocene (MPPF),60 (Ph3P=N-CH2-2-NC5H4)61 and [(Ph3P=N-CH2-2-NC5H4)AuCl2]ClO461 were prepared by reported methods. Purity of the compounds is based on elemental analysis and in all cases is ≥95%. Elemental analyses were performed on a Perkin Elmer 2400 CHNS/O Analyzer, Series II. NMR spectra were recorded in a Bruker AV400 (1H NMR at 400MHz, 13C NMR at 100.6 MHz, 31P NMR at 161.9 MHz). Chemical shifts (δ) are given in ppm using CDCl3 or d6-DMSO as solvent, unless otherwise stated. 1H and 13C chemical shifts were measured relative to solvent peaks considering TMS = 0 ppm; 31P{1H} was externally referenced to H3PO4 (85%). Infrared spectra (4000-250 cm−1) were recorded on a Nicolet 6700 FT-IR spectrophotometer from nujol mulls between polyethylene sheets. Mass spectra (ESI) were performed on an Agilent Analyzer or a Bruker Analyzer. Conductivity was measured in an OAKTON pH/conductivity meter in CH3CN solutions (10−3M). X-ray collection was performed at room temperature on a Kappa CCD diffractometer using graphite monochromated Mo-Kα radiation (λ=0.71073 Å). Electrophoresis experiments were carried out in a Bio-Rad Mini sub-cell GT horizontal electrophoresis system connected to a Bio-Rad Power Pac 300 power supply. Photographs of the gels were taken with an Alpha Innotech FluorChem 8900 camera. Fluorescence intensity measurements were carried out on a PTI QM-4/206 SE Spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence using a 1 cm path length quartz cuvette. Circular Dichroism spectra were recorded using a Chirascan CD Spectrometer equipped with a thermostated cuvette holder. UV-visible spectra have been recorded using a on a Perkin-Elmer Lambda 20 Bio spectrophotometer.
Synthesis
[{Cp-P(Ph2)=N-Ph}2Fe] (1)
A solution of DPPF (0.554 g, 1.0 mmol) in 15 mL of degassed CH2Cl2 and a solution of phenyl azide (0.238 g, 2.0 mmol) in 5 mL of degassed CH2Cl2 were mixed and refluxed for 3 hours and 40 minutes under nitrogen. After cooling the mixture to RT the solvent was evaporated to ca 2 mL and by addition of 6 mL of dry Et2O and 4 mL of dry n-hexane a precipitate formed which was separated by filtration under nitrogen. 1 was obtained as dark orange solid that was used without further purification. Yield: 0.483 g (61.3%). Anal. Calcd for [C46H38FeN2P2].0.6CH2Cl2 (787.57): C, 71.07; H, 5.02; N, 3.56. Found: C, 71.22; H, 4.90; N, 3.46. MS (ESI+) [m/z]: 737.1 [M + H]. 31P{1H} NMR (CDCl3): δ 2.7. 1H-NMR (CDCl3): δ 4.04 (4H, s, Cp), δ 4.53 (4H, s, Cp), δ 6.66 (2H, t, J = 6.9 Hz, Hpara, NAr), δ 6.75 (4H, d, J = 7.4 Hz, Hortho, NAr), δ 7.02 (4H, t, J = 7.2 Hz, Hmeta, NAr), δ 7.43 (8H, d, J = 5.5 Hz, Hmeta, PPh2), δ 7.51 (4H, d, J = 6.8 Hz, Hpara, PPh2), δ 7.64 (8H, dd, J = 11.8, 7.8 Hz, Hortho, PPh2). 13C{1H} NMR (CDCl3): δ 73.8 (d, J = 9.4 Hz, Cp), δ 74.4 (d, J = 11.3 Hz, Cp), δ 117.3 (s, Cpara, NAr), δ 123.4 (d, J = 17.9 Hz, Cortho, NAr), δ 128.4 (d, J = 12.1 Hz, Cmeta, PPh2), δ 128.6 (s, Cmeta, NAr), δ 131.5 (s, Cpara, PPh2), δ 132.1 (d, J = 9.7 Hz, Cortho, PPh2), δ 132.6 (s, PC), δ 151.1 (s, NC). IR (cm−1): ν 1264 (P=N).
[{Cp-P(Ph2)=N-CH2-2-NC5H4}2Fe] (2)
A solution of DPPF (0.886 g, 1.6 mmol) in 15 mL of degassed CH2Cl2 and a solution of 2-methylpyridine azide (0.968 g, 3.7 mmol) in 5 mL of degassed CH2Cl2 were mixed for 3.5 h at RT under nitrogen. The solvent was then evaporated to ca 2 mL and by addition of 6 mL of dry Et2O a precipitate formed which was separated by filtration under nitrogen. 2 was obtained as brown solid that was used without further purification. Yield: 0.577 g (66.7%). Anal. Calcd for C46H40FeN4P2 (766.6334): C, 72.07; H, 5.26; N, 7.31. Found: C, 71.81 ; H, 5.14 ; N, 7.12. MS (ESI+) [m/z]: 767.2151 [M+H]. 31P{1H} (CDCl3): δ 11.9. 1H NMR (CDCl3): δ 4.18 (4H, s, Cp), δ 4.48 (4H, s, Cp), δ 4.52 (4H, s, CH2), δ 7.07 (2H, t, J = 6.0 Hz, NCHCH), δ 7.38 (8H, d, J = 5.9 Hz, Hmeta), δ 7.47 (4H, d, J = 6.8 Hz, Hpara), δ 7.57 (8H, dd, J = 11.5, 7.5 Hz, Hortho), δ 7.69 (2H, t, J = 7.0 Hz, NC(CH2)CHCH), δ 8.00 (2H, d, J = 7.3 Hz, NC(CH2)CH), δ 8.44 (2H, d, J = 4.4 Hz, NCH). 13C{1H} NMR (CDCl3): δ 51.3 (s, CH2), δ 73.6 (d, J = 9.4 Hz, Cp), δ 120.8 (s, NCHCH), δ 121.4 (s, NC(CH2)CH), δ 128.1 (d, J = 10.5 Hz, Cmeta), δ 131.1 (s, Cpara), δ 132.0 (d, J = 9.0 Hz, Cortho), δ 136.3 (s, NC(CH2)CHCH), δ 148.2 (s, NCH) IR (cm−1): ν 1257 (P=N).
