

Abstract
The cannabinoid CB1 receptor is involved in complex physiological functions. The discovery of CB1 allosteric modulators generates new opportunities for drug discovery targeting the pharmacologically important CB1 receptor. 5-chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (ORG27569; 1) represents a new class of indole-2-carboxamides that exhibit allostery of CB1. To better understand the SAR, a group of indole-2-carboxamide analogs were synthesized and assessed for allostery of the CB1 receptor. We found that within the structure of indole-2-carboxamides, the presence of the indole ring is preferred for maintaining the modulator's high binding affinity for the allosteric site, but not for generating allostery on the orthosteric site. However, the C3 substituents of the indole-2-carboxamides significantly impact the allostery of the ligand. A robust CB1 allosteric modulator 5-chloro-N-(4-(dimethylamino)phenethyl)-3-pentyl-1H-indole-2-carboxamide (11j) was identified. It showed an equilibrium dissociation constant (KB) of 167.3 nM with a markedly high binding cooperativity factor (α=16.55) and potent antagonism of agonist-induced GTPγS binding.
Introduction
The endocannabinoid system is composed of at least two G-protein coupled receptors (GPCRs), CB1 and CB2, a group of endogenous lipid ligands and several catabolic and anabolic enzymes that are involved in the biosynthesis and degradation of the endogenous ligands.1 The CB1 receptors are mainly found on neurons of the central and peripheral nervous system2 where they attenuate neurotransmitter release. CB1 receptors are also present at much lower concentrations in some peripheral tissues, including spleen, tonsil, reproductive organs, endocrine glands, arteries, bone marrow, lungs and heart.3-5 Consistent with its widespread distribution, the CB1 receptor regulates a wide variety of physiological functions including neuronal development, neuromodulatory processes, energy metabolism, cardiovascular, respiratory as well as reproductive functions.6-8 In addition, it also modulates cell proliferation, motility, adhesion and apoptosis.9, 10
Like many GPCRs, the CB1 receptor displays a multiplicity of intracellular signal transduction mechanisms. It preferentially couples to Gi/o type G proteins while its interaction with Gs11 or Gq12 is also possible. Activation of the CB1 receptor primarily leads to the inhibition of adenylyl-cyclase thereby reducing cAMP levels in cells. The CB1 receptor also modulates the activation of mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase-1 and -2 (ERK1/2), p38 MAPK, p42/p44 MAPK and c-Jun N-terminal kinase (JNK).13 Additionally, CB1 can negatively couple to N- and P/Q-type voltage-gated Ca2+ channels and positively couple to A-type and inwardly rectifying K+ channels.14 CB1 also interacts with non-G protein partners such as β-arrestins, adaptor protein AP-3, GPCR-associated sorting protein 1 (GASP1) and the adaptor protein FAN to control receptor signaling or trafficking.15, 16
The activation of the CB1 receptor by endogenous and exogenous agonists such as anandamide and (-)-Δ9-tetrahydrocannabinol (Δ9-THC) has been linked to a wide range of pharmacological effects.17 Growing evidence suggests that the CB1 receptor exists in multiple active conformations, each of which may display distinct abilities to regulate individual signaling pathways.18 Thus, the assumption that there is only one active conformation of the CB1 receptor is overly simplistic and is inadequate to explain all cannabinoid-mediated responses. Recently, several small molecules (Figure 1) have been identified as allosteric modulators of the CB1 receptor. The representative members include ORG27569 (1),19 (1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea (2, PSNCBAM-1),20 3-(4-chlorophenyl)-5-(8-methyl-3-p-tolyl-8-azabicyclo[3.2.1]octan-2-yl)isoxazole (3, RTI-371)21 and the endogenous ligand (5S,6R,7E,9E,11Z,13E,15S)-5,6,15-trihydroxyicosa-7,9,11,13-tetraenoic acid (4, lipoxin A4).22 The identification of these ligands indicates the existence of allosteric site(s) on the CB1 receptor and suggests new opportunities for fine-tuning the signaling pathways of the CB1 receptor.
Figure 1. Structures of representative allosteric modulators of the CB1 receptor.

Indole-2-carboxamide 1 was the first allosteric modulator identified for the CB1 receptor.19 It increases specific binding of the tritium labeled CB1 agonist 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol ([3H]CP55,940) but decreases the binding of the tritium labeled CB1 inverse agonist 5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([3H]SR141716A) in membranes from cells expressing the CB1 receptor. Despite acting as an enhancer of agonist binding, it acts as an inhibitor of agonist-induced Gαi protein coupling.19, 23 It was further demonstrated that the 1-induced conformational change of the CB1 receptor leads to cellular internalization of the receptor and downstream activation of ERK signaling,23 both of which have been conventionally recognized as the consequences of CB1 receptor activation. More recent studies showed that the 1-induced activation of ERK1/2 signaling is Gi-independent24, 25 and mediated by β-arrestins.23 Hence, 1 not only acts as an allosteric agonist which is able to induce receptor internalization and ERK1/2 phosphorylation on its own, but also functions as an antagonist of CB1 agonist-induced [35S]GTPγS binding. This supports the notion that allosteric modulators can selectively regulate the functions of the CB1 receptor. Consequently, more functionally-defined pharmacological effects can be achieved for the physiologically and pharmacologically important CB1 receptors. Along with 1, a few other indole-2-carboxamides19, 24, 26 have also shown allosteric modulation of the CB1 receptor. To study the structure-activity relationships (SAR) of this class of compounds, we synthesized a series of analogs of 1 with the general structure shown in Figure 2, and individual structures shown in Schemes 1 and 2.
Figure 2. General structures of indole- and benzofuran-2-carboxamide analogs.

Scheme 1.

Syntheses of indole-2-carboxamide analogs (11a-l). Reagents and Conditions: (i) AlCl3, 1,2-dichloromethane, reflux, 2-3 h; (ii) (Et)3SiH, CF3COOH, 0 °C - rt, 4-12 h; (iii) NaOH, EtOH, reflux, 2 h; (iv) BOP, DIPEA, DMF, rt, 4-12 h.
Scheme 2.

Syntheses of benzofuran-2-carboxamide analogs 13a-d. Reagents and Conditions: (i) BOP, DIPEA, DMF, rt, 4-12 h.
Previous results from us24 and others26 indicated that the alkyl substituents at the C3 position have a profound influence on the ligand's allostery. For instance, replacing compound 1's C3 ethyl group with a n-pentyl group led to the enhancement of the allosteric effects, which is reflected by an improvement in the cooperativity factor α from 6.95 (1) to 17.6 (11a).24 The α cooperativity factor is a value that quantifies the magnitude by which the affinity of one ligand is changed by the other ligand when both are bound to the receptor simultaneously to form the ternary complex. When α is 1.0, the test modulator does not affect the binding of the orthosteric ligand. If α is less than 1.0, the test modulator reduces orthosteric ligand binding (negative allosteric modulation on orthosteric ligand binding). If α is greater than 1.0, the modulator increases orthosteric ligand binding (positive allosteric modulation).27 This prompted us to continue the investigation of the C3 substituents. Therefore, alkyl groups of different lengths, a cyclic ring and an aromatic ring were introduced at the C3 position to elucidate the SAR. Since we previously found that 11a displayed a remarkably enhanced binding cooperativity factor α24, we chose to use it as a template to vary the amines of the amide side, with the aim of improving the affinity of the allosteric modulator to its allosteric site, which is denoted by its equilibrium dissociation constant (KB).27 The above efforts yielded compounds 11b-11k (Scheme 1, Table 1 and Table 2). Next, to elucidate the significance of the indole ring in the indole-2-carboxamide structure, we replaced the indole ring of 11a with other bicyclic aromatic rings such as the benzofuran ring. This effort led to the compounds represented by the structure 13a-13d (Scheme 2, Table 3).
