

Abstract
Microglia polarization to the classical M1 activation state is characterized by elevated pro-inflammatory cytokines; however, a full profile has not been generated in the early stages of a sterile inflammatory response recruiting only resident microglia. We characterized the initial M1 state in a hippocampal injury model dependent upon tumor necrosis factor (TNF) receptor signaling for dentate granule cell death. Twenty-one-day-old CD1 male mice were injected with trimethyltin (TMT 2.3 mg/kg, i.p.) and the hippocampus was examined at an early stage (24-h post-dosing) of neuronal death. Glia activation was assessed using a custom quantitative nuclease protection assay (qNPA). We report elevated mRNA levels for glia response such as ionizing calcium-binding adapter molecule-1 and glial fibrillary acidic protein, (Gfap); Fas, hypoxia inducible factor alpha, complement component 1qb, TNF-related genes (Tnf, Tnfaip3, Tnfrsfla); interleukin -1 alpha, Cd44, chemokine (C-C motif) ligand (Ccl)2, Cc14, integrin alpha M, lipocalin (Lcn2), and secreted phosphoprotein 1 (Spp1). These changes occurred in the absence of changes in matrix metalloproteinase 9 and 12, neural cell adhesion molecule, metabotropic glutamate receptor (Grm)3, and Ly6/neurotoxin 1 (Lynx1), as well as, a decrease in neurotrophin 3, glutamate receptor subunit epsilon (Grin)-2b, and neurotrophic tyrosine kinase receptor, type 3. The M2 anti-inflammatory marker, transforming growth factor beta-1 (Tgfb1) was elevated. mRNAs associated with early stage of injury-induced neurogenesis including fibroblast growth factor 21 and Mki67 were elevated. In the “non-injured” temporal cortex receiving projections from the hippocampus, Lynx1, Grm3, and Grin2b were decreased and Gfap increased. Formalin fixed-paraffin-embedded tissue did not generate a comparable profile.
Keywords: M1 microglia, neuroinflammation, trimethyltin, tumor necrosis factor, microglia polarization
1. Introduction
As resident macrophages of the brain, microglia serve a key role in brain injury and neurodegenerative disorders. Within injured tissue, microglia exist in various states of reactivity/activation and retain their capability to shift their functional phenotype during specific stages of the inflammatory response (Stout et al., 2005; Graeber, 2010). This flexibility ensures a well-maintained glial inflammatory response and return to homeostasis. In vivo, the transformation of resident microglia into a phagocytic phenotype is strictly regulated and occurs in response to stimuli such as, cell death, accumulated debris, excess aberrant protein, or the presence of viral or bacterial pathogens (Kettenmann et al., 2011; Sierra et al., 2013). Upon the removal of such stimuli, or in the presence of a down-regulating signal, microglia commonly return to a ramified, non-reactive phenotype (Kettenmann et al., 2011). It is likely that the diverse morphological phenotypes of microglia observed over the course of an injury response are driven by the stage of participation in the cytotoxic response, immune regulation, or injured-tissue resolution. In efforts to characterize functional changes of microglia and activation state, staging, as based upon peripheral macrophage activation, has been proposed as a method to describe brain macrophage activation (Colton and Wilcock, 2010). For example, polarization of microglia to the classical activation state (M1) is characterized by high expression levels of the pro-inflammatory cytokines (interleukin (IL)-1β, IL-12, IL-23, and tumor necrosis factor alpha (TNFα), as well as inducible nitric oxide synthase (iNOS), CD40, major histocompatibility complex (MHC) I, and MHC II (Colton and Wilcock, 2010; Durafourt et al., 2012; Chhor et al., 2013; Jang et al., 2013). Transition or polarization to an M2 state, as experimentally induced by IL-4 and IL-10, is associated with repair and remodeling activities of macrophages (Colton and Wilcock, 2010).