[{Cp-P(Ph2)=N-CH2-2-NC5H4}Fe(Cp)] (3)
A solution of [(Cp-PPh2)Fe(Cp)] (0.738 g, 2 mmol) in 10 mL of degassed CH2Cl2 and a solution of 2-methylpyridine azide (0.2968 g, 2 mmol) in 5 mL of degassed CH2Cl2 were mixed for 3 hours and 30 minutes at RT under nitrogen. The solvent was then evaporated to ca 2 mL and by addition of 10 mL of dry Et2O a precipitate formed which was separated by filtration under nitrogen. 3 was obtained as brown solid that was used without further purification. Yield: 0.745 g (76.8%). Anal. Calcd for [C28H25FeN2P]0.1CH2Cl2 (484.83): C, 69.61; H, 5.24; N, 5.78. Found: C, 69.52; H, 5.23; N, 5.77. MS (ESI+) [m/z]: 477.1 [M+H]. 31P{1H} NMR (CDCl3): δ 13.1. 1H NMR (CDCl3): δ 4.16 (5H, s, Cp), δ 4.33 (2H, d, J = 1.6 Hz, Cp), δ 4.45 (2H, d, J = 1.6 Hz, Cp), δ 4.64 (2H, d, J = 18.0 Hz, CH2), δ 7.10 (1H, t, J = 6.0 Hz, NCHCH), δ 7.40–7.46 (4H, m, Hmeta), δ 7.50 (2H, dd, J = 7.2, 1.5 Hz, Hpara), δ 7.67–7.71 (4H, m, Hortho), δ 7.72–7.74 (1H, m, NC(CH2)CHCH), δ 8.06 (1H, d, J = 7.9 Hz, NC(CH2)CH), δ 8.46 (1H, d, J = 4.2 Hz, NCH). 13C{1H} NMR (CDCl3): δ 51.4 (s, CH2), δ 69.6 (s, Cp), δ 70.98 (d, J = 8.7 Hz, Cp), δ 72.7 (d, J = 10.9 Hz, Cp), δ 120.7 (s, NCHCH), δ 121.6 (s, NC(CH2)CH), δ 128.1 (d, J = 11.6 Hz, Cmeta), δ 131.1 (s, Cpara), 132.2 (d, J = 9.1 Hz, Cortho), δ 136.3 (s, NC(CH2)CHCH), δ 148.1 (s, NCH). IR (cm−1): ν 1303 (P=N).
[{Cp-P(Ph2)=NH-Ph}2Fe](AuCl4)2 4
A solution of H[AuCl4] (0.158 g, 0.4 mmol) in CH3CN (7 mL) was added over a solution of 1 (0.1472 g, 0.2 mmol) in degassed dry CH2Cl2. After 20 min stirring at RT the reaction mixture was concentrated under vacuum to ca. 2 mL. The addition of 8 mL of Et2O afforded dark brown oil, which was washed with water (2 × 5 mL). 4 was obtained as a light brown solid. Yield: 0.2053 g (66.5%). Anal. Calcd for [C46H40Au2Cl8FeN2P2]1.5CH2Cl2 (1543.56): C, 36.96; H, 2.81; N, 1.81. Found: C, 36.75; H, 2.75; N, 1.82. MS (ESI+) [m/z]: 737.1 [M-H]+. 31P{1H} NMR (d6-DMSO): δ 36.3. 1H-NMR (d6-DMSO): δ 4.58 (4H, s, Cp), δ 4.73 (4H, s, Cp), δ 6.89 (4H, d, J = 7.8 Hz, Hortho, NAr), δ 7.03 (2H, t, J = 7.2 Hz, Hpara, NAr), δ 7.20 (4H, t, J = 7.4 Hz, Hmeta, NAr), δ 7.78–7.84 (8H, m, Hmeta, PPh2), δ 7.88–7.91 (8H, m, Hortho, PPh2), δ 7.95 (4H, t, J = 6.8 Hz, Hpara, PPh2). 13C{1H} NMR (d6-DMSO): δ 76.2 (d, J = 13.4 Hz, Cp), δ 77.0 (d, J = 11.1 Hz, Cp), δ 121.9 (d, J = 5.7 Hz, Cortho, NAr), δ 124.7 (s, Cpara, NAr), δ 130.1 (s, Cmeta, NAr), δ 130.7 (d, J = 13.7 Hz, Cmeta, PPh2), δ 133.4 (d, J = 11.5 Hz, Cortho, PPh2), δ 136.0 (s, Cpara, PPh2). IR (cm−1): ν 1282, 963 (P=N). Conductivity Λ (MeCN) = 221.3 μS/cm (2 ions 1+/1−).
[({Cp-P(Ph2)=N-CH2-2-NC5H4}AuCl2)2Fe](ClO4)2 (5)
To a solution of K[AuCl4] (0.113 g; 0.3 mmol) in CH3CN (10 mL), AgClO4 (0.136 g, 0.66 mmol) was added. The resulting yellow reaction mixture was stirred at RT (protected from the light) during 30 min and subsequently filtered through a celite pad (to remove the AgCl formed). To the resulting yellow solution 2 (0.113 g, 0.14 mmol) was added. KClO4 precipitated immediately in the reaction media and after 1 h stirring at RT the reaction mixture was filtered through a celite pad. The resulting brown solution was concentrated under vacuum to ca. 2 mL. The addition of 2 mL of EtOH and 8 mL of Et2O afforded 5 as a black solid that was filtered, dried under vacuum and used without further purification. Yield: 0.1088 g (48.2%). Anal. Calcd for [C46H40Au2Cl6FeN4O8P2]1.5C4H10O (1612.46): C, 38.73; H, 3.44; N, 3.47. Found: C, 39.06; H, 3.10; N, 3.69. MS (ESI+) [m/z]: only ligand observed. 31P{1H} NMR (d6-DMSO): δ 41.7 ppm. 1H NMR (d6-DMSO): δ 4.31 (4H, d, J = 10.4 Hz, CH2), δ 4.69 (4H, s, Cp), δ 4.88 (4H, s, Cp), δ 7.44 (2H, d, J = 7.7 Hz, NCHCH), δ 7.48 (2H, s, NC(CH2)CH), δ 7.67–7.77 (12H, m, Hmeta + Hpara), δ 7.83–7.87 (8H, m, Hortho), δ 7.87–7.90 (2H, m, NC(CH2)CHCH), 8.51 (2H, d, J = 5.1 Hz, NCH). 13C{1H} NMR (d6-DMSO): δ 46.1 (s, CH2), δ 75.4 (d, J = 13.5 Hz, Cp), δ 76.8 (d, J = 11.0 Hz, Cp), δ 123.5 (s, NC(CH2)CH), δ 124.1 (s, NCHCH), δ 130.3 (d, J = 13.2 Hz, Cmeta), δ 133.2 (d, J = 11.5 Hz, Cortho), δ 135.9 (s, Cpara), δ 139.6 (s, NC(CH2)CHCH), δ 149.4 (s, NCH), δ 156.2 (s, NC). IR (cm−1): ν 382 (Au-Cl); 1071 (v br) and 623 (ClO4 −); 1291, 940 (P=N). Conductivity Λ (MeCN) = 412.8 μS/cm (3 ions, 12+/2−).