Table 1. Allostery of indole-2-carboxamide analogs 11a-f.
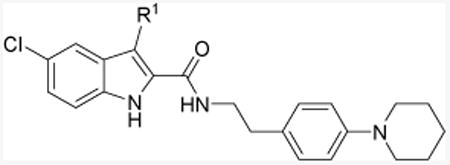
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | R1 | KB(nM)a | αb | [35S]GTPγS bindingyc |
| 1d | Et | 217.3 (170.3-277.2) | 6.95 | Antagonizing |
| 11ae | n-C5H11 | 469.9 (126.2-1750) | 17.60 | Antagonizing |
| 11b | Ph | ND | 1.0 | - |
| 11c | CH2Ph | 1067 (347.3-3344) | 11.37 | - |
| 11d | CH2-c-hexyl | 2408 (570.2-8151) | 9.84 | - |
| 11e | n-C7H15 | 651.2 (81.51-5203) | 7.38 | - |
| 11f | n-C9H19 | 259.7 (87.56-770) | 6.80 | Antagonizing |
KB: equilibrium disassociation constant.
α: binding cooperativity factor between CP55,940 and the tested allosteric modulators.
Effects on CP55,940-induced [35S]GTPγS binding in the presence of allosteric modulator.
Data cited for 1 is from our earlier report and given for comparison.23
Data cited for 11a is from our earlier report and given for comparison.24
ND: no detectable modulation of [3H]CP55,940 binding using up to 32 μM of test compound.
-: not performed.
Table 2. Allostery of indole-2-carboxamide analogs 11g-k.
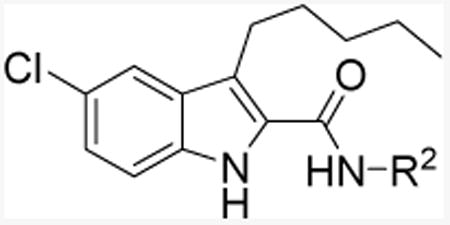
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | R2 | KB (nM)a | αb | [35S]GTPγS bindingc |
| 11ad |
|
469.9 (126.2-1750) | 17.06 | Antagonizing |
| 11g |
|
ND | 1.0 | - |
| 11h |
|
508.4 (96.34-2683) | 10.0 | Antagonizing |
| 11i |
|
2335 (695.3-7542) | 13.15 | - |
| 11j |
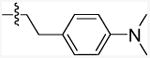
|
167.3 (23.39-1197) | 16.55 | Antagonizing |
| 11k |
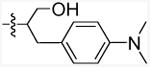
|
1018 (81.51-5203) | 13.27 | - |
KB: equilibrium disassociation constant.
α: binding cooperativity factor between CP55,940 and the tested allosteric modulators.
Effects on CP55,940-induced GTPγS binding in the presence of allosteric modulator.
Data cited for 11a is from our earlier report and given for comparison.24
ND: no detectable modulation of [3H]CP55,940 binding using up to 32 μM of test compound.
-: not performed.
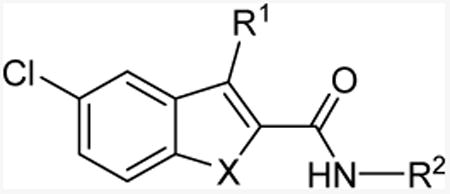
Table 3. Allostery of indole-/benzofuran-2-carboxamide analogs (11l and 13a-d).
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cmpd | X | R1 | R2 | KB(nM)a | αb | [35S]GTPγS bindingc |
| 1d | NH | Et |
|
217.3 (170.3-277.2) | 6.95 | Antagonizing |
| 11le | NH | Et |
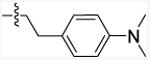
|
207.4 (155.9-2759) | 19.68 | - |
| 13a | O | H |
|
ND | 1.0 | - |
| 13b | O | Et |
|
2594 (1005-6696) | 18.04 | Antagonizing |
| 13c | O | Et |
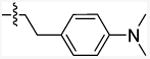
|
627.1 (151.5-2596) | 11.59 | - |
| 13d | O | H |
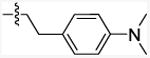
|
ND | 1.0 | - |
KB: equilibrium disassociation constant.
binding cooperativity factor between CP55,940 and the tested allosteric modulators.
Effects on CP55,940-induced GTPγS binding in the presence of allosteric modulator.
Data cited for 1 is from our earlier report and given for comparison.23
Compound 11l was reported earlier26 but was synthesized here and pharmacologically characterized for comparison with other analogs under the same conditions.
ND: no detectable modulation of [3H]CP55,940 binding using up to 32 μM of test compound.
-: not performed.
Chemistry
The syntheses of the indole-2-carboxamide analogs (11) were achieved through the methods illustrated in Scheme 1.
The C3 substituents were introduced through acylation of the commercially available ethyl 5-chloroindole-2-carboxylate (5) by Friedel-Crafts reaction catalyzed by aluminum chloride. Acylation of 5 with the selected acyl chloride (6) provided the desired 3-acyl-5-chloroindole-2-carboxylates (7). Reduction of the carbonyl group of 7 by triethylsilane generated the C3 alkylated 5-chloroindole-2-carboxylates (8), which were then hydrolyzed in basic conditions to yield the key intermediate acids (9). The final compounds (11a-11l) were prepared by coupling commercially available amines (10a-f) with the indole-2-carboxylic acids (9a-f) and commercially available 5-chloro-3-phenyl-2-carboxylic acid individually in the presence of BOP and diisopropylethyl amine (DIPEA) in anhydrous DMF at room temperature. Similarly, benzofuran-2-carboxamides (13a-d) were prepared according to the methods depicted in Scheme 2.
The commercially available 5-chlorobenzofuran-2-carboxylic acid 12a (AlfaAesar, Ward Hill, MA) or 5-chloro-3-ethyl-benzofuran-2-carboxylic acid 12b (Princeton BioMolecular Research, Monmouth Junction, NJ) was coupled with selected amines (10a or 10e) in the presence of BOP and DIPEA in anhydrous DMF at room temperature to provide the desired benzofuran-2-carboxamides (13a-13d). Most of the final compounds of the 11 and 13 series had acceptable solubility in organic solvents and were therefore purified through chromatography. However, compounds 11b, 11c and 11d had very poor solubility in most of the common organic solvents, such as acetone, ethyl acetate, tetrahydrofuran, dichloromethane, diethyl ether, ethanol and methanol. They were therefore purified using a combination of trituration, recrystallization and chromatography as detailed in the experimental section.
Results and Discussion
To investigate the allostery of the newly synthesized compounds against the CB1 receptor, we evaluated each compound's binding cooperativity factor α to quantify the magnitude of the allosteric effect on orthosteric ligand binding. Some of the compounds that showed α values comparable to that of 1 were selected for further testing their capability to modulate the agonist CP55,940-induced G protein coupling. Additionally, to evaluate the effectiveness of the modulator binding to the allosteric site, their equilibrium dissociation constants KB were also determined. The α and KB values were analyzed according to the allosteric ternary complex model.19 (See equation (1) in experimental section).
The results for compounds with structural variation at the C3 position are shown in Table 1. We found that replacing the C3 alkyl group of 1 with a phenyl ring completely abolished the allosteric effect of the ligand (11b compared to previously tested 123 and 11a24), whereas a benzyl group at C3 was somewhat tolerated but with a significantly increased KB value indicative of lower affinity (11c). Replacing the phenyl ring of the benzyl group in ligand 11c with a cyclohexyl group (11d) further increased KB although allosteric modulatory effects were retained. These data suggest that aliphatic or aromatic rings on the C3 alkyl group significantly decrease the affinity of the allosteric ligand. Therefore, linear alkyl chains as C3 substituents were employed for further exploration. The C3-n-heptyl (11e) and C3-n-nonyl (11f) analogs of 1 were synthesized. Although 11e and 11f displayed appreciable KB values, the α values substantially decreased relative to 11a, suggesting that a C3-n-pentyl is optimal for maximum allosteric modulatory effects.
