

Abstract
G protein-dependent signaling pathways control the activity of excitable cells of the nervous system and heart, and are the targets of neurotransmitters, clinically-relevant drugs, and drugs of abuse. G protein-gated inwardly-rectifying potassium (K+) (Girk/Kir3) channels are a key effector in inhibitory signaling pathways. Girk-dependent signaling contributes to nociception and analgesia, reward-related behavior, mood, cognition, and the heart rate regulation, and has been linked to epilepsy, Down syndrome, addiction, and arrhythmias. Here, we discuss recent advances in our understanding of Girk channel structure, organization in signaling complexes, and plasticity, as well as progress on the development of subunit-selective Girk modulators. These findings offer new hope for the selective manipulation of Girk channels to treat a variety of debilitating afflictions.
Introduction to Girk signaling
Signal transduction involving inhibitory (Gi/o) G proteins titrates the excitability of neurons, cardiac myocytes, and endocrine cells, actions crucial for regulating mood and cognition, nociception and antinociception, reward, energy homeostasis, motor activity and coordination, hormone secretion, and cardiac output. Not surprisingly, dysregulation of Gi/o-dependent signaling has been linked to a number of neurological and cardiac disorders. Given this, and since the efficacy of many clinically-relevant and abused drugs relates to their actions on Gi/o-dependent signaling, it is important that we understand with cellular, subcellular, and molecular detail how such signaling is organized, how it is regulated, and how and when it goes awry.
G protein-gated inwardly-rectifying potassium (K+) (Girk/Kir3) channels are a common effector for Gi/o-dependent signaling pathways in the heart and nervous system [1, 2]. Studies of mutant mice and a more limited set of linkage analyses have suggested that dysregulation of Girk signaling may contribute to certain human diseases and disorders (Table 1). While this work suggests that therapeutic approaches targeting Girk channels may prove beneficial in some settings, there is legitimate concern that manipulation of Girk signaling would trigger profound and widespread off-target effects. The goal of this review is to highlight recent developments related to our understanding of Girk channel diversity, compartmentalization, and plasticity. These studies suggest new opportunities for selective manipulation of Girk signaling, efforts that could eventually lead to novel treatments for debilitating human afflictions.
Table 1.
Physiological and pathophysiological relevance of Girk signaling
| Girk gene(s) | Mutation | Phenotype | References | |
|---|---|---|---|---|
| Cardiovascular physiology | ||||
| heart rate | Girk1, Girk4 | null | resting tachycardia | [86, 87] |
| parasympathetic regulation | Girk1, Girk4 | null | decreased chronotropic response | [86, 87] |
| heart rate variability | Girk4 | null | decreased variability | [86] |
| arrhythmia | GIRK1, GIRK4 | multiple | Long QT, atrial fibrillation | [88–90] |
| hypertension | GIRK4 | multiple | associated with aldosteronism | [91, 92] |
| Nociception | ||||
| thermal | Girk1, Girk2 | null | hyperalgesia | [93, 94] |
| mechanical | Girk2 | null | hyperalgesia | [93] |
| chemical | Girk2 | null | hyperalgesia | [93] |
| Antinociception | ||||
| opioid | Girk1, Girk2 | null | decreased analgesia | [93–97] |
| Girk3 | null | decreased sensitivity | [98] | |
| GIRK2 | polymorphism | increased dosing requirement | [99, 100] | |
| α2 adrenergic | Girk2,Girk3 | null | decreased analgesia | [93, 95, 98] |
| GABAB | Girk2 | null | decreased analgesia | [95] |
| cholinergic | Girk2 | null | decreased analgesia | [95] |
| cannabinoid | Girk2, Girk3 | null | decreased sensitivity | [95, 98] |
| Reward | ||||
| motor activity | Girk1, Girk2 | null | enhanced basal and cocaine-induced | [101–103] |
| natural rewards | Girk2, Girk4 | null | elevated responding for food | [104, 105] |
| Girk2 | triploid | enhanced sucrose intake | [58] | |
| self-administration | Girk2, Girk3 | null | decreased (cocaine) | [102] |
| Girk2 | null | enhanced consumption (ethanol) | [106] | |
| dependence/withdrawal | Girk2 & Girk3 | null | decreased opioid withdrawal | [97] |
| Girk3 | null | decreased sedative-hypnotic withdrawal | [107] | |
| GIRK2 | polymorphism | association with ethanol intake and stress | [108] | |
| Learning/memory | ||||
| spatial learning/memory | Girk4 | null | decreased recall | [109] |
| fear conditioning | Girk2 | triploid | decreased contextual recall | [58] |
| Anxiety | Girk2 | null | anxiolysis | [104, 110] |
| Schizophrenia | GIRK1 | polymorphism | genetic association | [111] |
| Seizure/epilepsy | Girk2 | null | increased spontaneous and PTZ-induced | [74] |
| Energy homeostasis | Girk4 | null | late-onset obesity | [105] |
| Thermoregulation | Girk2 | null | decreased drug-induced hypothermia | [112] |
| Neurodevelopment | Girk2 | weaver | loss of granule and dopamine neurons | [113] |
The table lists outcomes from behavioral studies involving mice harboring mutant Girk subunits (null/knockout, triploid, or weaver) or from human linkage studies (GIRK gene in all capital letters) that identified polymorphisms or mutations in GIRK genes.
Girk channel structure
Girk channels are tetramers formed by differential multimerization among the products of four genes: Girk1/Kir3.1/Kcnj3, Girk2/Kir3.2/Kcnj6, Girk3/Kir3.3/Kcnj9, and Girk4/Kir3.4/Kcnj5 [1, 2] (Figure 1A). Each Girk subunit possesses intracellular N- and C-terminal domains, and two transmembrane segments that flank a hydrophobic pore domain. Random assembly theoretically allows for the formation of many distinct Girk channel subtypes, and alternative splicing of the Girk1 and Girk2 genes potentially adds an additional spice of diversity (e.g., [3, 4]). There are, however, two observations that suggest that a more limited number of Girk channel subtypes exist in vivo: 1) Girk1 does not form functional homomultimeric channels [5, 6], an observation attributable at least in part to the absence of an endoplasmic reticulum (ER) export signal that is found in Girk2 and Girk4 [7]. Surprisingly, however, introduction of a single point mutation into the pore domain of Girk1 (F137S) is sufficient to yield functional homomeric channels [8]. Collectively, these findings suggest that while ER export signals are important, other factors such as protein folding and stability may also influence the trafficking of Girk channels. 2) The four Girk subunits exhibit overlapping but distinct expression patterns, which is particularly evident in the nervous system [9, 10]. Indeed, work in the last two decades has clarified the cell type-specific expression patterns of Girk subunits (Box 1). Moreover, recent crystallography studies have begun providing insight into the three-dimensional structure of Girk channels, with resolution of the membrane-spanning and large portions of the intracellular domains of homomeric channels [11–16] (Figure 1B).
Figure 1. Girk channel structure.