[({Cp-P(Ph2)=N-CH2-2-NC5H4}PdCl2)2Fe] (6)
A solution of [PdCl2(COD)] (0.074 g, 0.26 mmol) in 5 mL of degassed dry CH2Cl2 was added over a solution of 2 (0.0858 g, 0.13 mmol) in degassed dry CH2Cl2. The reaction mixture was stirred for one hour at RT after which the solvent was reduced to ca 2 mL. Addition of Et2O (15 mL) afforded 6 as a yellow solid that was collected by filtration and dried under vacuum. Yield: 0.072 g (49.5%). Anal. Calcd for C46H40Cl4FeN4P2Pd2 (1121.29): C, 49.27; H, 3.60; N, 5.00. Found: C, 49.56; H, 3.83; N, 3.27. MS (ESI+) [m/z]: 1084.9214 [M-Cl]+. 31P{1H} NMR (CDCl3): δ 40.8 ppm. 1H NMR (CDCl3): δ 4.20 (4H, s, Cp), δ 4.35 (4H, d, J = 10.6 Hz, CH2), δ 5.00 (4H, s, Cp), δ 7.23 (1H, d, J = 6.7 Hz, NC(CH2)CH), δ 7.39 (1H, t, J = 5.9 Hz, NCHCH), δ 7.57 (4H, d, J = 4.4 Hz, Hmeta), δ 7.61–7.67 (2H, m, Hpara), δ 7.74 (4H, dd, J = 12.4, 7.5 Hz, Hortho), δ 7.78 (1H, t, J = 8.4 Hz, NC(CH2)CHCH), δ 9.16 (1H, d, J = 5.3 Hz, NCH). 13C{1H} NMR (CDCl3): δ 58.7 (s, CH2), δ 73.9 (s, Cp), δ 76.9 (s, Cp), δ 119.7 (s, NC(CH2)CH), δ 123.5 (s, NCHCH), δ 129.0 (d, J = 12.7 Hz, Cmeta), δ 133.0 (d, J = 10.1 Hz, Cortho), δ 133.3 (s, Cpara), δ 138.9 (s, NC(CH2)CHCH), δ 150.7 (s, NCH), δ 164.4 (d, J = 12.1 Hz, NC(CH2)). IR (cm−1): ν 1287, 923 (P=N); 349, 331 (Pd-Cl). Conductivity Λ (MeCN) = 24.6 μs/cm (neutral).
[{(Cp-P(Ph2)=N-CH2-2-NC5H4)AuCl2}Fe(Cp)]ClO4 (7)
To a solution of K[AuCl4] (0.113 g; 0.3 mmol) in CH3CN (10 mL), AgClO4 (0.068 g, 0.33 mmol) was added. The resulting yellow reaction mixture was stirred at RT (protected from the light) during 30 min and subsequently filtered through a celite pad (to remove the AgCl formed). To the resulting yellow solution 3 (0.071 g, 0.15 mmol) was added. KClO4 precipitated immediately in the reaction media and after 1 h stirring at RT the reaction mixture was filtered through a celite pad. The solvent was reduced to ca. 2 mL and a precipitate was obtained by addition of 2 mL of EtOH and 8 mL of Et2O. The precipitate was filtered off and dried under vacuum to afford 7 as a dark brown solid. Yield: 0.100 g (71.8%). Anal. Calcd for [C28H25FeN2PCl3Au]CH2Cl2 (928.58): C, 37.51; H, 2.93; N, 3.02. Found: C, 37.82; H, 2.96; N, 3.12. MS (ESI+) [m/z]: 707.0361[(M - Cl) – ClO4]2+. 31P{1H} NMR (d6-DMSO): δ 43.1. 1H-NMR (DMSO): δ 4.26 (5H, s, Cp), δ 4.39 (2H, s, CH2), δ 4.71 (2H, s, Cp), δ 4.90 (2H, s, Cp), δ 7.42 (1H, s, NCHCH), δ 7.44–7.47 (6H, m, Hmeta + Hpara), δ 7.49 (1H, s NC(CH2)CH), δ 7.65–7.74 (4H, m, Hortho), δ 7.90 (1H, s, NC(CH2)CHCH), δ 8.62 (1H, d, J = 4.4 Hz, NCH). 13C{1H} NMR (d6-DMSO): δ 45.6 (s, CH2), δ 71.0 (s, Cp), δ 73.6 (s, Cp), δ 74.6 (s, Cp), δ 123.1 (s, NC(CH2)CH), δ 124.0 (s, NCHCH), δ 128.8 (d, J = 12.4 Hz, Cmeta), δ 131.3 (d, J = 10.9 Hz, Cortho), δ 131.8 (s, Cpara), δ 138.1 (NC(CH2)CHCH), δ 148.9 (s, NCH). IR (cm−1): ν 384 (Au-Cl); 1084 (v br) and 624 (ClO4−); 1289, 940 (P=N). Conductivity Λ (MeCN) =190.9 μs (2 ions 1+/1−).
[{(Cp-P(Ph2)=N-CH2-2-NC5H4)PdCl2}Fe(Cp)] (8)
A solution of [PdCl2(COD)] (0.057 g, 0.20 mmol) in 5 mL of degassed dry CH2Cl2 was added over a solution of 3 (0.103, 0.20 mmol) in degassed dry CH2Cl2. The reaction mixture was stirred for one hour at RT after which the solvent was reduced to ca 2 mL. Addition of Et2O (15 mL) afforded 6 as a yellow solid that was collected by filtration and dried under vacuum. Yield: 0.0806 g (61.8%). Anal. Calcd for C28H25FeN2PCl2Pd (653.66): C, 51.44; H, 3.86; N, 4.29. Found: C, 50.94; H, 3.96; N, 4.50. MS (ESI+) [m/z]: 653.1 [M]. 31P{1H} NMR (CDCl3): δ 41.0 ppm. 1H NMR (CDCl3): 4.03 (5H, s, Cp), δ 4.19 (2H, d, J = 8.1 Hz, CH2), δ 4.34 (2H, s, Cp), δ 4.59 (2H, s, Cp), δ 7.11 (1H, d, J = 7.6 Hz, NC(CH2)CH), 7.26 (1H, t, J = 6.5 Hz, NCHCH), δ 7.58–7.64 (4H, m, Hmeta), 7.64–7.70 (2H, m, Hpara), δ 7.74 (1H, t, J = 7.1 Hz, NC(CH2)CHCH), δ 8.13 (4H, dd, J = 12.7, 7.1 Hz, Hortho), δ 9.17 (1H, d, J = 5.3 Hz, NCH). 13C{1H} NMR (CDCl3): δ 58.1 (s, CH2), δ 70.2 (s, Cp), δ 72.4 (d, J = 9.8 Hz, Cp), δ 73.7 (d, J = 11.3 Hz, Cp), δ 119.0 (s, NC(CH2)CH), δ 122.7 (s, NCHCH), δ 128.5 (d, J = 13.0 Hz, Cmeta), δ 133.0 (d, J = 10.1 Hz, Cortho), δ 138.1 (s, NC(CH2)CHCH), δ 150.1 (s, NCH). IR (cm−1): ν 1286, 929 (P=N); 347, 331 (Pd-Cl). Conductivity Λ (MeCN) = 17.1 μS/cm (neutral).