Much of this proposed M1/M2 characterization of microglia is based upon classical activation by bacterial or viral fragments, invading microorganisms, or direct application of specific cytokines. As characterized in peripheral macrophages, inflammation, in the absence of microorganisms, has been termed “sterile inflammation” (Chen and Nunez, 2010). Such a response involves macrophage recruitment and production of the pro-inflammatory cytokines, TNFα and IL-1, and most likely involves activation of iNOS (Mantovani et al., 2005; Colton and Wilcock, 2010). Given the nature of a sterile inflammatory response, it is likely to be a primary form of neuroinflammation associated with neurodegenerative disease, trauma, or chemical-induced injury. Characterization of the early M1 sterile inflammatory response of microglia is currently in an infancy state; however, efforts to identifying activation state biomarkers are rapidly advancing. With regards to brain injury or neurodegenerative disorders, additional research is needed which focuses on the spectrum of events associated with a sterile inflammation as they occur in regions of cell death/degeneration and subsequent repair. Such information would contribute to the identification of targets and molecular triggers to regulate the inflammatory response.
Glial reactivity and neuroinflammation often accompany brain injury and neurodegenerative disorders and, while the causal nature of the response remains in question, the reactivity, per se, has been considered as a surrogate marker for neurotoxicity (Kreutzberg, 1996; Eng et al., 2000; O’Callaghan and Sriram, 2005; Panickar and Norenberg, 2005; Sofroniew, 2005; Streit et al., 2005). Results from studies examining the cascade of responses occurring across different brain regions with multiple types of injury suggest the occurrence of a common set of injury-induced changes associated with glial-inflammatory reactions (Sofroniew, 2009; Zhang et al., 2010; Little et al., 2012). However, exact characterization of a distinct M1 state of resident microglia is sparse. Using a model of chemical-induced brain injury localized to the hippocampus, we now provide data on the M1 polarization occurring during early stages of neuronal death and microglia activation.
In the mouse, an acute systemic injection of the hippocampal toxicant, trimethyltin (TMT), induces hippocampal damage that is characterized by localized dentate granule cell death with a sparing of Cornu Ammonis (CA) pyramidal neurons (Reuhl and Cranmer, 1984; Bruccoleri et al., 1998; Geloso et al., 2002; Harry et al., 2008). The associated resident microglia response coincides with an elevation of mRNα levels for the M1 associated cytokines, Tnfa and Il1a (Bruccoleri et al., 1998; Harry et al., 2002; Figiel and Dzwonek, 2007; Harry et al., 2008; Funk et al., 2011), and Ccl2 (Bruccoleri et al., 1998). This response is limited to resident microglia and occurs in the absence of infiltrating blood-borne monocytes (Funk et al., 2011), interferon-γ expression, or iNOS activation (Bruccoleri et al., 1998; Bruccoleri and Harry, 2000). However, a causal role for the pro-inflammatory cytokine, TNFα, and TNF receptor activation in the neuronal apoptosis induced by TMT has been demonstrated using various pharmacological interventions and genetically-modified mice (Harry and Lefebvre d’Hellencourt, 2003; Harry et al., 2008). With this injury model, a quantitative nuclease protection assay (qNPA) was used to assess gene changes in the mouse hippocampus occurring during the time of neuronal death and TNFα elevation as previously identified (Bruccoleri et al., 1998; Harry et al., 2002; 2008). A primary advantage of the qNPA is its multiplex format to hybridize specific mRNA probes to many different target transcripts within one sample aliquot without requiring RNA extraction, cDNA synthesis, or gene amplification. The assay allows for measurement of mRNA transcripts by direct 1:1 binding of a complementary DNA sequence and quantitation by chemiluminescence (Pechhold et al., 2009). A targeted qNPA containing mRNA probes for genes associated with various aspects of cellular responses to injury was developed for this study and included genes related to cell death, glutamate activation, inflammation, glial activation, and development/cell adhesion. The selection was made with the expectation that the profile generated would help to identify underlying mechanisms and affected cellular processes. With TMT-induced injury, the qNPA identified elevations in structural related genes associated with glia and in inflammatory genes associated with microglia reactivity that had not been previously characterized as M1-related. The profile of change observed in the hippocampus showed region specificity when compared to changes observed in the temporal cortex. Since the qNPA platform has been used for molecular classification of B-cell lymphomas in formalin-fixed paraffin embedded tissue (FFPE; Rimsza et al., 2011), we examined the feasibility of using this method for generating a molecular profile from fixed brain tissue. We found significant limitations that raised questions regarding the applicability of this method for such assessments.