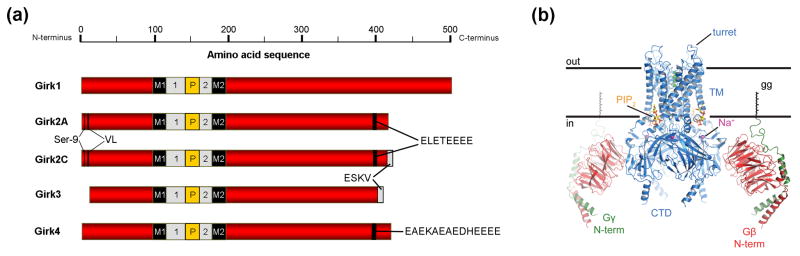
(a) Linear depiction of Girk channel subunits, including two prevalent Girk2 splice variants expressed in the mouse nervous system. The four Girk subunits exhibit a high degree of homology in the membrane-spanning (M1, M2), extracellular (1, 2), and pore (P) domains, with most inter-subunit differences observed in the distal N- and C-terminal domains. The Girk2 splice variants and Girk4 contain ER export signals (ELETEEEE and EAEKAEAEDHEEEE, respectively) not found in the other subunits [7]. Girk2 also exhibits an N-terminal internalization motif (VL), whose influence on channel trafficking is precluded by phosphorylation of Girk2(Ser9) [71]. Girk2C and Girk3 possess identical C-terminal PDZ interaction motifs (-ESKV). (b) Structure of the Girk-Gβγ complex from a side-view, with colors highlighting Girk2 (blue), Gβ (red), Gγ (green, with associated geranyl-geranyl (gg) lipid modification), PIP2 (yellow/orange sticks), Na+ ions (purple spheres), K+ ions (green spheres within the pore). Note that there are 4 Gβγ binding sites per channel, and the Gβγ facing the viewer has been removed for clarity [16].
Box 1. Cellular and sub-cellular diversity of Girk channels.
While Girk channel expression has been reported in neuroendocrine cells and more recently in some cancer cells (e.g., [114–117]), their expression and relevance is far better understood in the heart and nervous system. Girk1/4 heteromers comprise the muscarinic-gated atrial K+ channel IKACh, a critical mediator of the parasympathetic regulation of heart rate [5, 86]. While the prototypical neuronal Girk channel is thought to be the Girk1/2 heteromer, multiple Girk channel subtypes exist within the rodent nervous system. Indeed, Girk3 is expressed throughout the central nervous system [9], and while Girk4 expression is not prominent in the brain, it is found in a few regions including the hypothalamus and cerebellum [105, 118]. Girk1, Girk2, and Girk3 are co-expressed in many neuron populations, including hippocampal pyramidal neurons [9, 10]. In contrast, dopamine neurons of the VTA and substantia nigra pars compacta (SNc) display Girk2/3 and Girk2a/c heteromers, respectively [27, 119]. The cerebellum exemplifies the molecular diversity that can be achieved via differential subunit expression; seven distinct Girk expression patterns were discerned within the various neuronal subtypes in this brain region [118].
Girk channels are distributed mainly in the somato-dendritic compartment of neurons (e.g., [10, 96, 118, 120]). This distribution is consistent with most studies showing that Girk channels selectively mediate the postsynaptic inhibitory effects of neurotransmitters and related drugs, while making little or no contribution to their presynaptic inhibitory effects [121]. Some ultrastructural and functional data, however, support the contention that Girk channels contribute to presynaptic inhibition in some neurons [122–124]. Girk channels of distinct subunit composition can also exist within specific subcellular compartments of the same neuron. For example, Girk1 and Girk2 show extensive co-localization across the different dendritic layers of the hippocampus [120]. Their expression levels vary among dendritic regions innervated by distinct synaptic inputs in CA1 pyramidal cells, showing a significant increase from proximal to distal dendrites. In contrast, Girk3 is more uniformly distributed along the cell surface of pyramidal cells, including the presynaptic terminal [10]. In addition, Girk2 and Girk3 are evenly distributed along the postsynaptic density (PSD) of pyramidal cells and Purkinje cells, whereas Girk1 is absent from excitatory synapses and only observed at perisynaptic sites [10, 96, 120, 123]. Finally, in the sub-population of parvalbumin-expressing interneurons consisting mainly of basket and chandelier cells, Girk1, Girk2 and Girk3 share the same localization at very similar densities [125]. Collectively, these data argue that distinct Girk channel subtypes exist in a tissue/cell-type and subcellular compartment-dependent manner, a scenario that enhances the prospects for selective and efficacious therapeutic manipulation of Girk signaling.
Functional implications of Girk channel diversity
In expression systems, Girk channels of various composition – including heteromers (Girk1/2, Girk1/3, Girk1/4, Girk2/3, Girk2/4) and homomers (Girk2 and Girk4) – display K+ selectivity, inward rectification, and G protein-dependent gating [1, 2]. While it cannot form a functional homomer, Girk1 is an integral subunit of most neuronal Girk channels and the cardiac Girk channel IKACh [5, 17]. Girk1 confers robust receptor-dependent activity to Girk heteromers, attributable in part to unique residues found in the pore and second transmembrane helix that enhance single-channel conductance and open probability [8, 18–20]. The intracellular C-terminal domain also contributes to the potentiating influence of Girk1 on GPCR-dependent heteromeric channel activity, likely due to the presence of unique Gβγ, Gα, and PIP2 interaction domains and phosphorylation sites [20–26].
The functional relevance of Girk channel subunit composition is nicely illustrated in the ventral tegmental area (VTA). VTA dopamine neurons express Girk2 and Girk3, while VTA GABA neurons express Girk1, Girk2, and Girk3 [27, 28]. VTA dopamine neurons are significantly less sensitive than VTA GABA neurons to direct GABAB receptor-dependent inhibition [27, 28], consistent with the relatively low sensitivity of recombinant Girk2/3 heteromers to Gβγ-dependent activation [29]. Interestingly, ectopic expression of Girk1 or genetic ablation of Girk3 enhanced the sensitivity of the Girk channel in VTA dopamine neurons to GABAB receptor-dependent inhibition [28]. The negative influence of Girk3 on the sensitivity of Girk2/3 heteromers to Gβγ- and GABAB receptor-dependent activation may be linked to intrinsic structural elements that weaken its interaction with Gβγ or the coupling between Gβγ binding and channel gating, an explanation supported by the behavior of recombinant Girk2/3 heteromers [29]. Alternatively, Girk3-specific interactions with negative regulatory proteins expressed in VTA dopamine neurons (Rgs2 and/or sorting nexin 27 [28, 30], discussed below), interactions that are presumably precluded or mitigated by the presence of Girk1, may explain the differential sensitivity of Girk channels in VTA dopamine and GABA neurons to GABAB receptor activation. Regardless of the mechanism, the molecular and cellular diversity of Girk channels shapes the sensitivity of VTA dopamine output to GABAB receptor activation, and helps explain the intriguing pharmacodynamics differences between the GABAB receptor agonists baclofen (an anti-craving compound) and gamma hydroxybutyric acid (GHB, a drug of abuse) [28].
Macromolecular organization of Girk signaling
Girk channels are thought to exist in multi-protein complexes that include G protein-coupled receptors (GPCRs), G proteins, and regulatory proteins [1, 2, 31], a consensus that has emerged despite the fact that relatively few protein-protein interactions involving Girk channels have been demonstrated using classical biochemical approaches or native systems. Indeed, most reported interactions have involved over-expression of Girk subunits and putative binding partners in cell types that do not normally express Girk channels. Nevertheless, data obtained using these approaches has been valuable in supporting a conceptual model that aligns with key functional properties of GPCR-Girk signaling (e.g., Gi/o coupling specificity, signaling kinetics).