[(Ph3P=N-CH2-2-NC5H4)PdCl2] (11)
A solution of [PdCl2(COD)] (0.057 g, 0.2 mmol) in 7 mL of degassed dry CH2Cl2 was added over a solution of (Ph3P=N-CH2-2-NC5H4) 9 (0.074 g, 0.2 mmol) in degassed dry CH2Cl2. The reaction mixture was stirred for one hour at RT after which the solvent was reduced to ca 2 mL. Addition of Et2O (8 mL) afforded 11 as a yellow solid that was collected by filtration and dried under vacuum. Yield: 0.0675 g (61.8%). Anal. Calcd for C24H21N2PCl2Pd (545.74): C, 52.82; H, 3.88; N, 5.13. Found: C, 52.57; H, 3.94; N, 5.17. MS (ESI+) [m/z]: 510.2 [M-Cl]+. 31P{1H} NMR (CDCl3): δ 36.0 ppm. 1H NMR (CDCl3): δ 4.20 (2H, d, J = 5.9 Hz, CH2), δ 7.06 (1H, d, J = 7.7 Hz, NC(CH2)CH), δ 7.26 (1H, t, J = 6.4 Hz, NCHCH), δ 7.57 (6H, td, J = 7.5, 3.3 Hz, Hmeta), δ 7.64 (3H, td, J = 7.2, 1.3 Hz, Hpara), δ 7.71 (1H, td, J = 7.7, 1.4 Hz, NC(CH2)CHCH), δ 7.94 (6H, dd, J = 12.3, 7.2 Hz, Hortho), δ 9.22 (1H, d, J = 5.9 Hz, NCH). 13C{1H} NMR (CDCl3): δ 58.2 (s, CH2), δ 119.4 (s, NC(CH2)CH), δ 122.8 (s, NCHCH), δ 128.7 (d, J = 12.6 Hz, Cmeta), δ 132.9 (d, J = 2.8 Hz, Cpara), δ 133.9 (d, J = 9.8 Hz, Cortho), δ 138.0 (s, NC(CH2)CHCH), δ 150.7 (s, NCH), δ 165.0 (d, J = 17.7 Hz, NCNCH). IR (cm−1): ν 1287, 930 (P=N), 351, 331 (Pd-Cl). Conductivity Λ (MeCN) = 13.3 μS/cm (neutral).
X-Ray crystallography
Single crystals of 8 and 11 (see details in Table S1 in SI) were mounted on a glass fiber in a random orientation. Data collection was performed at RT on a Kappa CCD diffractometer using graphite monochromated Mo-Kα radiation (λ=0.71073 Å). Space group assignments were based on systematic absences, E statistics and successful refinement of the structures. The structures were solved by direct methods with the aid of successive difference Fourier maps and were refined using the SHELXTL 6.1 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were assigned to ideal positions and refined using a riding model. Details of the crystallographic data are given in Table S1 (SI). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. (CCDC 930539 (8) and 930540 (11)) or in the supporting information. Crystals of 8 (orange prisms with approximate dimensions 0.16 × 0.18 0.21 mm) were obtained from a solution of 8 in CH2Cl2 by slow diffusion of Et2O at RT. Crystals of 11 (deep yellow prisms with approximate dimensions 0.26 × 0.24 × 0.20 mm) were obtained from a solution of 11 in CHCl3 at 0 °C.
Computational Studies
All models were fully optimized with the Gaussian09 program package76 at the DFT level of theory. A hybrid density functional PBE077 was utilized together with the quasi-relativistic effective core potential basis set def2-TZVPPD78 for metal atoms (Au, Pd, Fe), and the standard all-electron basis sets 6–31G(d) for all other atoms. Solvent effects of the aqueous solvent were calculated with the conductor-like polarized continuum model (CPCM)79 for the gas phase optimized structures. Frequency analysis was done without scaling.
Cell culture and inhibition of cell growth
The human breast cancer cell line MCF7 and human ovarian cancer cell lines A2780 and A2780cisR (obtained from the European Centre of Cell Cultures ECACC, Salisbury, UK) were cultured respectively in DMEM (Dulbecco’s Modified Eagle Medium) and RPMI containing GlutaMaxI supplemented with 10% FBS and 1% penicillin/streptomycin (all from Invitrogen), at 37°C in a humidified atmosphere of 95% of air and 5% CO2 (Heraeus, Germany). Non-tumoral human embryonic kidney cells HEK293 were kindly provided by Dr. Maria Pia Rigobello (CNRS, Padova, Italy), and were cultivated in DMEM medium, added with GlutaMaxI (containing 10% FBS and 1% penicillin/streptomycin (all from Invitrogen) and incubated at 37°C and 5% CO2. For evaluation of growth inhibition, cells were seeded in 96-well plates (Costar, Integra Biosciences, Cambridge, MA) and grown for 24 h in complete medium. Solutions of the compounds were prepared by diluting a freshly prepared stock solution (in DMSO) of the corresponding compound in aqueous media (RPMI or DMEM for the A2780 and A2780cisR or MCF7 and HEK293, respectively). Afterwards, the intermediate dilutions of the compounds were added to the wells (100 μL) to obtain a final concentration ranging from 0 to 150 μM, and the cells were incubated for 72 h. DMSO at comparable concentrations did not show any effects on cell cytotoxicity. Following 72 h drug exposure, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the cells at a final concentration of 0.25 mg ml−1 incubated for 2 h, then the culture medium was removed and the violet formazan (artificial chromogenic precipitate of the reduction of tetrazolium salts by dehydrogenases and reductases) dissolved in DMSO. The optical density of each well (96-well plates) was quantified three times in tetraplicates at 540 nm using a multiwell plate reader, and the percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as a mean (± SE) of at least three independent experiments.
PARP-1 activity determinations
PARP-1 activity was determined using Trevigen’s HT Universal Colorimetric PARP Assay. This assay measures the incorporation of biotinylated poly(ADP-ribose) onto histone proteins in a 96 microtiter strip well format. Recombinant human PARP-1 (High Specific Activity, purified from E.coli containing recombinant plasmid harboring the human PARP gene, supplied with the assay kit) was used as the enzyme source. 3-Aminobenzamide (3-AB), provided in the kit, was used as control inhibitor. Two controls were always performed in parallel: a positive activity control for PARP-1 without inhibitors, that provided the 100% activity reference point, and a negative control, without PARP-1 to determine background absorbance. PARP-1 was incubated with different concentrations of compounds for 1 h at RT prior deposition on the plate wells. The final reaction mixture (50 μL) was treated with TACS-Sapphire™, a horseradish peroxidase colorimetric substrate, and incubated in the dark for 30 min. Absorbance was read at 630 nm after 30 min. The data correspond to means of at least three experiments performed in triplicate ± S.D.