Overall, this series of analogs indicates that the type and size of the substituents at the C3 position of indole-2-carboxamides have a critical role in the allosteric effects on the CB1 receptor. Our results support the hypothesis of the presence of a hydrophobic sub-pocket in close proximity of the C3-position of 1 when it is bound to the allosteric site. This sub-site could only be occupied by linear alkyl groups but not cyclic structures.
Among the C3-alkyl analogs of 1, the previously tested compound 11a24 exhibited the most robust allostery (α=17.6) of the CB1 receptor. In an earlier communication, we reported that 11a promoted the phosphorylation of CB1 downstream effectors ERK1/2 in a Gi-independent but β-arrestin-1 mediated manner.24 Although a markedly high α factor was achieved, 11a exhibited moderately weak affinity for CB1, as reflected by its relatively high KB value (469.9 nM). Hence, this compound was selected for further modification on the amide side with the aim of exploring SAR and improving the affinity to the allosteric site on CB1. This effort led to the synthesis of the compounds presented in Table 2.
When a methylene group of the ethylene linker between the piperidinylphenyl group and the amide bond was removed, it resulted in complete elimination of the allostery of the ligand (11g vs 11a). This indicates that the substituted phenyl ring on the amide side must be separated from the amide bond by at least a 2-carbon linkage. This ethylene moiety was therefore retained for the rest of this series of compounds (11h-11k). When the piperidinyl group was replaced by a structurally related but more hydrophilic morpholinyl group (11h), the KB value increased and the cooperativity factor α decreased. This suggests that the hydrophilic substitution on the amino group decreased the affinity of the ligand for the allosteric site and reduced the allosteric modulation of the orthosteric site. Replacing the piperidinyl group with a phenoxy group (11i) resulted in a significant increase in the KB value, but the high binding cooperativity (α) was retained. Next, swapping the cyclic piperidinyl group with an acyclic dimethyl amino group on the phenyl ring led to 11j which exhibited the most robust allostery (KB = 167.3 nM; α= 16.55) reported so far for the CB1 receptor. A low KB value and a high α factor, as achieved in 11j, are significant and indicate the low concentration of compound 11j that is needed to achieve the increase in affinity this allosteric modulator has on CP55,940 binding; i.e. a smaller concentration of 11j (compared to all CB1 allosteric modulators) is needed to achieve its robust allosteric effect on agonist binding. This is a key factor that is taken into consideration in therapeutic drug treatments. Moving forward to introduce a hydroxymethyl group at the α position of the ethylene linker of 11j reduced its affinity, but a relatively good binding cooperativity (α value) was maintained (11k). Overall, in this series of compounds, we found that structural modifications on the amide side generally do not significantly affect the cooperativity factor α, whereas the influence on the KB of the allosteric modulators is much more profound.
To date, the majority of structural modifications on 1 have been focused on the substitution of the indole ring19, 24 and the variation of the amide side.26 To unveil the importance of the indole ring on the KB and α values, we synthesized several benzofuran-2-carboxamides and compared them with their indole-2-carboxamide counterparts. The results are shown in Table 3.
We learned that replacing the indole ring of 1 with a benzofuran ring (13b) resulted in a significant loss of the affinity of the ligand (KB = 2594 nM), however, the binding cooperativity with the orthosteric ligand was markedly enhanced (13b vs 1). Similarly, replacing the indole ring of a potent CB1 allosteric modulator reported earlier (11l)26 with a benzofuran ring (13c) led to a 3-fold increase in KB while the high binding cooperativity (α=11.59) was well-preserved. These results suggest that the bicyclic aryl moiety (i.e. the indole ring, Figure 2) significantly impacts the ligand's affinity compared to its impact on the binding cooperativity. The indole ring, therefore, accounts more for the ligand's capability to bind to the allosteric site, but is less influential on the ligand's capability to modulate the orthosteric site. In the benzofuran-2-carboxamides, removal of the C3 alkyl group (i.e. 13c vs 13d) eliminated the allosteric effects completely.
To further characterize the nature of the compounds that show positive allosteric modulation on the binding of the CB1 orthosteric agonist [3H]CP55,940, compounds 11f, 11h, 11j and 13b were chosen for their strong allosteric properties (i.e. low KB value or high α value) as representatives from each series of analogs and further tested for their effects on CP55,940-induced and basal G protein coupling using a [35S]GTPγS binding assay. Figure 3 shows that 11f, 11h, 11j and 13b decrease agonist-induced G protein coupling to the CB1 receptor in a concentration-dependent manner similar to the effects exhibited by the previously reported 1, 2, 11a and 4.19, 20, 22-24 Remarkably, compound 11j exhibited a robust inhibitory effect in CP55,940-induced GTPγS binding (Figure 3C) evident by a progressive decrease in the Emax values with an increasing concentration of 11j. For example, at 0.1 μM of 11j, the CP55,940-induced [35S]GTPγS binding was substantially reduced (Emax = 53.4 ± 3.1 fmol/mg) compared with that in the absence of 11j (Emax = 113.0 ± 2.6 fmol/mg) while compounds 11f, 11h and 13b exhibited comparable levels of reduction at 3.2 μM and higher (Emax = 61.4 ± 2.0, 58.2 ± 1.07 and 59.5 ± 1.4 fmol/mg, respectively; Figure 3A, B and D). Treatment with 1 μM of 11j showed complete inhibition of CP55,940-induced [35S]GTPγS binding (Emax = 36.04 ± 2.0 fmol/mg) while at least 10 μM of 11f, 11h and 13b was needed to achieve a similar effect. Because of the robust allostery that compound 11j displayed, the basal levels of G protein coupling in the absence of CP55,940 (Figure 3E) were also examined. Compound 11j also antagonized the basal level of [35S]GTPγS in a concentration-dependent manner. Treatment with 10 μM of 11j resulted in complete inhibition of this basal activity. While this pattern of G protein coupling is consistent with an inverse agonist's profile in its ability to inhibit basal and CP55,940-induced GTPγS binding, when collectively taken with its enhancement of the orthosteric agonist CP55,940 binding, it indicates that these compounds are allosteric modulators that are likely able to selectively regulate the functions of the CB1 receptor. To date, compound 11j is the most effective allosteric antagonist of CB1 G protein coupling and surpasses both 1 and 11a which we also found inhibited G protein coupling at 3.2 μM.23, 24 Moreover, this pattern is consistent with the robust allostery of compound 11j as reflected by its binding parameters (Table 2).
Figure 3.

Dose-response curves for CP55,940-induced [35S]GTPγS binding to human embryonic kidney (HEK293) cell membranes expressing the CB1 wild-type receptor in the absence and presence of compounds 11f (A), 11h (B), 11j (C) and 13b (D) at the indicated concentrations. The basal level of [35S]GTPγS binding was also measured in the absence of any orthosteric ligand and the inhibition of it was tested at the indicated concentration of compound 11j (E). Data are presented as specific binding of GTPγS to the membranes. Non-specific binding was determined in the presence of 10 μM unlabeled GTPγS. Each data point represents the mean ± S.E. (error bars) of at least three independent experiments performed in duplicate. Statistical significance of the differences in each concentration of 11j compared to basal (i.e. no ligand) in (E) was assessed using one-way analysis of variance and Bonferroni's post-hoc test; ***, p < 0.005. The dotted line in (E) indicates the level of non CB1-mediated GTPγS binding obtained from [35S]GTPγS binding to the mock-transfected membrane sample.
A recent study of the CB1 receptor explored the mechanism of action of 1 using a site-directed fluorescence labeling approach.28 The results showed that the agonist CP55,940 induces a movement in the cytoplasmic end of TM6, which accompanies G protein coupling. Upon co-treatment with 1, this agonist-induced movement is blocked. This state of CB1 may explain how 1 and its analogs can elicit differential effects on CB1 agonist affinity and efficacy (i.e. a positive allosteric modulator of binding but an antagonist of G protein coupling activity).