A core signaling complex: GPCR-Gαβγ-Girk
The interaction with Gβγ represents, from a structural and functional perspective, the best understood of the protein-protein interactions involving Girk channels. Gβγ binds to Girk channels, strengthening the interaction between the channel and phosphatidylinositol-4,5-bisphosphate (PIP2), a required co-factor for channel gating [32, 33]. While biochemical and structure-function approaches have suggested multiple interaction domains for Gβγ in all four Girk subunits [1, 34], clear resolution of this critical interaction was obtained recently with the crystallization of a complex formed by Gβγ and the Girk2 homomer [16] (Figure 1B). Girk2 homomers possess four binding sites for Gβγ found at the well-conserved cytoplasmic interfaces between adjacent subunits that contribute to formation of the extended cytoplasmic pore; Gβγ promotes a “pre-open” state of the channel that is intermediate between the closed state of the channel and the open conformation of a constitutively-active Girk2 homomer [15, 16].
Girk channels also interact with Gαi/o. The inactive heterotrimeric G protein (Gαi/o-GDP/Gβγ), as well as Gαi/o-GDP and Gαi/o-GTP, can bind to intracellular domains of Girk channels [25, 26, 35–38]. Interactions between Girk channels and Gαi/o-GDP (either alone or in the context of the inactive heterotrimeric G protein) suppress basal activity of Girk channels, while enhancing their G protein-dependent activation [25, 39, 40]. Moreover, the selective association between Girk channels and Gαi/o likely explains in part the strict coupling specificity between Girk channels and Gi/o-dependent signaling pathways in vivo [36, 41]. This specificity may be reinforced further by selective pre-coupling between certain GPCRs and Gαi/o-GDP/Gβγ [42], and/or by direct GPCR-Girk interactions [31]. Indeed, several GPCRs that couple to Gi/o G proteins, including D2 and D4 dopamine receptors and GABAB receptors, have been shown to interact directly with Girk channels [31, 43, 44].
Collectively, these studies support the vision of a compact core Girk signaling complex, wherein minor conformational changes triggered by agonist binding to GPCR unveils key protein-protein interaction interfaces [45]. Organization of signaling elements within a multi-protein complex affords several advantages over random, collision-based designs. The close spatial proximity of the relevant molecules allows for fast and efficient signaling, and the noise or “cross-talk” that might otherwise occur with non-specific collision events is minimized, creating a tailored intracellular response to an external stimulus. Moreover, the strength or sensitivity of the signaling pathway can theoretically be titrated, by altering macrocomplex composition, to suit the needs of the cell under specific circumstances. With respect to this latter point, two Girk-interacting proteins warrant further discussion.
Sorting nexin 27 (SNX27)
Girk2c and Girk3 possess identical C-terminal Class 1 interaction motifs for PDZ domain-containing proteins (-ESKV) (Figure 1A). Using the distal C-terminal domain of Girk3 as bait in an unbiased proteomic screen, sorting nexin 27 (SNX27) was identified as a Girk-interacting protein [30]. SNX27 regulates the trafficking between cell surface and endosome of an array of neuronal signaling proteins [46], and has been implicated in Down syndrome and addiction [47, 48]. SNX27 is the only member of the sorting nexin family that has a PDZ domain, and it recognizes class I PDZ-binding motifs [49].
Amino acids immediately upstream from the ESKV motif in Girk3 are crucial for promoting the Girk3-SNX27 interaction, and preclude its interaction with PSD95 [30, 50]. SNX27 also contains a Ras association (RA) and a lipid-binding phox-homology (PX) domain. All three functional domains are critical for the proper function of SNX27, which in the context of Girk signaling involves targeting Girk3-containing channels to early endosomes, effectively reducing the surface expression of Girk channels and enhancing the excitability of host neurons [30, 50, 51] (Figure 2). Interestingly, while SNX27 can bind to Girk2c, the surface distribution of homomeric Girk2c channels is unaffected by SNX27 expression, suggesting that another factor(s) may influence the trafficking fates of Girk channels [52].
Figure 2. Subunit-dependent regulation of Girk channel trafficking by SNX27.

Neuronal Girk channels are thought to internalize into early endosomes (EE), at which point they can be recycled back to the cell surface via recycling endosomes (RE) or diverted to late endosomes/lysosomes (LE-Ly) for degradation. Via a selective physical and functional association with Girk3, SNX27 enhances the trafficking of Girk3-containing channels to early endosomes, leading ultimately to increased lysosomal degradation and consequently, reduced numbers of Girk channels on the cell surface (right side of schematic). In contrast, SNX27 does not influence the trafficking of Girk2 homomers or Girk1/2 heteromers (left side of schematic).
The up-regulation of b isoform of SNX27 (SNX27b) in response to in vivo exposure to the psychostimulants cocaine and amphetamine is particularly intriguing in light of recent observations that GABAB-Girk signaling is weakened by acute psychostimulant exposure in VTA dopamine and GABA neurons, and following chronic cocaine treatment in Layer 5/6 pyramidal neurons of the medial prefrontal cortex (mPFC) [47, 53–55] (discussed below). Similarly, the association between loss of SNX27 and Down syndrome [48] is interesting given that Girk signaling is enhanced in the hippocampus and cortex of a mouse model of Down syndrome [56, 57], and since Girk2 triploid mice recapitulated many of the neurological phenotypes associated with this syndrome [58]. While altered SNX27 expression and/or function will certainly impact a wide array of cell signaling pathways, these studies argue that SNX27-dependent alterations in Girk signaling may contribute to some of the cellular and behavioral deficits linked to psychostimulant addiction and Down syndrome.
R7 RGS proteins
GPCR-Girk signaling is negatively-modulated by Regulator of G protein Signaling (RGS) proteins [1, 2]. RGS proteins enhance the GTPase activity of Gα subunits, accelerating the deactivation of G protein signaling following agonist removal [59]. Accordingly, RGS proteins accelerate receptor-induced Girk current deactivation kinetics, among other influences [60]. Discrete protein modules confer unique functionality to specific RGS proteins [59], and these – together with their unique cell/tissue expression patterns – appear to promote interactions with Girk channels. For example, the selective expression of Rgs2 in VTA dopamine neurons, and its preferential association with Girk3, weakens the coupling between GABAB receptors and the Girk2/3 heteromer [28]. Interestingly, changes in the expression of Rgs2 in response to chronic GHB (and morphine) treatment correlated with altered GABAB-Girk coupling in VTA dopamine neurons, and this neuroadaptation may contribute to the development of tolerance to GHB and other drugs of abuse.