Interaction of compounds 5–8, 10, 11 and cisplatin with plasmid (pBR322) DNA by Electrophoresis (Mobility Shift Assay)
10 μL aliquots of pBR322 plasmid DNA (20 μg/mL) in buffer (5 mM Tris/HCl, 50 mM NaClO4, pH = 7.39) were incubated with different concentrations of the compounds (5–8, 10, 11) (in the range 0.25 and 4.0 metal complex:DNAbp) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin-DNA were prepared as controls. After the incubation period, the samples were loaded onto the 1 % agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterwards, the gel was stained for 30 min with a solution of GelRed Nucleic Acid stain.
Interaction of compounds 5, 7 and 8 with HSA by Fluorescence Spectroscopy
A solution of each compound (8 mM) in DMSO was prepared and ten aliquots of 2.5 μL were added successively to a solution of HSA (10 μM) in phosphate buffer (pH = 7.4) to achieve final metal complex concentrations in the range 10–100 μM. The excitation wavelength was set to 295 nm, and the emission spectra of HSA samples were recorded at room temperature in the range of 300 to 450 nm. The fluorescence intensities of the metal compounds 5, 7 and 8, the buffer and the DMSO are negligible under these conditions. The fluorescence was measured 240 s after each addition of compound solution. The data were analyzed using the classical Stern-Volmer equation F0/F = 1 + KSV[Q].
Supplementary Material
Acknowledgments
Funding Sources
National Institute of General Medical Sciences (NIGMS), SC2GM082307 (M.C.) and the University of Groningen (Rosalind Franklin Fellowship to A.C.)
We thank the financial support of a grant from the National Institute of General Medical Sciences (NIGMS), SC2GM082307 (M.C.) and the University of Groningen (Rosalind Franklin Fellowship) for funding (A.C.). We thank undergraduate MARC student Claribel Nunez for the initial preparation of ligand 1 and LSAMP student Farrah Benoit for her assistance with some of the fluorescence spectroscopy measurements. EU COST Actions CM1105 and CM0902 are gratefully acknowledged for providing opportunities of discussion and for financial support. The computational work has been made possible by the use of the Finnish Grid Infrastructure resources. This paper is dedicated to Prof. Gerard van Koten on the occasion of his 70th birthday.
ABBREVIATIONS
- COD
cyclooctadiene
- DFT
density functional theory
- DMSO
dimethylsulfoxide
- DPPF
1,1′-Bis-diphenylphosphino-ferrocene
- EDTA
ethylenediaminetetracaetic acid
- HSA
human serum albumin
- HOMO
highest occupied molecular orbital
- IM
iminophosphorane
- ITC
isothermal titration calorimetry
- LUMO
lowest unoccupied molecular orbital
- MPPF
diphenylphosphino-ferrocene
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PARP-1
Poly(ADP-ribose) Polymerase-1
- PBS
phosphate buffer saline
- TAE
Tris-acetate/EDTA buffer
- T-Jurkat
human acute lymphoblastic leukemia cells
- T-Jurkat sh Bak
human acute lymphoblastic leukemia cells which do not express the Bak gene
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Supporting information available: Table with the crystal data and structure refinement for complexes 8 and 11. CIF file for the X-ray crystal structures of compounds 8 and 11. Details on DFT calculations. 1H and 13C{1H} NMR spectra of compounds 1, 3, 4 and 5. Stability of compounds 4–8, 10 and 11 in d6-DMSO solution overtime assessed by 31P{1H} NMR spectroscopy and selected 31P{1H} NMR spectra of the decomposition of compounds 5, 6 and 10 overtime. UV-vis spectra of compounds 5 and 10 in PBS overtime. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Thayer AM. Platinum drugs take their roll. Chem Eng News. 2010;88(26):24–28. [Google Scholar]
- 2.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 3.Alessio E. Bioinorganic Medicinal Chemistry. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 4.Noffke AL, Habtemarian A, Pizarro AM, Sadler PJ. Designing organometallic compounds for catalysis and therapy. Chem Commun. 2012;48:5219–5246. doi: 10.1039/c2cc30678f. [DOI] [PubMed] [Google Scholar]
- 5.Aris SM, Farrell NP. Towards Antitumor Active trans-Platinum Compounds. Eur J Inorg Chem. 2009:1293–1302. doi: 10.1002/ejic.200801118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berners-Price SJ, Filipovska A. Gold compounds as therapeutic agents for human diseases. Metallomics. 2011;(41):279–304. doi: 10.1039/c1mt00062d. [DOI] [PubMed] [Google Scholar]
- 7.Komeda S, Casini A. Next-generation anticancer metallodrugs. Curr Top Med Chem. 2012;12:219–223. doi: 10.2174/156802612799078964. [DOI] [PubMed] [Google Scholar]
- 8.Bergamo A, Gaiddon C, Schellens JHM, Beijnen JH, Sava G. Approaching tumour therapy beyond platinum drugs: Status of the art and perspectives of ruthenium drug candidates. J Inorg Biochem. 2012;106:90–99. doi: 10.1016/j.jinorgbio.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 9.Casini A. Exploring the mechanisms of metal-based pharmacological agents via an integrated approach. J Inorg Biochem. 2012;109:97–106. doi: 10.1016/j.jinorgbio.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Gold compounds as anticancer agents: chemistry, celular pharmacology, and preclinical studies. Med Res Rev. 2010;30:550–580. doi: 10.1002/med.20168. [DOI] [PubMed] [Google Scholar]
- 11.Casini A, Hartinger C, Gabbiani C, Mini E, Dyson PJ, Keppler BK, Messori L. Gold(III) compounds as anticancer agents: relevance of gold-protein interactions for their mechanism of action. J Inorg Biochem. 2008;102:564–575. doi: 10.1016/j.jinorgbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Bergamo A, Sava G. Ruthenium anticancer compounds: myths and realities of the emerging metal-based drugs. Dalton Trans. 2011;40:7817–7823. doi: 10.1039/c0dt01816c. [DOI] [PubMed] [Google Scholar]