Conclusion
We synthesized a group of analogs of the CB1 allosteric modulator 1 to understand the structure activity relationship of indole-2-carboxamides in allosteric modulation of the CB1 receptor. Within the structure of indole-2-carboxamides, the presence of the indole ring seems to be more influential on the ligand's binding affinity to the allosteric site than on the binding cooperativity (α) between the allosteric site and the orthosteric site. The C3 alkyl groups on the indole-2-carboxides profoundly impacted the allostery of the ligand. Through structural modification, we identified a robust CB1 allosteric modulator 11j which showed an equilibrium dissociation constant KB of 167.3 nM with a markedly high binding cooperativity factor (α=16.55) and potent inhibition of GTPγS binding. Although a small binding constant KB for the allosteric modulator is not necessary for maximum binding cooperativity with the CB1 orthosteric ligand, CB1 modulators with high binding affinities to the allosteric site and high cooperativity towards the orthosteric site are desirable for therapeutic applications.
In recent years, drug discovery targeting allosteric sites of GPCRs has gained momentum.29 This approach offers a number of potential advantages over drugs targeting the orthosteric site, such as improved selectivity across receptor subtypes and the capability to maintain the spatial and temporal signaling profile of the endogenous ligand. The nature of biased signaling of some CB1 allosteric modulators such as 123, 25, 11a,24 and 422 suggested a possibility to selectively regulate specific functions of the CB1 receptor. The activation and inactivation of the CB1 receptor have been linked to therapeutic implications for many diseases such as drug addiction, anxiety, depression, obesity, neurodegeneration, cancer and inflammation. Compound 1 has been the prototype to date for allosteric modulators of the CB1 receptor. Growing evidence indicates that 123, 25 and its analogs24 are functionally positive allosteric modulators of the CB1 receptor although they antagonize Gi-protein coupling activity to CB1. This class of compounds may selectively transduce the orthosteric agonist signals through the ERK1/2 phosphorylation pathway. Very recently, Pamplona et al demonstrated using in vivo experiments that the CB1 positive allosteric modulator 4 is protective against β-amyloid-induced neurotoxicity.22 The therapeutic potential of indole-2-carboximides represented by 1 as neuroprotective agents remains to be explored. Only recently, the explicit search for CB1 allosteric modulators has been initiated. Our work provides new insight into the structural requirements of CB1 allosteric modulators of the class of indole-2-carboxamides. It will aid the future design and synthesis of more robust allosteric modulators for the pharmacologically and physiologically vital CB1 receptor.
Experimental Section
Compounds
Tested compounds (11a-11l and 13a-13d) were synthesized for this study except 1, which was purchased from Tocris Bioscience (Minneapolis, MN). Compounds 11a19 and 11l26 have previously been reported and were used in this study for comparison.
CB1 Expression and Membrane Preparation
HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 3.5 mg/ml glucose at 37 °C in 5% CO2. For transient receptor expression, HEK293 cells were plated at 800,000 cells/100-mm dish on the day prior to transfection. The cells were transfected using the calcium phosphate precipitation method.30 24 h post-transfection, membranes of transfected cells were prepared as described previously.31
Radioligand Binding Assay
Membranes from HEK293 cells expressing the CB1 wild-type receptor were prepared as previously described.31 In assays used to determine the cooperativity between allosteric and orthosteric ligands, approximately 4 μg of membrane was incubated at 30 °C for 60 min with a fixed tracer ([3H]CP55,940; 147.9 Ci/mmol, PerkinElmer Life Sciences (Boston, MA)) concentration typically at its Kd (2.5 nM) which was determined from a saturation binding isotherm. Binding assays were performed with at least nine concentrations of unlabeled allosteric compound (ranging between 100 pM and 100 μM) as described previously.23 Nonspecific binding was determined in the presence of 1 μM unlabeled CP55,940. Reactions were terminated by adding 250 μL of TME buffer containing 5% BSA followed by filtration with a Brandel cell harvester through Whatman GF/C filter paper. Radioactivity was measured using liquid scintillation counting. The total assay volume and the amount of membrane samples were adjusted to avoid ligand depletion by keeping the bound ligand less than 10% of the total.
GTPγS Binding Assay
GTPγS binding assays were performed as described previously. Briefly, 7.5 μg of membranes were incubated for 60 min at 30°C in a total volume of 200 μL GTPγS binding assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl) with unlabeled CP55,940 (at least nine different concentrations were used ranging between 100 pM and 100 μM), 0.1 nM [35S]GTPγS (1250 Ci/mmol; PerkinElmer Life Sciences, Boston, MA), 10 μM GDP (Sigma, St. Louis, MO), and 0.1% (w/v) BSA in the absence and presence of varying concentrations of the allosteric compounds as indicated. The basal GTPγS binding was measured in the absence of orthosteric ligand and with 5 μM GDP to increase the window of basal activity. Nonspecific binding was determined with 10 μM unlabeled GTPγS (Sigma, St. Louis, MO). The reaction was terminated by rapid filtration through Whatman GF/C filters. The radioactivity trapped in the filters was determined by liquid scintillation counting.
Ligand and GTPγS Binding Data Analysis
All ligand binding assays and GTPγS binding assays were carried out in duplicate. Data are presented as the mean ± S.E. value or the mean with the corresponding 95% confidence limits from at least three independent experiments. The interactions between the orthosteric radiolabeled agonist [3H]CP55,940 and the test modulators were analyzed using Prism 6.0 (GraphPad Software Inc., San Diego, CA) according to the following allosteric ternary complex model of interaction.19
| Equation 1 |
Where Y denotes the specific bound orthosteric ligand divided by the total concentration of orthosteric ligand [A]. [B] denotes the concentration of the allosteric ligands. KA and KB are equilibrium dissociation constants for their respective ligands, and α is a cooperativity factor that quantifies the allosteric interaction in terms of magnitude and direction between the two ligands when they simultaneously occupy the receptor. When α is 1.0, the tested modulator does not change orthosteric ligand binding. If α is less than 1.0, the tested modulator reduces orthosteric ligand binding (negative allosteric modulation on orthosteric ligand binding). If α is greater than 1.0, the modulator increases orthosteric ligand binding (positive allosteric modulation). For this analysis, the value of [A] was kept constant and was the average of radioactive orthosteric ligand concentration employed in the binding assays. KA was also fixed as a constant and was determined from previous CP55, 940 saturation binding assays, whereas values of α and KB were determined by nonlinear regression.
Synthesis
All chemical reagents and solvents were purchased from Sigma-Aldrich Chemical Company unless specified otherwise and used without further purification. All anhydrous reactions were performed under a static argon atmosphere in dried glassware using anhydrous solvents. Organic phases in work up were dried over anhydrous Na2SO4, and removed by evaporation under reduced pressure. The crude compounds were purified by a Combiflash Rf chromatography system (Teledyne Technologies, Inc, Thousand Oaks, CA) unless specified otherwise. Purities of the intermediates were established by Thin-layer Chromatography (TLC), melting point, 1HNMR, and mass spectrometry. Analytical Thin-layer Chromatography (TLC) was run on pre-coated silica gel TLC aluminum plates (Whatman®, UV254, layer thickness 250 μm), and the chromatograms were visualized under ultraviolet (UV) light. Melting points were determined on a capillary Electrothermal melting point apparatus and are uncorrected. 1HNMR spectra of intermediates were recorded on a Bruker Avance DPX-300 spectrometer operating at 300 MHz. 1HNMR spectra of final compounds were recorded on a Bruker AV-500 spectrometer operating at 500 MHz. All NMR spectra were recorded using CDCl3 or DMSO-d6 as solvent unless otherwise stated and chemical shifts are reported in ppm (parts per million) relative to tetramethylsilane (TMS) as an internal standard. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broadened singlet) and coupling constants (J) are reported in hertz (Hz). Low resolution mass spectra were performed at the School of Chemical Sciences, University of Illinois at Urbana-Champaign. The purity of each tested compound was analyzed by combustion elemental analysis and was confirmed to be greater than 99%. Corresponding combustion elemental analysis was conducted in Roberson Microlit laboratories (Madison, NJ).