2. METHODS
2.1. Animals and Dosing
At postnatal day 20, pathogen-free CD-1 male mice (Charles River Laboratories, Raleigh, NC) were singular housed in plastic cages with shaved hardwood bedding that included Nestlett® nesting material and one-third of original bedding to minimize stress. Animals were maintained within a semi-barrier animal facility (21°±2°C; 50%±5% humidity; 12-h light/dark cycle; 6:00-18:00). Food (NIH 31) and deionized, distilled drinking water were available ad libitum. On PND 21, mice received a single intraperitoneal (i.p.) injection of either vehicle (sterile 0.9% NaCl) or 2.3 mg/kg trimethyltin hydroxide (TMT; Alfa Products, Danvers, MA) in a dosing volume of 2 ml/kg body weight using a 50μl Hamilton syringe. The injection was delivered 4 h into the light period of a 12-h light/dark cycle. This dose level of TMT has been well characterized to induce apoptotic death of dentate granule neurons and microglia activation within 24 h post-injection (Harry et al., 2008). Thus, at this time point (24 h post-injection) mice were deeply anesthetized with CO2, decapitated, and the brain excised. No morbidity was observed under these dosing conditions. All procedures complied with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the National Institute of Environmental Health Sciences.
2.2. Confirmation of Hippocampal Damage and Glial Response
The TMT model of hippocampal damage has been well established in the literature. However, to confirm the severity of damage and the associated glia response occurring in the same cohort as animals used for molecular analysis, a randomly-selected subset of the dosing cohort (2 vehicle control and 6 TMT dosed) was selected for histological examination at 24 h post-injection. The excised brains were dissected in the mid-sagittal plane and immersion fixed in 4% paraformaldehyde (PFA)/phosphate buffer (PB; pH 7.2) overnight at room temperature (RT). After rinsing in PB, tissue was processed for paraffin embedding. Rehydrated and ethanol cleared 8μm paraffin sections through the hippocampus were collected and mounted on Superfrost® Plus slides (Fisher Scientific; Pittsburgh, PA).
Cells undergoing apoptotic cell death were identified by immunohistochemistry of cleaved caspase 3 (active caspase 3). Rehydrated and ethanol cleared sections were incubated with 3% H2O2 for 15 min at RT, washed at RT in two changes of 1X Wash Buffer (Biocare Medical; Concord, CA) for 5 min each, and subjected to heat induced epitope retrieval (HIER) in 1X citrate buffer using a decloaking chamber (Biocare Medical). Slides were then washed at RT in 2 changes of 1X Wash Buffer for 5 min each and incubated in 10% normal donkey serum (Jackson ImmunoResearch; West Grove, PA) for 20 min at RT. Sections were incubated for 1 h at RT with a rabbit polyclonal antibody for cleaved caspase 3 (1:100; Biocare Medical; 1% BSA in tris buffered salts) and rinsed in 2 changes of 1X Wash Buffer (5 min RT). Donkey anti-rabbit IgG secondary antibody (1:500; Jackson ImmunoResearch) was applied to each section for 30 min at RT then rinsed with 2 changes of 1X Wash Buffer. Vectastain R.T.U. Elite label (Vector Laboratories, Burlingame, CA) was applied to each section for 30 min at RT then rinsed with 2 changes of 1X Wash Buffer. Active Caspase 3 positive cells were visualized with 3,3′-diaminobenzidine (DAB)-H2O2 (Dako Corporation, Carpinteria, CA) following a 5-min incubation.
Astrocytes were labeled with an antibody to glial fibrillary acidic protein (GFAP). Sections were treated with 3% H2O2 for 10 min then subjected HIER in 1X Reveal Decloaker using a decloaking chamber (Biocare Medical), allowed to cool to RT, and washed with PB for 20 min at RT. Sections were blocked with 2% normal goat serum/PB for 30 min at RT prior to 1 h incubation at RT to a polyclonal rabbit anti-cow GFAP (1:200; Dako Corporation). Sections were washed for 20 min with PB at RT, incubated with a secondary IgG antibody (20 min RT), rinsed, incubated for 30 min at RT in avidin-biotin complex (ABC) reagent (Vectastain Elite, Vector Laboratories), and rinsed with PB for 20 min at RT. The reaction was developed with DAB-H2O2 for 5 min at RT.