- 13.Pizarro AM, Habtemarian A, Sadler P. Activation mechanisms for organometallic anticancer complexes. Topics Organomet Chem. 2010;32:21–56. [Google Scholar]
- 14.Casini A, Reedijk J. Interactions of anticancer Pt compounds with proteins: an overlooked topic in medicinal inorganic chemistry? Chem Sci. 2012;3:3135. [Google Scholar]
- 15.Schuh E, Pfluger C, Citta A, Folda A, Rigobello MP, Bindoli A, Casini A, Mohr F. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J Med Chem. 2012;55:5518–5528. doi: 10.1021/jm300428v. and refs. therein. [DOI] [PubMed] [Google Scholar]
- 16.Berners-Price SJ, Filipovska A. Gold compounds as therapeutic agents for human diseases. Metallomics. 2011;3:863–873. doi: 10.1039/c1mt00062d. [DOI] [PubMed] [Google Scholar]
- 17.Dalla Via L, Nardon C, Fregona D. Targeting the ubiquitin-proteasome pathway with inorganic compounds to fight cancer: a challenge for the future. Future Med Chem. 2012;4:525–543. doi: 10.4155/fmc.11.187. [DOI] [PubMed] [Google Scholar]
- 18.Serratrice M, Edafe F, Mendes F, Scopelliti R, Zakeeruddin SM, Gratzel M, Santos I, Cinellu MA, Casini A. Cytotoxic gold compounds: synthesis, biological characterization and investigation on their inhibition properties of the zinc finger protein PARP-1. Dalton Trans. 2012;41:3287–3293. doi: 10.1039/c2dt11913g. [DOI] [PubMed] [Google Scholar]
- 19.Mendes F, Groessl M, Nazarov AA, Tsybin YO, Sava G, Santos I, Dyson PJ, Casini A. Metal-based inhibition of poly(ADP-ribose)polymerase-the guardian angel of DNA. J Med Chem. 2011;54:2196–2206. doi: 10.1021/jm2000135. [DOI] [PubMed] [Google Scholar]
- 20.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 21.Jeggo PA. DNA-repair: PARP-another guardian angel? Curr Biol. 1997;8:R49–R51. doi: 10.1016/s0960-9822(98)70032-6. [DOI] [PubMed] [Google Scholar]
- 22.Donzello MP, Viola E, Ercolani C, Fu Z, Futur D, Kadish KM. Tetra-2,3-pyrazinoporphyrazines with Externally Appended Pyridine Rings. 12. New Heteropentanuclear Complexes Carrying Four Exocyclic Cis-platin-like Functionalities as Potential Bimodal (PDT/Cis-platin) Anticancer Agents. Inorg Chem. 2012;51:12548–12559. doi: 10.1021/ic301989a. and refs. therein. [DOI] [PubMed] [Google Scholar]
- 23.González-Pantoja JF, Stern M, Jarzecki AA, Royo E, Robles-Escajeda E, Varela-Ramirez A, Aguilera RJ, Contel M. Titanocene-Phosphine Derivatives as Precursors to Cytotoxic Heterometallic TiAu2 and TiM (M = Pd, Pt) Compounds. Studies of their Interactions with DNA. Inorg Chem. 2011;50:11099–11110. doi: 10.1021/ic201647h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenzel M, Bertrand B, Eymin MJ, Comte V, Harvey JA, Richard P, Groessl M, Zava O, Amrouche H, Harvey PD, Le Gendre P, Picquet M, Casini A. Multinuclear Cytotoxic Metallodrugs: Physicochemical Characterization and Biological Properties of Novel Heteronuclear Gold-Titanium Complexes. Inorg Chem. 2011;50:9472–9480. doi: 10.1021/ic201155y. [DOI] [PubMed] [Google Scholar]
- 25.Pelletier F, Comte V, Massard A, Wenzel M, Toulot S, Richard P, Picquet M, Le Gendre P, Zava O, Edafe F, Casini A, Dyson PJ. Development of bimetallic titanocene-ruthenium- arene complexes as anticancer agents: relationships between structural and biological properties. J Med Chem. 2010;53:6923–6933. doi: 10.1021/jm1004804. [DOI] [PubMed] [Google Scholar]
- 26.CG, Phillips AD, Nazarov AA. Polynuclear Ruthenium Osmium and Gold Complexes. The Quest for Innovative Anticancer Chemotherapeutics. Curr Top Med Chem. 2011;11:2688–2702. doi: 10.2174/156802611798040769. [DOI] [PubMed] [Google Scholar]
- 27.Hillard EA, Vessieres A, Jaouen G. Ferrocene functionalized endocrine modulators as anticancer agents. Top Organomet Chem. 2010;32:81–117. [Google Scholar]
- 28.Snegur LV, Babin VN, Simenel AA, Nekrasov YS, Ostrovskaya LA, Sergeeva NS. Antitumor activities of ferrocene compounds. Russ Chem Bull, Int Ed. 2010;59:2167–2178. [Google Scholar]
- 29.Recent relevant example: Plazuk D, Wieczorek A, Blauz A, Rychlik B. Synthesis and biological activities of ferrocenyl derivatives of paclitaxel. Med Chem Commun. 2012;3:498–501.
- 30.Bjelosevic H, Guzei IA, Spencer LC, Persson T, Kriel FH, Hewer R, Nell MJ, Gut J, van Rensburg CEJ, Rosenthal PJ, Coates J, Darkwa J, Elmroth SKC. Platinum(II) and gold(I) complexes based on 1,1-bis(diphenylphosphino)metallocene derivatives: synthesis, characterization and biological activity of the gold complexes. J Organomet Chem. 2012;720:52–59. [Google Scholar]
- 31.Gimeno MC, Goitia H, Laguna A, Luque ME, Villacampa MD, Sepulveda C, Meireles M. Conjugates of ferrocene with biological compounds. Coordination to gold complexes and antitumoral properties. J Inorg Biochem. 2011;105:1373–1382. doi: 10.1016/j.jinorgbio.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Fourie E, Erasmus E, Swarts JC, Jakob A, Lang H, Joone GK, van Rensburg CEJ. Cytotoxicity of ferrocenyl-ethynyl phosphine metal complexes of gold and platinum. Anticancer Res. 2011;31:825–830. [PubMed] [Google Scholar]
- 33.Horvath UEI, Bentivoglio G, Hummel M, Schottenbergm H, Wurst K, Nell MJ, van Rensburg CEJ, Cronje S, Raubenhemier HC. A cytotoxic bis(carbene)gold(I) complex of ferrocenyl complexes: synthesis and structural characterization. New J Chem. 2008;32:533–539. [Google Scholar]
- 34.Viotte M, Gautheron B, Ninfant’ev I, Kuz’mina LG. New potentially cytotoxic thiolatogold(I) complexes of 1,1′-bis(diphenylphopshino)ferrocene. Inorg Chim Acta. 1996;253:71–76. [Google Scholar]