General procedure A: Preparation of ethyl 3-actyl-5-chloro-1H-indole-2-carboxylates (7a-7f)
The solution of ethyl 5-chloro-1H-indole-2-carboxylate (5; 10 mmol) in anhydrous 1,2-dichloroethane (25 mL) was added anhydrous aluminum chloride (10 mmol) powder and the selected acyl chloride liquid (6; 11.5 mmol) dropwise at room temperature. The reaction mixture was stirred and refluxed under argon atmosphere for 2-3 h. The completion of the reaction was monitored by TLC (25-33% ethyl acetate in hexane). Upon completion, the reaction mixture was poured into ice-cold water (150 mL) then treated with 4N HCl to pH 2. The mixture was extracted with ethyl acetate twice. The combined organic phase was washed with water and brine successively. The organic layer was separated and dried over anhydrous sodium sulfate. Filtration and removal of solvent in vacuo provided the corresponding crude product, which was purified by Combiflash chromatography (0-40% of ethyl acetate in hexane) and yielded the desired products 7a-7f.
General procedure B: Preparation of ethyl 3-alkyl-5-chloro-1H-indole-2-carboxylates (8a-8f)
Liquid triethylsilane (1.18 mL, 7.40 mmol) was added dropwise to the solution of ethyl 3-actyl-5-chloro-1H-indole-2-carboxylate (7; 3.76 mmol) in trifluoroacetic acid (2.88 mL) at 0 °C. The reaction mixture was then gradually warmed up to room temperature and stirred for 4-12 h. The reaction is monitored by TLC (30% of ethyl acetate in hexane). Upon reaction completion, the mixture was poured onto ice and treated with saturated sodium carbonate aqueous solution to pH 7, and was then extracted with ethyl acetate. The combined organic phase was washed with water and brine, and dried over anhydrous sodium sulfate. Filtration and removal of solvent provided the crude product, which was purified either by Combiflash silica gel column chromatography (0-30% of ethyl acetate in hexane) or recrystallization in the mixture of acetone and ethyl acetate (1:4) to yield the corresponding products 8a-8f.
General procedure C: Preparation of 3-alkyl-5-chloro-1H-indole-2-carboxylic acids (9a-9f)
The solution of ethyl 5-chloro-3-alkyl-1H-indole-2-carboxylate (8; 1.9 mmol) in anhydrous ethanol (20 mL) was added 3 N sodium hydroxide aqueous solution (3.8 mL, 11.44 mmol). The reaction mixture was stirred and refluxed for 2 h. Upon completion of the hydrolysis monitored by TLC (30% ethyl acetate in hexane), it was cooled to room temperature and treated with 0.1 N HCl to pH 2. White solid precipitated and was filtered. The cake was washed with a minimal amount of cold water and hexane (2 mL) and then dried in a vacuum oven over night to provide the white solid products (9a-9f).
General procedure D: Preparation of 5-chloroindole-2-carboxamides (11a-11l) or 5-chloro-benzofuran-2-carboxamides (13a-13d)
To the solution of 5-chloroindole-2-carboxylic acid (9; 0.68 mmol) or 5-chlorobenzofuran-2-carboxlic acid (12; 0.68 mmol), BOP (502 mg, 1.02 mmol), and N,N-diisopropylethylamine (0.71 mL, 4.08 mmol) in anhydrous DMF (4 mL) were added the solution of appropriate amine (10; 0.82 mmol) in 1 mL of anhydrous DMF. The reaction mixture was stirred at room temperature for 4-12 h. Upon completion of the coupling reaction, which was monitored by TLC (30% ethyl acetate in hexane), the mixture was poured into cold water (40 mL) and extracted with ethyl acetate twice. The organic layer was separated and washed with water, brine and dried over anhydrous sodium sulfate. Filtration and removal of solvent in vacuo provided the crude product, which was then purified either by trituration followed by recrystallization in the solvent specified in the supporting information or by flash chromatography to generate the final compounds (11a-11l) or (13a-13d) respectively.
5-Chloro-3-pentyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11a)
The title compound was reported earlier by us.24 It was resynthesized by the modified methods reported here to be used as a template for further modifications, and was obtained from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a, 214 mg, 0.81 mmol) and commercially available 2-(4-piperidin-1-yl-phenyl)-ethylamine (10a, Oakwood Chemical, 200 mg, 0.98 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-50% of ethyl acetate in hexane) and yielded 120 mg (32.8 %) of white solid. mp: 218-219 °C. 1H NMR (500 MHz, DMSO-d6): δ 11.32 (s, 1H), 7.94 (t, J = 5.5 Hz, 1H), 7.61 (bs, 1H), 7.39 (d, J = 8.6 Hz, 1H), 7.17 (dd, J = 8.6, 1.0 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 3.53 (m, 2H), 3.13 (t, J = 5.6 Hz, 4H), 3.00 (t, J = 7.5 Hz, 2H), 2.75 (t, J = 7.5 Hz, 2H), 1.69-1.64 (m, 4H), 1.59-1.54 (m, 4H), 1.36-1.28 (m, 4H), 0.90 (t, J = 7.5 Hz, 3H). MS (EI): m/z = 451.2 (M+). Anal. calcd. for (C27H34ClN3O): C, 71.74; H, 7.58; N, 9.30; found: C, 71.78; H, 7.57; N, 9.04.
5-Chloro-3-phenyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11b)
The title compound was prepared from 5-chloro-3-phenyl-1H-indole-2-carboxylic acid (9b; 200 mg, 0.74 mmol) and 2-(4-piperidin-1-yl-phenyl)-ethylamine (10a, Oakwood Chemical, 180 mg, 0.88 mmol) according to the general procedure D. The crude product (470 mg) was triturated sequentially with acetone (5 mL) and diethyl ether (5 mL) to yield 200 mg of light-yellow colored solid which was then purified by Combiflash chromatography (0-50% of ethyl acetate in hexane) to yield 57 mg of light-yellow colored solid. The mother solution was concentrated and purified by Combiflash chromatography (0-50 % of ethyl acetate in hexane) to yield 86 mg of light-yellow colored solid. Hence, a total of 143 mg (42.2 %) of solid was collected. mp: 183-185 °C. 1H NMR (500 MHz, Chloroform-d): δ 9.54 (bs, 1H), 7.46-7.42 (m, 3H), 7.40-7.32 (m, 4H), 7.25 (dd, J = 9.0 Hz, 1.5 Hz, 1H), 6.86 (d, J = 10.0 Hz, 2H), 6.81 (d, J = 10.0 Hz, 2H), 6.02 (t, J = 5.3 Hz, 1H), 3.54 (q, J = 6.4 Hz, 2H), 3.12 (t, J = 6.5 Hz, 4H), 2.63 (t, J = 6.7 Hz, 2H), 1.74-1.69 (m, 4H), 1.60-1.55 (m, 2H). MS (EI): m/z = 457.2 (M+). Anal. Calcd. for (C28H28ClN3O·½H2O): C, 72.01; H, 6.26; N, 9.00; found: C, 72.27; H, 6.07; N, 8.67.