Recent data show that Girk channels are modulated by RGS proteins in the R7 subfamily, which includes Rgs6, Rgs7, Rgs9, and Rgs11. The R7 RGS proteins possess a domain resembling the G protein Gγ subunit (G gamma-like domain, or GGL) that promotes association with the atypical 5th member of the G protein Gβ family, Gβ5 [61]. The crystal structure of the Rgs9-Gβ5 complex reveals that the interaction between GGL and Gβ5 resembles that observed in conventional Gβγ dimers [62]. Not surprisingly, therefore, complexes formed between R7 RGS proteins and Gβ5 (Rgs/Gβ5) bind to Girk channels and modulate the kinetics of m2 muscarinic receptor/Girk signaling in atrial cardiomyocytes (Rgs6/Gβ5) [63, 64] and GABAB receptor/Girk signaling in hippocampal CA1 pyramidal neurons (Rgs7/Gβ5) [44, 65]. Moreover, Gβ5 ablation enhanced the sensitivity of Girk channels to GABAB receptor stimulation in hippocampal neurons, indicating that Rgs/Gβ5 complexes can also negatively influence GPCR-Girk coupling efficiency [65]. Interestingly, Rgs6/Gβ5 complexes appear to modulate GABAB-Girk signaling in cerebellar granule cells [66], showing that complexes containing either Rgs6 or Rgs7 are relevant to neuronal Girk signaling. In addition, the Rgs/Gβ5-Girk channel interaction may be controlled by the R7 RGS-binding protein R7BP [67], providing another layer of fine regulation of GPCR-Girk signaling and another potential therapeutic target.
Loss of the Rgs6/Gβ5-dependent modulation of Girk signaling in the heart correlates with bradycardia and enhanced parasympathetic influence [63, 64]. As dysregulation of the parasympathetic control of cardiac output has been linked to sick sinus syndrome, heart failure, and arrhythmia [68], selective targeting of the Rgs6/Gβ5-Girk axis may prove beneficial in clinical settings involving cardiac disorders. While the relevance of the Rgs7/Gβ5-dependent modulation of GABAB-Girk signaling in the hippocampus is not understood, mice lacking Gβ5 were more sensitive to the sedative effect of the GABAB agonist baclofen [65]. Moreover, mice lacking R7BP exhibited enhanced thermal nociceptive thresholds and augmented analgesic effects of opioids and baclofen, consistent with its proposed role as a facilitator of the Rgs/Gβ5-dependent regulation of GPCR-Girk signaling [67]. As enhanced Girk signaling has been linked to depotentiation [69], a particular form of excitatory synaptic plasticity, and cognitive deficits associated with Down syndrome [58], it will be particularly important to explore the relationship between the Rgs7/Gβ5-regulation of Girk signaling and hippocampal-dependent learning and memory. Moreover, it will be interesting to probe the involvement of Rgs/Gβ5 complexes in other neuronal GPCR-Girk signaling pathways.
Plasticity of Girk signaling
Recent work has shown that the strength and sensitivity of neuronal Girk signaling is titratable and subject to regulation by multiple stimuli. The first clear example of Girk signaling plasticity came with the demonstration that stimulation protocols that evoked NMDA receptor-dependent long-term potentiation (LTP) of glutamatergic neurotransmission in hippocampal CA1 neurons also strengthened synaptic GABAB-Girk signaling [70]. Subsequent work in cultured hippocampal pyramidal neurons revealed that neuronal activity triggered by NMDA receptor activation lead to a rapid increase in the levels of Girk channels (Girk1/2) on the somatodendritic membrane, and enhanced Girk signaling via the A1 adenosine receptor [71]. Pharmacologic or genetic ablation of Girk signaling suggested that this neuroadaptation is critical for the depotentiation of excitatory LTP [69].
Drugs of abuse
As documented in Table 1, Girk signaling shapes many of the behavioral effects of drugs of abuse, including opioids, psychostimulants (cocaine), and ethanol. Exposure to drugs of abuse can alter neuronal Girk signaling in durable fashion in the reward circuitry, the core of which consists of interconnected neurons in the VTA, mPFC, and nucleus accumbens (NAc) [72]. For example, a single non-contingent exposure to cocaine suppressed GABAB-Girk signaling by 50% in VTA dopamine (but not SNc dopamine) neurons for several days [53], paralleling the better-understood enhancement of glutamatergic neurotransmission occurring in the same neurons [72]. Acute psychostimulant exposure also persistently suppressed GABAB-Girk signaling in VTA GABA neurons [54]. Finally, repeated cocaine exposure suppressed GABAB-Girk signaling in Layer 5/6 glutamatergic pyramidal neurons of the mPFC [55], the main source of glutamatergic input to the VTA and NAc. This neuroadaptation was specific for pyramidal neurons in the prelimbic cortex (as compared to pyramidal neurons in the infralimbic and motor cortices), and persisted for more than a month after the final cocaine injection. Moreover, persistent suppression of Girk signaling in Layer 5/6 of the mPFC pre-sensitized mice to the motor-stimulatory effects of acute cocaine. While more work is required to understand the behavioral relevance of drug-induced adaptations in Girk signaling in the reward circuitry, these early insights, along with the anatomic and cellular specificity of the neuroadaptations, and their durability, suggest that they drive and/or contribute to the persistent expression of addictive behaviors including drug-seeking, craving, and relapse.
Mechanism(s) of plasticity
Plasticity of Girk signaling triggered by neuronal activity and drugs of abuse involves the redistribution of Girk2-containing channels to and from the surface membrane (Figure 3). Enhanced Girk signaling in hippocampal pyramidal neurons triggered by NMDA receptor activation was linked to enhanced trafficking of Girk2-containing channels from recycling endosomes to the cell surface [71]. In contrast, increased intracellular labeling at the expense of surface labeling was seen for Girk2 in VTA dopamine and GABA neurons following acute cocaine and methamphetamine treatment [53, 54], respectively, and in Layer 5/6 pyramidal neurons following repeated cocaine [55]. Interestingly, a similar redistribution of the GABAB receptor from the surface to inside the cell was observed in VTA GABA and Layer 5/6 mPFC pyramidal neurons, but not in VTA dopamine neurons, suggesting that different mechanisms mediate Girk signaling plasticity in different cell types. Consistent with this premise, the methamphetamine-induced adaptation in VTA GABA neurons and cocaine-induced adaptation in Layer 5/6 mPFC pyramidal neurons were both blocked by pretreatment with a D1 dopamine receptor antagonist, while the cocaine-induced adaptation in VTA dopamine neurons was D2 dopamine receptor-dependent. Moreover, GABAB-Girk signaling (but not somatodendritic inhibitory signaling via D2 dopamine or α2 adrenergic receptors, or somatodendritic GABAB-dependent signaling that did not involve Girk channels) was suppressed by repeated cocaine in Layer 5/6 mPFC pyramidal neurons, while Girk signaling via both the GABAB and D2 dopamine receptor was suppressed by acute cocaine in VTA dopamine neurons. These observations are reminiscent of the selective enhancement by neuronal activity of Girk signaling via the A1 adenosine (but not GABAB) receptor [69], and suggest that some forms of Girk signaling plasticity are compartmentalized within neurons, and presumably driven by the GPCR or other proteins within the signaling macrocomplex.
Figure 3. Plasticity of neuronal Girk signaling.

Cocaine-induced suppression of Girk signaling in VTA (a–c) and mPFC (d–f). a) A single cocaine injection reduced baclofen-induced (GABAB receptor-dependent) Girk currents in VTA dopamine neurons, an adaptation that persisted for up to 5 days, required activation of D2 dopamine receptors, and correlated with a redistribution of Girk2-containing channels (but not GABABR) from the cell surface to intracellular sites (b,c) [53]. (d) Repeated cocaine administration suppressed baclofen-induced Girk currents in Layer 5/6 pyramidal neurons, an adaptation that persisted for more than a month, required activation of D1 dopamine receptors, and correlated with a phosphorylation-dependent redistribution of Girk2-containing channels and GABAB receptors from the cell surface to intracellular sites (e,f) [55]. Blue line in the current traces shows that the baclofen-induced current was reversed by a GABAB receptor antagonist.