- 35.Viotte M, Gautheron B, Kubicki MM, Ninfant’ev I, Fricker SP. Synthesis, structure of nitrogen-containig phosphinogold(I) ferrocenes. In vitro activity against bladder and colon carcinoma cell lines. Metal-based Drugs. 1995;2:311–326. doi: 10.1155/MBD.1995.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viotte M, Gautheron B, Kubicki MM, Mugnier Y, Parish RV. New iron(II)- and gold(I)-containing metallocenes. X-ray structure of a three-coordinate gold(I) ferrocenophane-type representative. Inorg Chem. 1995;34:3465–3473. [Google Scholar]
- 37.Nieto D, Gonzalez-Vadillo AM, Bruna S, Pastor CJ, Rios-Luci C, Leon LC, Padron JM, Navarro-Ranninger C, Cuadrado I. Heterodimetallic platinum(II) compounds with β-aminoethylferrocenes: synthesis, electrochemical behavior and anticancer activity. Dalton Trans. 2012;41:432–441. doi: 10.1039/c1dt11358e. [DOI] [PubMed] [Google Scholar]
- 38.Chellan P, Land KM, Shokar A, Au A, An SH, Clavel CM, Dyson PJ, de Kock C, Smith PJ, Chibale K, Smith GS. Exploring the versatility of cycloplatinated thiosemicarbazones as antitumor and antiparasitic agents. Organometallics. 2012;31:5791–5799. [Google Scholar]
- 39.Samouei H, Rashidi M, Heinemann FW. A cyclometalated diplatinum complex containing 1,1′-bis(diphenylphoshino)ferrocene as spacer ligand: antitumor study. J Organomet Chem. 2011;696:3764–3771. [Google Scholar]; Schulz J, Renfrew AK, Cisarova I, Dyson PJ, Stepnicka P. Synthesis and anticancer activity of chalcogenide derivatives and platinum(II) and palladium(II) complexes derived from a polar ferrocene-phosphanyl-carboxamide. Appl Organomet Chem. 2010;24:392–397. [Google Scholar]
- 40.Rajput J, Moss JR, Hutton AT, Hendricks DT, Arendse CE, Imrie C. Synthesis, characterization and cytotoxicity of some palladium(II), platinum(II), rhodium(I) and iridium(I) complexes of ferrocenylpyridine and relataed ligands. Crystal and molecular structure of trans-dichlorobis(3-ferrocenylpyridine)palladium(II) J Organomet Chem. 2004;689:1553–1568. [Google Scholar]
- 41.Henderson W, Alley SR. Platinum(II) complexes containing ferrocene-derived phosphonate ligands; synthesis, structural characterization and antitumor activity. Inorg Chim Acta. 2001;322:106–112. [Google Scholar]
- 42.Mason RW, McGrouter K, Ranatunge-Bandarage PRR, Robinson BH, Simpson J. Toxicology and antitumour activity of ferrocenylamines and platinum derivatives. Appl Organomet Chem. 2001;13:163–173. [Google Scholar]
- 43.Bincoletto C, Tersariol ILS, Oliveira CR, Dreher S, Fausto DM, Soufen AS, Nascimento FD, Caires ACF. Chiral cyclopalladated complexes derived from N,N-dimethyl-1-phenethylamine with bridging bis(diphenylphosphine)ferrocene ligand as inhibitors of the cathepsin B activity and as antitumor agents. Bioorg Med Lett. 2005;13:3047–3055. doi: 10.1016/j.bmc.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 44.Barbosa CMV, Oliveira CR, Nascimiento FD, Smith MCM, Fausto DM, Soufen MA, Sena E, Araujo RC, Tersariol ILS, Bincoletto C, Caires ACF. Biphosphinic palladacycle complex mediates lysosomal-membrane permeabilization and cell death in K562 leukaemia cells. Eur J Pharmacol. 2006;542:37–47. doi: 10.1016/j.ejphar.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Oliveira CR, Barbosa CMV, Nascimento FD, Lanetzki CS, Meneghin MB, Pereira FEV, Paredes-Gamero EJ, Ferreira AT, Rodrigues T, Queiroz MLS, Caires ACF, Tersariol ILS, Bincoletto C. Pre-clinical antitumour evaluation of Biphosphinic Palladacycle Complex in human leukaemia cells. Chem Biol Interact. 2009;177:181–189. doi: 10.1016/j.cbi.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 46.Tauchman J, Suss-Fink G, Stepnicka P, Zava O, Dyson PJ. Arene ruthenium complexes with phosphinoferrocene amino acid conjugates: synthesis, characterization and cytotoxicity. J Organomet Chem. 2013;723:233–238. [Google Scholar]
- 47.Ott I, Kowalski K, Gust K, Maurer J, Mucke P, Winter RF. Comparative biological evaluation of two ethylene linked mixed binuclear ferrocene/ruthenium organometallic species. Bioorg Med Chem Lett. 2010;20:866–869. doi: 10.1016/j.bmcl.2009.12.080. [DOI] [PubMed] [Google Scholar]
- 48.Auzias M, Gueniat J, Therrien B, Suss-Fink G, Renfrew AK, Dyson PJ. Arene-ruthenium complexes with ferrocene-derived ligands: synthesis and characterization of complexes of the type [Ru(η6-arene)(NC5H4CH2NHOCOC5H4FeC5H5)Cl2] and [Ru(η6-arene)(NC3H3N(CH2)2O2C-C5H4FeC5H5)Cl2] J Organomet Chem. 2009;694:855–861. [Google Scholar]
- 49.Wu CH, Wu DH, Liu X, Guoyiqibayi G, Guo DD, Lv G, Wang XM, Yan H, Jiang H, Lu ZH. Ligand-based neutral ruthenium(II) arene complex: selective anticancer action. Inorg Chem. 2009;48:2352–2354. doi: 10.1021/ic900009j. [DOI] [PubMed] [Google Scholar]
- 50.Von Poelhsitz G, Bogado AL, Peres de Araujo M, Selistre-de-Araujo HS, Ellena J, Castellano EE, Batista AA. Synthesis, characterization, X-ray structure and preliminary in vitro antitumor activity of the nitrosyl complex fac-[RuCl3(NO)(dppf)], dppf = 1,1′-bis(diphenylphopshine)ferrocene. Polyhedron. 2007;26:4707–4712. [Google Scholar]
- 51.Conradie J, Swarts JC. Relationship between electrochemical potentials and substitution reaction rates of ferrocene-containing B-diketonato rhodium(I) complexes; cytotoxicity of [Rh(FcCOCHCOPh)(cod)] Dalton Trans. 2011;40:5844–5851. doi: 10.1039/c1dt00013f. [DOI] [PubMed] [Google Scholar]
- 52.Weber B, Serafin A, Michie J, van Rensburg C, Swarts JC, Bohm L. Cytotoxicity and cell death pathways invoked by two new rhodium-ferrocene complexes in bening and malignant prostatic cell lines. Anticancer Res. 2004;24:763–770. [PubMed] [Google Scholar]
- 53.Sathyadevi P, Krishnamoorthy P, Burotac RR, Cowley AH, Dharmaraj N. Synthesis of novel heterometallic copper(I) hydrazone Schiff base complexes: a comparative study on the effect of heterocyclic hydrazides towards interaction with DNA/protein, free radical scavenging and cytotoxicity. Metallomics. 2012;4:498–511. doi: 10.1039/c2mt00004k. [DOI] [PubMed] [Google Scholar]