3-Benzyl-5-chloro-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11c)
The title compound was synthesized from 3-benzyl-5-chloro-1H-indole-2-carboxylic acid (9c; 200 mg, 0.7 mmol) and commercially available 2-(4-(piperidin-1-yl)phenyl)-ethylamine (10a, Oakwood Chemical, 172 mg, 0.84 mmol) according to the general procedure D. The obtained crude product was triturated with methanol to provide 100 mg of off-white solid. The mother solution was then concentrated and yielded a light-red solid, which was then purified by Combiflash chromatography (0-40% of ethyl acetate in hexane) and yielded 36 mg of white solid, which was recrystallized in hot ethyl acetate (6.5 mL) to yield 23 mg of white solid. Therefore, a total of 123 mg (37.2 %) of the desired product was isolated. mp: 180-182 °C. 1HNMR (500 MHz, DMSO-d6): δ 11.4 (s, 1H), 8.04 (t, J = 5.5 Hz, 1H), 7.46 (s, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.19-7.14 (m, 4H), 7.11 (dd, J = 8.7 Hz, 2 Hz, 1H), 7.07-7.03 (m, 1H), 7.0 (d, J = 8.2 Hz, 2H), 6.76 (d, J = 7.9 Hz, 2H), 4.30 (s, 2H), 3.41(q, J = 6.8 Hz, 2H), 2.98 (t, J = 5.7 Hz, 4H), 2.67 (t, J = 7.3 Hz, 2H), 1.50-1.55 (m, 4H), 1.41-1.45 (m, 2H). MS (EI): m/z = 471.2 (M+). Anal. Calcd. for (C29H30ClN3O): C, 73.79; H, 6.41; N, 8.90; found: C, 73.76; H, 6.46; N, 8.83.
5-Chloro-3-(cyclohexylmethyl)-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11d)
The title compound was synthesized from 5-chloro-3-(cyclohexylmethyl)-1H-indole-2-carboxylic acid (9c; 200 mg, 0.69 mmol), and commercially available 2-(4-Piperidin-1-yl-phenyl)-ethylamine (10a, Oakwood Chemical, 168 mg, 0.82 mmol) according to the general procedure D. The obtained solid crude product was recrystallized in a hot solvent mixture of ethyl acetate and acetone (6:1) and provided 132 mg of white crystals. The mother solution was concentrated, and purified by Combiflash chromatography (0-50% of ethyl acetate in hexane) and yielded 46 mg of white solid. Thus, a total of 178 mg (54.0 %) of product was isolated. mp: 218-220 °C. 1HNMR (500 MHz, DMSO-d6): δ 11.35 (s, 1H), 7.94 (t, J = 5.5 Hz, 1H), 7.60 (s, 1H), 7.38 (d, J = 8.9 Hz, 1H), 7.17 (dd, J = 8.9 Hz, 2.0 Hz, 1H), 7.10 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.2 Hz, 2H), 3.48 (q, J = 6.8 Hz, 2H), 3.07 (t, J = 5.0 Hz, 4H), 2.83 (d, J = 7 Hz, 2H), 2.75 (t, J = 7.3 Hz, 2H), 1.62-1.58 (m, 6H), 1.56-1.48 (m, 6H), 1.13-1.02 (m, 3H), 0.99-0.88 (m, 2H). MS (EI): m/z = 477.2 (M+). Anal. Calcd. for (C29H36ClN3O) C, 72.86; H, 7.59; N, 8.79; found: C, 72.75; H, 7.56; N, 8.69.
5-Chloro-3-heptyl-N-(4-piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11e)
The title compound was prepared from ethyl-5-chloro-3-heptyl-1H-indole-2-carboxylic acid (9d; 200 mg, 0.62 mmol) and commercially available 2-(4-piperidin-1-ylphenyl)-ethylamine (10a; Oakwood Chemical, 208 mg, 1.02 mmol) according to the general procedure D. 115 mg (38.6 %) of white solid was isolated as product by Combiflash chromatography (0-40% of ethyl acetate in hexane). mp:178-180 °C. 1HNMR (500 MHz, Chloroform-d): δ 9.11 (bs, 1H), 7.52 (d, J = 1.5 Hz, 1H), 7.28 (d, J = 8.5 Hz, 1H), 7.19 (dd, J = 7.5 Hz, 1.5 Hz, 1H), 7.11 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 8.2 Hz, 2H), 6.03 (t, J = 5.3 Hz, 1H), 3.76 (q, J = 6.4 Hz, 2H), 3.11 (t, J = 5.5 Hz, 4H), 2.86 (t, J = 6.6 Hz, 2H), 2.68 (t, J = 7.8 Hz, 2H), 1.70 (quin, J = 11.0 Hz, 4H), 1.49-1.41 (m, 2H), 1.30-1.16 (m, 10H), 0.87 (t, J = 7.5 Hz, 3H). MS (EI): m/z = 479.2 (M+); Anal. Calcd. for (C29H38ClN3O): C, 72.55; H, 7.98; N, 8.75; found: C, 72.31; H, 8.01; N, 8.55.
5-Chloro-3-nonyl-N-(4-piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (11f)
The title compound was prepared from ethyl-5-chloro-3-nonyl-1H-indole-2-carboxylic acid (9e; 200 mg, 0.57 mmol) and commercially available 2-(4-piperidin-1-yl-phenyl)ethylamine (10a, Oakwood Chemical, 190 mg, 0.93 mmol) according to general procedure D. 116 mg (40 %) of white solid was isolated as product. mp: 148-150 °C. 1HNMR (500 MHz, Chloroform-d): δ 9.09 (bs, 1H), 7.54 (d, J = 1.5 Hz, 1H), 7.30 (d, J = 8.5 Hz, 1H), 7.21 (dd, J = 7.5 Hz, 1.5 Hz, 1H), 7.14 (d, J = 7.9 Hz, 2H), 6.99-6.91 (m, 2H), 6.03 (bs, 1H), 3.78 (q, J = 6.4 Hz, 2H), 3.20-3.09 (m, 4H), 2.88 (t, J = 6.6 Hz, 2H), 2.70 (t, J = 7.8 Hz, 2H), 1.80-1.66 (m, 4H), 1.65-1.54 (m, 2H), 1.52-1.42 (m, 2H), 1.32-1.15 (m, 12H), 0.89 (t, J = 7.5 Hz, 3H). MS (EI): m/z = 507.2 (M+); Anal. Calcd. for (C31H42ClN3O): C, 73.27; H, 8.33; N, 8.27; found: C, 73.49; H, 8.57; N, 8.09.
5-Chloro-3-pentyl-N-(4-(piperidin-1-yl)benzyl)-1H-indole-2-carboxamide (11g)
The title compound was prepared from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a, 232 mg, 0.87 mmol) and commercially available (4-(piperidin-1-yl)phenyl)methanamine (10b, 200 mg, 1.05 mmol) according to the general procedure D. 105 mg (27.6%) of white solid product was isolated by Combiflash chromatography (0-30% ethyl acetate in hexane). mp:196-198 °C; 1H NMR (500 MHz, Chloroform-d): δ 9.26 (bs, 1H), 7.56 (s, 1H), 7.31 (d, J = 8.2 Hz, 1H), 7.24 (d, J = 8.2 Hz, 2H), 7.23 (dd, J = 8.9, 2.1 Hz, 1H), 6.94 (d, J = 8.2 Hz, 2H), 6.27 (bs, 1H), 4.59 (d, J = 5.5 Hz, 2H), 3.16 (t, J = 5.5 Hz, 4H), 2.85 (t, J = 5.5 Hz, 2H), 1.79-1.67 (m, 4H), 1.65-1.55 (m, 2H), 1.33-1.20 (m, 6H), 0.84 (t, J = 5.5 Hz, 3H). MS (EI): m/z = 437.2 (M+). Anal. Calcd. for (C26H32ClN3O): C, 71.30; H, 7.36; N, 9.59; found: C, 71.06; H, 7.40; N, 9.37.