The trafficking of Girk channels and GPCR-Girk complexes to and from the cell surface that underlies the forms of plasticity described above appears to be regulated by phosphorylation. For example, the potentiation of synaptic GABAB-Girk signaling triggered in parallel with LTP of glutamatergic neurotransmission was dependent on CaMKII activity [70]. And in cultured hippocampal neurons, CaMKII activation (via prolonged morphine treatment, activation of metabotropic glutamate receptors, or a constitutively-active CaMKII mutant) shifted the distribution of Girk2 from dendritic shafts to spines, leading to enhanced Girk signaling via 5-HT receptors and decreased GABAB-Girk signaling [73]. While the direct molecular target of CaMKII was not determined in these studies, Girk2(Ser9) is a reasonable candidate. Phosphorylation of Girk2(Ser9), which sits upstream of a unique internalization motif (VL) [7], suppresses surface trafficking of Girk2-containing channels [71]. Conversely, dephosphorylation of Girk2(Ser9) via protein phosphatase 1 (PP1) promotes surface trafficking of Girk2-containing channels from recycling endosomes, explaining the activity-dependent potentiation of Girk signaling linked to the depotentiation of LTP [69, 71]. While dephosphorylation of Girk2(Ser9) promotes enhanced surface distribution of Girk channels, psychostimulant-induced suppression of GABAB-Girk signaling in VTA GABA and Layer 5/6 mPFC neurons is more likely related to the phosphorylation status of the GABAB receptor [54, 55]. Surprisingly, despite the durable nature of the drug-induced suppression of GABAB-Girk signaling in these pyramidal neurons, acute intracellular treatment with the PP1/PP2a inhibitor okadaic acid restored normal Girk signaling.
Pharmacologic manipulation of Girk channels
Given the broad distributions and roles of Girk channels in the nervous system, and their contributions to cardiac and endocrine physiology, global and direct pharmacologic manipulation of Girk signaling should evoke an array of consequences, many undesirable (Table 1). Indeed, global constitutive ablation of Girk2 triggers an array of phenotypes, most notably a shortened lifespan due to spontaneous lethal seizures [74]. While constitutive ablation of other Girk subunits is correlated with less severe phenotypes, the full therapeutic potential associated with inhibiting or enhancing Girk signaling will likely not be achieved without regional and/or Girk subunit-selective manipulation. For example, an agonist with selectivity for Girk2/3 heteromers, the Girk channel subtype that appears to be uniquely expressed in VTA dopamine neurons [28], could be useful as an anti-craving compound. In addition, drugs that selectively activate or inhibit Girk1/4 heteromers, and which cannot pass the blood-brain barrier, could represent efficacious therapies for certain types of arrhythmias. Indeed, two drugs that can inhibit Girk1/4 heteromers (NTC-801 and NIP-151) showed promise in preclinical studies in the treatment of atrial fibrillation [75, 76].
While Girk channels are activated in a G protein-independent manner by ethanol [77, 78], volatile anesthetics [79, 80], and the flavonoid naringin [81], and are blocked by an array of psychoactive compounds (many of which are clinically-relevant) (e.g., [82]), most of the compounds have other primary molecular targets and/or there is little evidence for Girk subtype specificity or pharmacokinetic advantages. Recently, however, a new class of subunit-selective, efficacious, potent, and direct-acting Girk channel agonists and antagonists was identified [83–85]. The prototype (ML297) is strikingly selective for Girk1-containing heteromers, and was efficacious in mice in delaying seizure onset in a maximal electroshock model of epilepsy, and preventing convulsions and lethality in a chemically-induced epilepsy model [84]. Accordingly, this family of compounds and derivatives should afford an excellent opportunity to investigate the potential therapeutic benefits associated with direct activation or inhibition of Girk1-containing channels, and may serve as a platform for the identification of compounds that can discriminate across a wide range of channel sub-types.
Conclusions
Efforts by many research groups over the last two decades have revealed key functional, structural, and regulatory features of Girk channels. This body of evidence, combined with our evolving understanding of Girk channel contributions to physiology and disease, and a continually-improving capacity for pharmacologic manipulation, sets the stage for an exciting future of investigations into the therapeutic potential of this interesting and important channel class. Such efforts hold the promise of yielding novel therapeutic approaches to the treatment of many forms of neurological, cardiovascular, and neuroendocrine disorders.
OUTSTANDING QUESTIONS.
Pharmacology
The recent identification of the subunit-selective, efficacious, and potent direct modulators of Girk signaling represents an important step forward in this field, and should provide a foundation on which efforts in synthetic chemistry, molecular modeling/simulations, and crystallography synergize to yield new compounds with optimized pharmacodynamic and pharmacokinetic properties. When available, these compounds will greatly facilitate efforts directed at understanding the physiological relevance of Girk channels, permitting us to move beyond studies in mutant mice.
Macrocomplex formation
While a large body of evidence supports the existence of discrete signaling complexes containing Girk channels in vivo, gaps in our understanding of Girk channel trafficking and regulation argue that there are additional proteins that influence Girk function - either via direct or indirect physical interaction - remaining to be discovered. Moreover, understanding the molecular determinants of macrocomplex formation will be helpful as it can potentially provide the means to selectively manipulate Girk signaling in a subtle manner.
Physiological relevance
Further investigation into the physiological relevance of Girk channels is required to better understand opportunities for beneficial therapeutic manipulation. The development of novel pharmacological tools that can discriminate among the various Girk channel subtypes existing in vivo will facilitate this process, and will complement next-generation genetic approaches that can give region and cell-type dependent insights into the function of Girk channels formed by distinct combinations of subunits (including specific splice isoforms). In addition, efforts that build on the growing evidence linking mutations or polymorphisms in GIRK genes to human disease will be invaluable.
Plasticity
We are just beginning to understand the triggers and mechanisms underlying the plasticity in Girk signaling. Going forward, it will be important to understand more about the mechanisms involved in the dynamic modulation of Girk channel trafficking, including differences that may exist across distinct drugs of abuse, neuron populations, Girk channel subtypes, and GPCR-Girk combinations. It will be particularly interesting to explore the potential relationship between SNX27, the phosphoregulation of Girk trafficking, and the drug-induced adaptations in Girk signaling seen in the reward circuitry. Of course, understanding the physiological (and perhaps pathophysiological) relevance of the adaptations is the ultimate goal.
Highlights.
Girk channels are novel targets for therapeutic interventions in a broad array of human nervous system disorders.
Girk channels exist in multi-protein complexes whose molecular composition can differ to suit the cell needs under specific circumstances.
Girk signaling shapes many of the behavioral effects of drugs of abuse, including opioids, cocaine, methaamphetamine, and ethanol.
Plasticity of Girk signaling triggered by neuronal activity and drugs of abuse involves the subcellular redistribution of the channel.