Microglia were identified by lectin binding using the method of Streit (Streit, 1990) with slight modification. HIER sections were pre-incubated for 20 min at RT with PB containing CaCl2 MgCl2, MnCl2 (0.1 mm each) and 0.1% Triton X-100 (Sigma-Aldrich; St. Louis, MO). Sections were incubated overnight at 4°C with horseradish peroxidase-conjugated lectin (1:100; Bandeiraea simplifolia BS-I, IB4; Sigma-Aldridge) in PB containing cations and Triton-X 100. Following overnight incubation, sections were rinsed in PB for 20 min at RT and sites containing bound peroxidase-lectin conjugates were developed with DAB-H2O2 following incubation for 5 min at RT. Following DAB, sections were rinsed in dH2O for 3 min at RT and counterstained with Harris hematoxylin for 30 sec, rinsed in tap water, dehyrated by an ethanol gradient, and coverslipped using Vecta Mount™ permanent mounting medium (Vector Laboratories). Immunohistochemical stained tissues were imaged at 40x magnification using an Aperio Scanscope T2 Scanner (Aperio Technologies, Inc. Vista, CA) and viewed using Aperio Imagescope v. 6.25.0.1117.
2.3. Tissue collection and preparation for HTG qNPA
Hippocampi were dissected from each hemisphere of excised brains from mice previously injected with either saline vehicle or TMT (n=7 per group). From one hemisphere, the dissected hippocampi were frozen and stored at −80°C. The temporal cortex was collected to represent a brain region in which TMT-induced neuronal death was not observed but one that receives projections from the hippocampus. Samples were shipped to High Throughput Genomics (HTG; Tucson, AZ) for analysis by qNPA. Tissue was weighed, placed in lysis buffer for a final tissue concentration of 50 mg/ml and ground using a plastic pestle. The tissue was heated to 95°C for 15-30 min until the tissue lysate was no longer viscous. Samples were diluted to a concentration of 4 ng/ml and subjected to HTG Molecular’s qNPA™ (Tucson, AZ).
From the contralateral hemisphere of the same brain used for fresh tissue analysis, the dissected hippocampi was immersion fixed in 10% formalin (in DEPC treated RNase-free water; Phoenix Biotechnologies; Encinitas, CA) at RT for 18 h, rinsed in PB, and processed for paraffin embedding using a RHS-1 Microwave Vacuum Histo-Processor (Milestone Medical Technologies, Inc, Kalamazoo MI). Serial 5μm paraffin sections through the entire hippocampus were collected on non-coated microscope slides from a 38°C waterbath. Prior to sectioning, the water-bath was washed with RNase Away (Life Technologies, Durham, NC), maintained under a UV light for 12 h, and filled with RNase-free water for section collection. The technician maintained RNase-free conditions throughout tissue sectioning, including the use of personal items such as, latex gloves, facial mask, and hair covering. A cleaning and decontamination procedure was conducted each day. Slides were shipped to HTG for processing and analysis. For each section, paraffin around the tissue was etched and sections were scraped from the slides into a microcentrifuge tube in a manner to minimize collection of paraffin. Multiple sections from each individual animal were pooled into a tube for a final concentration of 0.12 cm2 of the 5μm tissue sections/ml. A 25 μl aliquot was added to each well. Denaturation oil (500 μl) was added to each sample and incubated at 95°C for 15 min. Proteinase K (Sigma-Aldridge) was added to a final concentration of 1 mg/ml of lysate and incubated at 50°C for 30 min, followed by incubation at 95°C for 15 min to destroy the Proteinase K activity.
2.4. qNPA Array
A specialized array of 50-mer probes was designed for the detection of 44 selected genes of interest in neural tissue samples using a 96-well plate format for a quantitative nuclease protection assay (qNPA; Table 1). The qNPA assay provides a multiplex format to hybridize specific mRNA probes to many different target transcripts within one sample aliquot (High Throughput Genomics, Tucson, AZ). This method is based upon the direct ribonuclease protection assay approach and allows for optimization of conditions to detect low-copy signals as can occur in the brain with inflammatory related genes. To identify genes of interest for inclusion on the qNPA array, various sources of data were evaluated: 1) gene expression data from other models of brain injury, 2) protein and gene expression studies conducted on hippocampal tissue from TMT-dosed animals, and 3) pilot data from an Agilent Mouse Oligo array (Agilent Technologies, Palo Alto, CA) of the hippocampus at 24 h post-TMT. The qNPA probe selection represented numerous aspects of inflammatory responses, both pro-inflammatory and anti-inflammatory, and included many of the interleukins, chemokines, matrix metalloproteinases (MMP), and growth factors. Aspects of the cell-injury response were represented with the inclusion of probes for genes associated with glia, heat shock proteins, oxidative stress, hypoxia, and apoptosis. For a number of these genes, our previous work provided and existing database on mRNA and protein changes to allow for comparison.