5-Chloro-N-(4-morpholinophenethyl)-3-pentyl-1H-indole-2-carboxamide (11h)
The title compound was synthesized from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a; 200 mg, 0.75 mmol), and commercially available 2-(4-morpholinophenyl)ethanamine (10c, Matrix Scientific, 186 mg, 0.9 mmol). The product was purified by Combiflash chromatography (0-60% ethyl acetate in hexane) and yielded 110 mg (32.3 %) of white solid. mp: 180-182 °C. 1H NMR (500 MHz, DMSO-d6): δ 11.25 (s, 1H), 7.87 (t, J = 5.5 Hz, 1H), 7.54 (s, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.10 (dd, J = 8.6 Hz, 1.6 Hz, 1H), 7.06 (d, J = 8.2 Hz, 2H), 6.81 (d, J = 8.2 Hz, 2H), 3.65 (t, J = 4.0 Hz, 4H), 3.41 (q, J = 6.9 Hz, 2H), 2.98 (t, J = 4.3 Hz, 4H), 2.87 (t, J = 7.5 Hz, 2H), 2.70 (t, J = 7.3 Hz, 2H), 1.43 (quin, J = 7.3 Hz, 2H), 1.25-1.12 (m, 4H), 0.77 (t, J = 7.3 Hz, 3H). MS (EI): m/z = 453.2 (M+). Anal. Calcd. for (C26H32ClN3O2): C, 68.78; H, 7.10; N, 9.26; found: C, 68.79; H, 7.01; N, 9.11.
5-Chloro-3-pentyl-1H-indole-2-carboxylic acid [2-(4-phenoxy-phenyl)-ethyl]-amide (11i)
The title compound was synthesized from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a; 200 mg, 0.75 mmol) and commercially available 2-(4-phenoxy-phenyl)ethylamine (10d, Matrix Scientific, 219 mg, 1.03 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-30% ethyl acetate in hexane) and yielded 122 mg (35 %) of white solid. mp: 143-145 °C. 1H NMR (500 MHz, Chloroform-d): δ 9.06 (bs, 1H), 7.55 (s, 1H), 7.34-7.29 (m, 2H), 7.38-7.34 (m, 2H), 7.23 (d, J = 8.2 Hz, 2H), 7.12 (td, J = 7.4, 1.1 Hz, 1H), 7.00 (t, J = 8.2 Hz, 4H), 6.02 (bs, 1H), 3.82 (q, J = 6.3 Hz, 2H), 2.96 (t, J = 6.3 Hz, 2H), 2.73 (t, J = 7.4 Hz, 2H), 1.51 (quin, J = 7.4 Hz, 2H), 1.35-1.19 (m, 4H), 0.88 (t, J = 7.4 Hz, 3H). MS (EI): m/z = 460.2 (M+). Anal. Calcd. for (C28H29ClN2O2): C, 72.95; H, 6.34; N, 6.08; found: C, 72.96; H, 6.31; N, 6.06.
5-Chloro-N-(4-(dimethylamino)phenethyl)-3-pentyl-1H-indole-2-carboxamide (11j)
The title compound was synthesized from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a; 200 mg, 0.75 mmol) and commercially available 4-(2-aminoethyl)-N,N-dimethylaniline (10e, ChemBridge, 148 mg, 0.90 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-20% ethyl acetate in hexane) and provided 119 mg (38.5 %) of white solid. mp: 183-185 °C; 1H NMR (500 MHz, CDCl3-d) δ 9.17 (bs, 1H), 7.53 (d, J = 1.2 Hz, 1H), 7.31 (d, J = 8.5 Hz, 1H), 7.22 (dd, J = 8.9 Hz, 1.2 Hz, 1H), 7.13 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 6.04 (bs, 1H), 3.79 (q, J = 6.2 Hz, 2H), 2.95 (s, 6H), 2.88 (t, J = 6.2 Hz, 2H), 2.70 (t, J = 7.8 Hz, 2 H), 1.47 (p, J = 7.8 Hz, 2H), 1.29-1.14 (m, 4H), 0.86 (t, J = 7.8 Hz, 3H); MS (EI): m/z = 411.3 (M+). Anal. calcd. for (C24H30ClN3O): C, 69.97; H, 7.34; N, 10.20; found: C, 69.82; H, 7.08; N, 10.08.
5-Chloro-N-(1-(4-(dimethylamino)phenyl)-3-hydroxypropan-2-yl)-3-pentyl-1H-indole-2-carboxamide (11k)
The title compound was synthesized from 5-chloro-3-pentyl-1H-indole-2-carboxylic acid (9a; 200 mg, 0.75 mmol) and 2-amino-3-(4-(dimethylamino)phenyl)propan-1-ol (10f, Matrix Scientific, 175 mg, 0.90 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-40% ethyl acetate in hexane) and provided 112 mg (33.8 %) of white solid. mp: 163-165 °C. 1H NMR (500 MHz, CDCl3-d): δ 8.97 (bs, 1H), 7.57 (d, J = 1.8 Hz, 1H), 7.29 (d, J = 8.9 Hz, 1H), 7.22 (dd, J = 8.9 Hz, 1.8 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 6.32 (d, J = 6.4 Hz, 1H), 4.43-4.35 (m, 1H), 3.88-3.75 (m, 2H), 3.01-2.96 (m, 2H), 2.94 (s, 6H), 2.93-2.83 (m, 2H), 2.82-2.68 (m, 4H), 1.31-1.25 (m, 2H), 1.18 (t, J = 7.8 Hz, 3H). MS (EI): m/z = 441.2 (M+). Anal. calcd. for (C25H32ClN3O2·¼H2O): C, 67.25; H, 7.34; N, 9.41; found: C, 67.39; H, 6.98; N, 9.14.
5-Chloro-N-(4-(dimethylamino)phenethyl)-3-ethyl-1H-indole-2-carboxamide (11l)
The title compound was reported earlier26 but was synthesized here and used for comparison with other analogs under the same assay conditions. It was prepared from 5-chloro-3-ethyl-indole-2-carboxylic acid (9f; 200 mg, 0.90 mmol) and commercially available 4-(2-aminoethyl)-N,N-dimethylaniline (10e, ChemBridge, 176 mg, 1.07 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-20% ethyl acetate in hexane) and yielded 140 mg (42 %) white solid. mp 150-152 °C. 1H NMR (500 MHz, Chloroform-d): δ 9.19 (bs, 1H), 7.55 (s, 1H), 7.31 (d, J = 8.5 Hz, 1H), 7.21 (dd, J = 8.5 Hz, 5.0 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 8.5 Hz, 2H), 6.02 (bs, 1H), 3.79 (q, J = 6.3 Hz, 2H), 2.94 (s, 6H), 2.89 (t, J = 6.3 Hz, 2H), 2.72 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H). MS (EI): m/z = 369.2 (M+). Anal. calcd for (C21H24ClN3O): C, 68.19; H, 6.54; N, 11.36. Found: C, 68.08; H, 6.43; N, 11.17.
5-Chloro-N-(4-(piperidin-1-yl)phenethyl)benzofuran-2-carboxamide (13a)
The title compound was prepared from commercially available 5-chlorobenzofuran-2-carboxylic acid (12a, AfarAesa, 192 mg, 0.98 mmol) and 2-(4-(piperidin-1-yl)phenyl)ethanamine (10a; Oakwood Chemical, 241 mg, 1.18 mmol) according to the general procedure D. The crude product was purified by Combiflash chromatography (0-40% ethyl acetate in hexane) and yielded 56.2 mg (15 %) of white solid. mp: 184-186 °C. 1H NMR (500 MHz, Chloroform-d): δ 7.65 (d, J = 1.8 Hz, 1H), 7.43-7.34 (m, 2H), 7.36 (dd, J = 10 Hz, 1.5 Hz, 1H), 7.14 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.2 Hz, 2H), 6.64 (bs, 1H), 3.71 (q, J = 6.7 Hz, 2H), 3.23-3.10 (m, 4H), 2.87 (t, J = 6.7 Hz, 2H), 1.79-1.67 (m, 4H), 1.66-1.45 (m, 2H). MS (EI): m/z = 382.0 (M+). (C22H23ClN2O2·½H2O): C, 67.43; H, 6.17; N, 7.15; found: C, 67.46; H, 6.51; N, 6.79.