Acknowledgments
This work was supported by NIH grants to KW (MH061933, HL105550, and DA034696) and the Spanish Ministry of Science and Innovation BFU2012-38348 and CONSOLIDER-Ingenio CSD2008-0000 (RL). The authors thank members of the Wickman and Luján laboratories for their suggestions for the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci. 2010;11:301–315. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hibino H, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 3.Wei J, et al. Characterization of murine Girk2 transcript isoforms: structure and differential expression. Genomics. 1998;51:379–390. doi: 10.1006/geno.1998.5369. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L, et al. Cloning and characterization of G protein-gated inward rectifier K+ channel (GIRK1) isoforms from heart and brain. J Mol Neurosci. 2001;16:21–32. doi: 10.1385/JMN:16:1:21. [DOI] [PubMed] [Google Scholar]
- 5.Krapivinsky G, et al. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy ME, et al. GIRK4 confers appropriate processing and cell surface localization to G-protein-gated potassium channels. J Biol Chem. 1999;274:2571–2582. doi: 10.1074/jbc.274.4.2571. [DOI] [PubMed] [Google Scholar]
- 7.Ma D, et al. Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron. 2002;33:715–729. doi: 10.1016/s0896-6273(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 8.Chan KW, et al. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc Natl Acad Sci USA. 1996;93:14193–14198. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karschin C, et al. IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci. 1996;16:3559–3570. doi: 10.1523/JNEUROSCI.16-11-03559.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez-Alacid L, et al. Developmental regulation of G protein-gated inwardly-rectifying K+ (GIRK/Kir3) channel subunits in the brain. Eur J Neurosci. 2011;34:1724–1736. doi: 10.1111/j.1460-9568.2011.07886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida M, MacKinnon R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell. 2002;111:957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 12.Inanobe A, et al. Structural diversity in the cytoplasmic region of G protein-gated inward rectifier K+ channels. Channels. 2007;1:39–45. [PubMed] [Google Scholar]
- 13.Nishida M, et al. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inanobe A, et al. A structural determinant for the control of PIP2 sensitivity in G protein-gated inward rectifier K+ channels. J Biol Chem. 2010;285:38517–38523. doi: 10.1074/jbc.M110.161703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whorton MR, MacKinnon R. X-ray structure of the mammalian GIRK2-betagamma G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao YJ, et al. Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci. 1996;16:7137–7150. doi: 10.1523/JNEUROSCI.16-22-07137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slesinger PA, et al. Identification of structural elements involved in G protein gating of the GIRK1 potassium channel. Neuron. 1995;15:1145–1156. doi: 10.1016/0896-6273(95)90102-7. [DOI] [PubMed] [Google Scholar]
- 19.Chan KW, et al. Specific regions of heteromeric subunits involved in enhancement of G protein-gated K+ channel activity. J Biol Chem. 1997;272:6548–6555. doi: 10.1074/jbc.272.10.6548. [DOI] [PubMed] [Google Scholar]
- 20.Wydeven N, et al. Structural elements in the Girk1 subunit that potentiate G protein-gated potassium channel activity. Proc Natl Acad Sci USA. 2012;109:21492–21497. doi: 10.1073/pnas.1212019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medina I, et al. A switch mechanism for G beta gamma activation of I(KACh) J Biol Chem. 2000;275:29709–29716. doi: 10.1074/jbc.M004989200. [DOI] [PubMed] [Google Scholar]
- 22.Ivanina T, et al. Mapping the Gbetagamma-binding sites in GIRK1 and GIRK2 subunits of the G protein-activated K+ channel. J Biol Chem. 2003;278:29174–29183. doi: 10.1074/jbc.M304518200. [DOI] [PubMed] [Google Scholar]
- 23.Thomas AM, et al. Differential phosphoinositide binding to components of the G protein-gated K+ channel. J Membr Biol. 2006;211:43–53. doi: 10.1007/s00232-006-0014-5. [DOI] [PubMed] [Google Scholar]
- 24.Rusinova R, et al. Mass spectrometric analysis reveals a functionally important PKA phosphorylation site in a Kir3 channel subunit. Pflugers Arch. 2009;458:303–314. doi: 10.1007/s00424-008-0628-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinstein M, et al. Divergent regulation of GIRK1 and GIRK2 subunits of the neuronal G protein gated K+ channel by GalphaiGDP and Gbetagamma. J Physiol. 2009;587:3473–3491. doi: 10.1113/jphysiol.2009.173229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berlin S, et al. G alpha(i) and G betagamma jointly regulate the conformations of a G betagamma effector, the neuronal G protein-activated K+ channel (GIRK) J Biol Chem. 2010;285:6179–6185. doi: 10.1074/jbc.M109.085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruz HG, et al. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- 28.Labouebe G, et al. RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007;10:1559–1568. doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- 29.Jelacic TM, et al. Functional and biochemical evidence for G-protein-gated inwardly rectifying K+ (GIRK) channels composed of GIRK2 and GIRK3. J Biol Chem. 2000;275:36211–36216. doi: 10.1074/jbc.M007087200. [DOI] [PubMed] [Google Scholar]
- 30.Lunn ML, et al. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci. 2007;10:1249–1259. doi: 10.1038/nn1953. [DOI] [PubMed] [Google Scholar]
- 31.Doupnik CA. GPCR-Kir channel signaling complexes: defining rules of engagement. J Recept Signal Tr R. 2008;28:83–91. doi: 10.1080/10799890801941970. [DOI] [PubMed] [Google Scholar]
- 32.Huang CL, et al. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 33.Sui JL, et al. Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokogawa M, et al. NMR analyses of the Gbetagamma binding and conformational rearrangements of the cytoplasmic pore of G protein-activated inwardly rectifying potassium channel 1 (GIRK1) J Biol Chem. 2011;286:2215–2223. doi: 10.1074/jbc.M110.160754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang CL, et al. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 36.Clancy SM, et al. Pertussis-toxin-sensitive Galpha subunits selectively bind to C-terminal domain of neuronal GIRK channels: evidence for a heterotrimeric G-protein-channel complex. Mol Cell Neurosci. 2005;28:375–389. doi: 10.1016/j.mcn.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Rebois RV, et al. Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. J Cell Sci. 2006;119:2807–2818. doi: 10.1242/jcs.03021. [DOI] [PubMed] [Google Scholar]
- 38.Mase Y, et al. Structural basis for modulation of gating property of G protein-gated inwardly rectifying potassium ion channel (GIRK) by i/o-family G protein alpha subunit (Galphai/o) J Biol Chem. 2012;287:19537–19549. doi: 10.1074/jbc.M112.353888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peleg S, et al. G(alpha)(i) controls the gating of the G protein-activated K+ channel, GIRK. Neuron. 2002;33:87–99. doi: 10.1016/s0896-6273(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 40.Rubinstein M, et al. Galphai3 primes the G protein-activated K+ channels for activation by coexpressed Gbetagamma in intact Xenopus oocytes. J Physiol. 2007;581:17–32. doi: 10.1113/jphysiol.2006.125864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rusinova R, et al. Specificity of Gbetagamma signaling to Kir3 channels depends on the helical domain of pertussis toxin-sensitive Galpha subunits. J Biol Chem. 2007;282:34019–34030. doi: 10.1074/jbc.M704928200. [DOI] [PubMed] [Google Scholar]