- 54.Maity B, Roy M, Banik B, Majumdar R, Dighe RR, Chakravarty AR. Ferrocene-promoted photoactivated DNA cleavage and anticancer activity of terpyridyl copper(II) phenantroline complexes. Organometallics. 2010;29:3632–3641. [Google Scholar]
- 55.Shaik N, Martínez A, Augustin I, Giovinazzo H, Varela A, Aguilera R, Sanaú M, Contel M. Synthesis of Apoptosis-Inducing Iminophosphorane Organogold(III) Complexes Study of Their Interactions with Biomolecular Targets. Inorg Chem. 2009;48:1577–1587. doi: 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vela L, Contel M, Palomera L, Azaceta G, Marzo I. Iminophosphorane-organogold(III) complexes induce cell death through mitochondrial ROS production. J Inorg Biochem. 2011;105:1306–1313. doi: 10.1016/j.jinorgbio.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carreira M, Calvo-Sanjuán R, Sanaú M, Zhao X, Magliozzo RS, Marzo I, Contel M. Cytotoxic Hydrophilic iminophosphorane coordination compounds of d8 metals. Studies of their Interactions with DNA and HAS. J Inorg Biochem. 2012;116:204–214. doi: 10.1016/j.jinorgbio.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carreira M, Calvo-Sanjuán R, Sanaú M, Marzo I, Contel M. Organometallic Palladium Complexes with a Water-Soluble Iminophosphorane Ligand as Potential Anticancer Agents. Organometallics. 2012;31:5772–5781. doi: 10.1021/om3006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staudinger H, Meyer JJ. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv Chim Acta. 1919;2:635–646. [Google Scholar]
- 60.Sollott GP, Mertwoy HE, Portnoy S, Snead JL. Unsymmetrical tertiary phosphines of ferrocene by Friedel-Crafts reactions. I. Ferrocenylphenylphosphines. J Org Chem. 1963;28:1090–1092. [Google Scholar]
- 61.Aguilar D, Contel M, Navarro R, Soler T, Urriolabeitia EP. Gold(III) iminophosphorane complexes as catalysts in C-C and C-O bond formations. J Organomet Chem. 2009;694:486–493. [Google Scholar]
- 62.Vicente J, Abad JA, Clemente R, Lopez-Serrano J, Ramirez de Arellano MC, Jones PG, Butista D. Mercurated and Palladated Iminophosphoranes. Synthesis and Reactivity. Organometallics. 2003;22:4248–4259. [Google Scholar]
- 63.Falvello LR, Gracia MM, Lazaro I, Navarro R, Urriolabeitia EP. Different coordinating behaviour of the imino-phosphoranes Ph3P=NC(O)CH2Cl and Ph3P=NC(O)-2-NC5H4 towards MII complexes (M=Pd, Pt) New J Chem. 1999;35:227–235. [Google Scholar]
- 64.Avis MW, Elsevier CJ, Veldman N, Kooijman H, Spek AL. Monodentate σ-N and Bidentate σ-N,σ-N′ Coordination of 1,1-Bis((N-p-tolylimino)diphenylphosphoranyl)ethane, CHCH3(PPh2=NC6H4-4-CH3)2, to Platinum(II) Inorg Chem. 1996;35:1518–1528. doi: 10.1021/ic950748w. [DOI] [PubMed] [Google Scholar]
- 65.Baker J, Jarzecki AA, Pulay P. Direct Scaling of Primitive Valence Force Constants: An Alternative Approach to Scaled Quantum Mechanical Force Fields. J Phys Chem A. 1998:1412–1424. [Google Scholar]
- 66.Dabrowiak JC. Metals in medicine. Ch 4. John Wiley and Sons, Ltd; Chichester, UK: 2009. pp. 109–14. [Google Scholar]
- 67.Liu HK, Sadler P. Metal complexes as DNA intercalators. Acc Chem Res. 2011;44:349–359. doi: 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
- 68.For example: Ruíz J, Cutillas N, Vicente C, Villa MD, López G, Lorenzo J, Avilés FX, Moreno V, Bautista D. New palladium(II) and platinum(II) complexes with the model nucleobase 1-methylcytosine: antitumor activity and interactions with DNA. Inorg Chem. 2005;44:7365–7376. doi: 10.1021/ic0502372.
- 69.Gao E, Zhu M, Liu L, Huang Y, Wang L, Shi S, Chuyue ZW, Sun Y. Impact of the Carbon Chain Length of Novel Palladium(II) Complexes on Interaction with DNA Cytotoxic Activity. Inorg Chem. 2010;49:3261–3270. doi: 10.1021/ic902176e. [DOI] [PubMed] [Google Scholar]
- 70.Quiroga A, Pérez JM, López-Solera I, Masaguer JR, Luque A, Román P, Edwards A, Alonso C, Navarro-Ranninguer C. Novel tetranuclear orthometalated complexes of Pd(II) and Pt(II) derived from p-isopropylbenzaldehyde thiosemicarbazone with cytotoxic activity in cis-DDP resistant tumor cell lines. Interaction of these complexes with DNA. J Med Chem. 1998;41:1399–1408. doi: 10.1021/jm970520d. [DOI] [PubMed] [Google Scholar]
- 71.Gao EJ, Wang KH, Zhu MC, Liu L. Hairpin-shaped tetranuclear palladium(II) complex: synthesis, crystal structure, DNA binding and cytotoxicity activity studies. Eur J Med Chem. 2010;45:2784–2790. doi: 10.1016/j.ejmech.2010.02.061. [DOI] [PubMed] [Google Scholar]
- 72.Fox K. Protocols Methods in Mol Biol. Humana Press Inc; Totowa, NJ: 1997. Drug-DNA Interact. [Google Scholar]
- 73.Gumus F, Eren G, Acik L, Celebi A, Ozturk F, Yilmaz S, Sagkan RI, Gur S, Ozkul A, Elmali A, Elerman Y. Synthesis cytotoxicity and DNA interactions of new cisplatin analogues containing substituted benzimidazole ligands. J Med Chem. 2009;52:1345–1357. doi: 10.1021/jm8000983. [DOI] [PubMed] [Google Scholar]
- 74.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem Rev. 2006;106:2224–2248. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 75.Lacowicz JR. Principles of Fluorescence Spectroscopy. Ch 8. Kluver Academic/Plenum Publishers; New York: 1999. pp. 238–264. [Google Scholar]
- 76.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. M1. Gaussian 09, Revision C.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 77.Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 78.Andrae D, Häußermann U, Dolg M, Stoll H, Preuß P. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor Chim Acta. 1990;77:123–141. [Google Scholar]
- 79.Cossi M, Rega N, Scalmani G, Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.