5-Chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)benzofuran-2-carboxamide (13b)
The title compound was synthesized from commercially available 5-chloro-3-ethylbenzofuran-2-carboxylic acid (12b, Princeton BioMolecular Research, 200 mg, 0.89 mmol) and 2-(4-(piperidin-1-yl)phenyl)ethanamine (10a, Oakwood Chemical, 270 mg, 1.32 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-40% ethyl acetate in hexane) and provided 223 mg (61.0 %) of white solid. mp: 135-137 °C. 1H NMR (500 MHz, Chloroform-d): δ 7.61 (d, J = 1.5 Hz, 1H), 7.41-7.34 (m, 2H), 7.15 (d, J = 7.3 Hz, 2H), 6.99-6.86 (m, 2H), 6.66 (t, J = 5.5 Hz, 1H), 3.68 (q, J = 6.7 Hz, 2H), 3.23-3.09 (m, 6H), 2.87 (t, J = 7.0 Hz, 2H), 1.80-1.66 (m, 4H), 1.64-1.50 (m, 2H), 1.31 (t, J = 7.5 Hz, 3H). MS (EI): m/z = 410.1 (M+). Anal. calcd. for (C24H27ClN2O2): C, 70.15; H, 6.62; N, 6.82; found: C, 70.14; H, 6.58; N, 6.79.
5-Chloro-N-(4-(dimethylamino)phenethyl)-3-ethylbenzofuran-2-carboxamide (13c)
The title compound was prepared from commercially available ethyl-5-chloro-3-ethyl-1H-benaofuran-carboxylic acid (12b, Princeton BioMolecular Research, 200 mg, 0.89 mmol) and 4-(2-aminoethyl)-N,N-dimethylaniline (10e, ChemBridge, 175 mg, 1.07 mmol) according to the general procedure D. The product was purified by Combiflash chromatography (0-30% ethyl acetate in hexane) and provided 118 mg (35.8 %) of white solid. mp: 112-114 °C. 1H NMR (500 MHz, Chloroform-d): δ 7.61 (d, J = 1.2 Hz, 1H), 7.35-7.33 (m, 2H), 7.14 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 6.68 (bs, 1H), 3.67 (q, J = 6.7 Hz, 2H), 3.13 (q, J = 7.6 Hz, 2H), 2.94 (s, 6H), 2.85 (t, J = 6.6 Hz, 2H), 1.3 (t, J = 7.6 Hz, 3H). MS (EI): m/z =370.2 (M+). Anal. calcd. for (C21H23ClN2O2): C, 68.01; H, 6.25; N, 7.55; found: C, 67.95; H, 6.18; N, 7.45.
5-Chloro-N-(4-(dimethylamino)phenethyl)benzofuran-2-carboxamide (13d)
The title compound was prepared from commercially available 5-chlorobenzo[b]furan-2-carboxylic acid (12a, AlfaAesar, 200 mg, 1.02 mmol) and 4-(2-aminoethyl)-N,N-dimethylaniline (10e, ChemBridge, 201 mg, 1.22 mmol). The product was purified by Combiflash chromatography (0-30% ethyl acetate in hexane) and provided 77 mg (22.0 %) of white solid. mp: 177-181 °C. 1H NMR (500 MHz, Chloroform-d): δ 7.65 (d, J = 1.8 Hz, 1H), 7.38-7.42 (m, 2H), 7.36 (dd, J = 10 Hz, 1.8 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 8.5 Hz, 2H), 6.66 (bs, 1H), 3.71 (q, J = 6.6 Hz, 2H), 2.95 (s, 6H), 2.87 (t, J = 6.6 Hz, 2H). MS (EI): m/z = 342.2 (M+). Anal. calcd. for (C19H19ClN2O2): C, 66.57; H, 5.59; N, 8.17; found: 66.81; H, 5.44; N, 7.97.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant DA020763 (to D.A.K.) and Faculty Development Fund by Texas A&M Health Sciences Center (to D.L.).
Abbreviations
- CB1
cannabinoid receptor type 1
- CB2
cannabinoid receptor type 2
- SAR
structure activity relationship
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)triphosphate
- ERK
extracellular signal-regulated kinases
- cAMP
3′-5′-cyclic adenosine monophosphate
- BOP
(benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- DIPEA
diisopropylethyl amine
- DMF
dimethylformamide
- rt
room temperature
- HEK293
Human Embryonic Kidney 293 cells
- BSA
Bovine serum albumin
- TME
Tris-Mg2+- EDTA
- EGTA
ethylene glycol tetraacetic acid
- TLC
thin-layer chromatography
- MS
mass spectrometry
Footnotes
Supporting Information Available: Details of synthesis and compound characterization of intermediates 7a-f to 9a-f can be found in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Battista N, Di Tommaso M, Bari M, Maccarrone M. The endocannabinoid system: an overview. Front Behav Neurosci. 2012;6:1–7. doi: 10.3389/fnbeh.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerard CM, Mollereau C, Vassart G, Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J. 1991;279:129–134. doi: 10.1042/bj2790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straiker AJ, Maguire G, Mackie K, Lindsey J. Localization of cannabinoid CB1 receptors in the human anterior eye and retina. Invest Ophthalmol Vis Sci. 1999;40:2442–2448. [PubMed] [Google Scholar]
- 5.Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 6.Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(1):345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 7.Pertwee RG. Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. 2006;147(1):S163–171. doi: 10.1038/sj.bjp.0706406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 9.Rajesh M, Mukhopadhyay P, Hasko G, Pacher P. Cannabinoid CB1 receptor inhibition decreases vascular smooth muscle migration and proliferation. Biochem Biophys Res Commun. 2008;377:1248–1252. doi: 10.1016/j.bbrc.2008.10.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzman M, Sanchez C, Galve-Roperh I. Cannabinoids and cell fate. Pharmacol Ther. 2002;95:175–184. doi: 10.1016/s0163-7258(02)00256-5. [DOI] [PubMed] [Google Scholar]
- 11.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 14.Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- 15.Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17:1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;160:454–466. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pertwee RG. Pharmacology of cannabinoid receptor ligands. Curr Med Chem. 1999;6:635–664. [PubMed] [Google Scholar]
- 18.Hudson BD, Hebert TE, Kelly ME. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- 19.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 20.Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pamplona FA, Ferreira J, Menezes de Lima O, Jr, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R, Monory K, Lutz B, Canetti C, Matias I, Calixto JB, Marsicano G, Guimaraes MZ, Takahashi RN. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci U S A. 2012;109:21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn KH, Mahmoud MM, Samala S, Lu D, K DA. Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J Neurochem. 2013;124:584–589. doi: 10.1111/jnc.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, Ross RA. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piscitelli F, Ligresti A, La Regina G, Coluccia A, Morera L, Allara M, Novellino E, Di Marzo V, Silvestri R. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB(1) receptor. J Med Chem. 2012;55:5627–5631. doi: 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- 27.Christopoulos A, Kenakin TG. protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 28.Fay JF, Farrens DL. A key agonist-induced conformational change in the cannabinoid receptor CB1 is blocked by the allosteric ligand Org 27569. J Biol Chem. 2012;287:33873–33882. doi: 10.1074/jbc.M112.352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller CE, Schiedel AC, Baqi Y. Allosteric modulators of rhodopsin-like G protein-coupled receptors: Opportunities in drug development. Pharmacol Ther. 2012;135:292–315. doi: 10.1016/j.pharmthera.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn KH, Bertalovitz AC, Mierke DF, Kendall DA. Dual role of the second extracellular loop of the cannabinoid receptor 1: ligand binding and receptor localization. Mol Pharmacol. 2009;76:833–842. doi: 10.1124/mol.109.057356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.