- 42.Nobles M, et al. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci USA. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciruela F, et al. Evidence for oligomerization between GABAB receptors and GIRK channels containing the GIRK1 and GIRK3 subunits. Eur J Neurosci. 2010;32:1265–1277. doi: 10.1111/j.1460-9568.2010.07356.x. [DOI] [PubMed] [Google Scholar]
- 44.Fajardo-Serrano A, et al. Association of Rgs7/Gbeta5 complexes with Girk channels and GABA receptors in hippocampal CA1 pyramidal neurons. Hippocampus. 2013 doi: 10.1002/hipo.22161. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raveh A, et al. Elucidation of the gating of the GIRK channel using a spectroscopic approach. J Physiol. 2009;587:5331–5335. doi: 10.1113/jphysiol.2009.180158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeffer SR. A nexus for receptor recycling. Nat Cell Biol. 2013;15:446–448. doi: 10.1038/ncb2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kajii Y, et al. A developmentally regulated and psychostimulant-inducible novel rat gene mrt1 encoding PDZ-PX proteins isolated in the neocortex. Mol Psychiatr. 2003;8:434–444. doi: 10.1038/sj.mp.4001258. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlton JG, Cullen PJ. Sorting nexins. Curr Biol. 2005;15:R819–820. doi: 10.1016/j.cub.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Balana B, et al. Mechanism underlying selective regulation of G protein-gated inwardly rectifying potassium channels by the psychostimulant-sensitive sorting nexin 27. Proc Natl Acad Sci USA. 2011;108:5831–5836. doi: 10.1073/pnas.1018645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balana B, et al. Ras-association domain of sorting Nexin 27 is critical for regulating expression of GIRK potassium channels. PloS One. 2013;8:e59800. doi: 10.1371/journal.pone.0059800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nassirpour R, Slesinger PA. Subunit-specific regulation of Kir3 channels by sorting nexin 27. Channels. 2007;1:331–333. doi: 10.4161/chan.5191. [DOI] [PubMed] [Google Scholar]
- 53.Arora D, et al. Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci. 2011;31:12251–12257. doi: 10.1523/JNEUROSCI.0494-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Padgett CL, et al. Methamphetamine-evoked depression of GABA(B) receptor signaling in GABA neurons of the VTA. Neuron. 2012;73:978–989. doi: 10.1016/j.neuron.2011.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hearing M, et al. Repeated cocaine weakens GABA-Girk signaling in Layer 5/6 pyramidal neurons in the prelimbic cortex. Neuron. 2013;80:159–170. doi: 10.1016/j.neuron.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harashima C, et al. Abnormal expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in hippocampus, frontal cortex, and substantia nigra of Ts65Dn mouse: a model of Down syndrome. J Comp Neurol. 2006;494:815–833. doi: 10.1002/cne.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Best TK, et al. Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. J Neurophysiol. 2007;97:892–900. doi: 10.1152/jn.00626.2006. [DOI] [PubMed] [Google Scholar]
- 58.Cooper A, et al. Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc Natl Acad Sci USA. 2012;109:2642–2647. doi: 10.1073/pnas.1109099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sjogren B. Regulator of G protein signaling proteins as drug targets: current state and future possibilities. Adv Pharmacol. 2011;62:315–347. doi: 10.1016/B978-0-12-385952-5.00002-6. [DOI] [PubMed] [Google Scholar]
- 60.Doupnik CA, et al. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson GR, et al. The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem Biophys. 2009;54:33–46. doi: 10.1007/s12013-009-9052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheever ML, et al. Crystal structure of the multifunctional Gbeta5-RGS9 complex. Nat Struct Mol Biol. 2008;15:155–162. doi: 10.1038/nsmb.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Posokhova E, et al. RGS6/Gβ5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ Res. 2010;107:1350–1354. doi: 10.1161/CIRCRESAHA.110.224212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang J, et al. RGS6, a modulator of parasympathetic activation in heart. Circ Res. 2010;107:1345–1349. doi: 10.1161/CIRCRESAHA.110.224220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xie K, et al. Gbeta5 recruits R7 RGS proteins to GIRK channels to regulate the timing of neuronal inhibitory signaling. Nat Neurosci. 2010;13:661–663. doi: 10.1038/nn.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maity B, et al. Regulator of G protein signaling 6 (RGS6) protein ensures coordination of motor movement by modulating GABAB receptor signaling. J Biol Chem. 2012;287:4972–4981. doi: 10.1074/jbc.M111.297218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou H, et al. GIRK channel modulation by assembly with allosterically regulated RGS proteins. Proc Natl Acad Sci USA. 2012;109:19977–19982. doi: 10.1073/pnas.1214337109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stewart A, et al. RGS proteins in heart: brakes on the vagus. Front Physiol. 2012;3:95. doi: 10.3389/fphys.2012.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chung HJ, et al. G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc Natl Acad Sci USA. 2009;106:635–640. doi: 10.1073/pnas.0811685106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang CS, et al. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell. 2005;123:105–118. doi: 10.1016/j.cell.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 71.Chung HJ, et al. Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels. Proc Natl Acad Sci USA. 2009;106:629–634. doi: 10.1073/pnas.0811615106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nassirpour R, et al. Morphine- and CaMKII-dependent enhancement of GIRK channel signaling in hippocampal neurons. J Neurosci. 2010;30:13419–13430. doi: 10.1523/JNEUROSCI.2966-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Signorini S, et al. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci USA. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hashimoto N, et al. Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IKACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J Cardiovasc Pharm. 2008;51:162–169. doi: 10.1097/FJC.0b013e31815e854c. [DOI] [PubMed] [Google Scholar]
- 76.Machida T, et al. Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:94–102. doi: 10.1161/CIRCEP.110.951608. [DOI] [PubMed] [Google Scholar]
- 77.Kobayashi T, et al. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat Neurosci. 1999;2:1091–1097. doi: 10.1038/16019. [DOI] [PubMed] [Google Scholar]
- 78.Lewohl JM, et al. G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci. 1999;2:1084–1090. doi: 10.1038/16012. [DOI] [PubMed] [Google Scholar]
- 79.Weigl LG, Schreibmayer W. G protein-gated inwardly rectifying potassium channels are targets for volatile anesthetics. Mol Pharmacol. 2001;60:282–289. doi: 10.1124/mol.60.2.282. [DOI] [PubMed] [Google Scholar]
- 80.Yamakura T, et al. Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels. Anesthesiology. 2001;95:144–153. doi: 10.1097/00000542-200107000-00025. [DOI] [PubMed] [Google Scholar]
- 81.Yow TT, et al. Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br J Pharmacol. 2011;163:1017–1033. doi: 10.1111/j.1476-5381.2011.01315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kobayashi T, et al. Inhibition of G protein-activated inwardly rectifying K+ channels by different classes of antidepressants. PloS One. 2011;6:e28208. doi: 10.1371/journal.pone.0028208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wen W, et al. Discovery of ‘molecular switches’ within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg Med Chem Lett. 2013;23:4562–4566. doi: 10.1016/j.bmcl.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaufmann K, et al. ML297 (VU0456810), the First Potent and Selective Activator of the GIRK Potassium Channel, Displays Antiepileptic Properties in Mice. ACS Chem Neurosci. 2013;4:1278–1286. doi: 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramos-Hunter SJ, et al. Discovery and SAR of a novel series of GIRK1/2 and GIRK1/4 activators. Bioorg Med Chem Lett. 2013;23:5195–5198. doi: 10.1016/j.bmcl.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wickman K, et al. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 87.Bettahi I, et al. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem. 2002;277:48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- 88.Yang Y, et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jabbari J, et al. Common polymorphisms in KCNJ5 [corrected] are associated with early-onset lone atrial fibrillation in Caucasians. Cardiology. 2011;118:116–120. doi: 10.1159/000323840. [DOI] [PubMed] [Google Scholar]
- 90.Wang F, et al. The phenotype xharacteristics of Type-13 Long QT Syndrome with mutation in KCNJ5 (Kir3.4-G387R) Heart rhythm. 2013;10:1500–1506. doi: 10.1016/j.hrthm.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 91.Scholl UI, Lifton RP. New insights into aldosterone-producing adenomas and hereditary aldosteronism: mutations in the K+ channel KCNJ5. Curr Op Nephrol Hy. 2013;22:141–147. doi: 10.1097/MNH.0b013e32835cecf8. [DOI] [PubMed] [Google Scholar]
- 92.Mulatero P, et al. Role of KCNJ5 in familial and sporadic primary aldosteronism. Nat Rev Endocrinol. 2013;9:104–112. doi: 10.1038/nrendo.2012.230. [DOI] [PubMed] [Google Scholar]
- 93.Mitrovic I, et al. Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci USA. 2003;100:271–276. doi: 10.1073/pnas.0136822100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marker CL, et al. Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci. 2004;24:2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blednov YA, et al. A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc Natl Acad Sci USA. 2003;100:277–282. doi: 10.1073/pnas.012682399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marker CL, et al. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cruz HG, et al. Absence and rescue of morphine withdrawal in GIRK/Kir3 knockout mice. J Neurosci. 2008;28:4069–4077. doi: 10.1523/JNEUROSCI.0267-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith SB, et al. Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenet Genom. 2008;18:231–241. doi: 10.1097/FPC.0b013e3282f55ab2. [DOI] [PubMed] [Google Scholar]
- 99.Nishizawa D, et al. Association between KCNJ6 (GIRK2) gene polymorphisms and postoperative analgesic requirements after major abdominal surgery. PloS One. 2009;4:e7060. doi: 10.1371/journal.pone.0007060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lotsch J, et al. A KCNJ6 (Kir3.2, GIRK2) gene polymorphism modulates opioid effects on analgesia and addiction but not on pupil size. Pharmacogenet Genom. 2010;20:291–297. doi: 10.1097/FPC.0b013e3283386bda. [DOI] [PubMed] [Google Scholar]
- 101.Blednov YA, et al. Hyperactivity and dopamine D1 receptor activation in mice lacking Girk2 channels. Psychopharmacology. 2002;159:370–378. doi: 10.1007/s00213-001-0937-6. [DOI] [PubMed] [Google Scholar]
- 102.Morgan AD, et al. Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice. Neuropsychopharmacology. 2003;28:932–938. doi: 10.1038/sj.npp.1300100. [DOI] [PubMed] [Google Scholar]
- 103.Arora D, et al. Altered neurotransmission in the mesolimbic reward system of Girk mice. J Neurochem. 2010;114:1487–1497. doi: 10.1111/j.1471-4159.2010.06864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pravetoni M, Wickman K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008;7:523–531. doi: 10.1111/j.1601-183X.2008.00388.x. [DOI] [PubMed] [Google Scholar]
- 105.Perry CA, et al. Predisposition to late-onset obesity in GIRK4 knockout mice. Proc Natl Acad Sci U A. 2008;105:8148–8153. doi: 10.1073/pnas.0803261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blednov YA, et al. Potassium channels as targets for ethanol: studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice. J Pharmacol Exp Ther. 2001;298:521–530. [PubMed] [Google Scholar]
- 107.Kozell LB, et al. Mapping a barbiturate withdrawal locus to a 0.44 Mb interval and analysis of a novel null mutant identify a role for Kcnj9 (GIRK3) in withdrawal from pentobarbital, zolpidem, and ethanol. J Neurosci. 2009;29:11662–11673. doi: 10.1523/JNEUROSCI.1413-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clarke TK, et al. KCNJ6 is associated with adult alcohol dependence and involved in gene x early life stress interactions in adolescent alcohol drinking. Neuropsychopharmacology. 2011;36:1142–1148. doi: 10.1038/npp.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wickman K, et al. Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4. J Neurosci. 2000;20:5608–5615. doi: 10.1523/JNEUROSCI.20-15-05608.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Blednov YA, et al. GIRK2 deficient mice. Evidence for hyperactivity and reduced anxiety. Physiol Behav. 2001;74:109–117. doi: 10.1016/s0031-9384(01)00555-8. [DOI] [PubMed] [Google Scholar]
- 111.Yamada K, et al. Association study of the KCNJ3 gene as a susceptibility candidate for schizophrenia in the Chinese population. Hum Genet. 2012;131:443–451. doi: 10.1007/s00439-011-1089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Costa AC, et al. G-protein-gated potassium (GIRK) channels containing the GIRK2 subunit are control hubs for pharmacologically induced hypothermic responses. J Neurosci. 2005;25:7801–7804. doi: 10.1523/JNEUROSCI.1699-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patil NCD, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- 114.Gregerson KA, et al. Identification of G protein-coupled, inward rectifier potassium channel gene products from the rat anterior pituitary gland. Endocrinology. 2001;142:2820–2832. doi: 10.1210/endo.142.7.8236. [DOI] [PubMed] [Google Scholar]
- 115.Plummer HK, 3rd, et al. Expression of G-protein inwardly rectifying potassium channels (GIRKs) in lung cancer cell lines. BMC Cancer. 2005;5:104. doi: 10.1186/1471-2407-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwanir S, Reuveny E. Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels. Pflugers Arch. 2008;456:1097–1108. doi: 10.1007/s00424-008-0479-4. [DOI] [PubMed] [Google Scholar]
- 117.Wagner V, et al. Cloning and characterisation of GIRK1 variants resulting from alternative RNA editing of the KCNJ3 gene transcript in a human breast cancer cell line. J Cell Biochem. 2010;110:598–608. doi: 10.1002/jcb.22564. [DOI] [PubMed] [Google Scholar]
- 118.Aguado C, et al. Cell type-specific subunit composition of G protein-gated potassium channels in the cerebellum. J Neurochem. 2008;105:497–511. doi: 10.1111/j.1471-4159.2007.05153.x. [DOI] [PubMed] [Google Scholar]
- 119.Inanobe A, et al. Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koyrakh L, et al. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Luscher C, et al. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 122.Ladera C, et al. Pre-synaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem. 2008;107:1506–1517. doi: 10.1111/j.1471-4159.2008.05712.x. [DOI] [PubMed] [Google Scholar]
- 123.Fernandez-Alacid L, et al. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J Neurochem. 2009;110:1363–1376. doi: 10.1111/j.1471-4159.2009.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Michaeli A, Yaka R. Dopamine inhibits GABA(A) currents in ventral tegmental area dopamine neurons via activation of presynaptic G-protein coupled inwardly-rectifying potassium channels. Neuroscience. 2010;165:1159–1169. doi: 10.1016/j.neuroscience.2009.11.045. [DOI] [PubMed] [Google Scholar]
- 125.Booker SA, et al. Differential GABAB-receptor-mediated effects in perisomatic- and dendrite-targeting parvalbumin interneurons. J Neurosci. 2013;33:7961–7974. doi: 10.1523/JNEUROSCI.1186-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]