Table 1. Hippocampal qNPA mRNA expression levels.
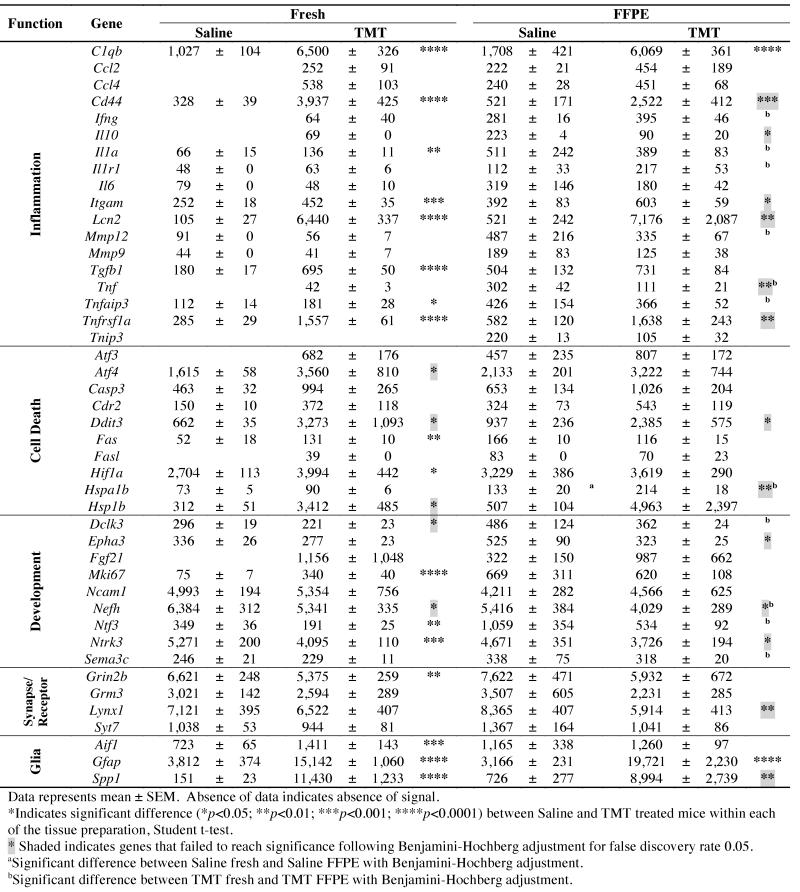
|
The assays were conducted at High Throughput Genomics (Tucson, AZ) using methods as previously reported (Rimsza et al., 2011). Briefly, 25 μl sample lysates representing 100 ng tissue were incubated with a mixture of gene specific probes to form probe-mRNA duplexes. Non-hybridized probes were digested by S1 nuclease followed by alkaline hydrolysis to eliminate mRNA in duplexes. The remaining intact qNPA probes were proportional to the abundance of specific mRNA in neural lysates. Samples were transferred for probe detection to array plates (96 well) in which each well contained plate-bound 50-mer oligonucleotides specifically designed to capture each specific gene probe. The plate contained both fresh and fixed hippocampus from both saline and TMT injected mice (n=7 per group) for equal representation of all conditions.
Each well contained an array of 47 probes consisting of 44 genes of interest, 2 housekeeping genes, and 1 negative control gene (a plant gene, ANT). After binding of the detection probes with horseradish peroxidase and the addition of F substrate, a chemluminescent signal was detected for quantification. Signals were simultaneously imaged from the plate bottom with a CCD-based Omix Imager (HTG) and analyzed using Vuescript software (HTG). Based upon a pilot study examining 30 potential housekeeping genes, Actb (beta actin) and Ppia (peptidylprolyl isomerase A (cyclophilin A) were selected on the basis of expression level and low variance in brain tissue across various ages. mRNA levels of the genes of interest were normalized to housekeeping genes for fold-change comparisons. The mean, standard deviation, and coefficient of variance (CV) were determined across triplicate wells of each sample. Coefficient of variance for each gene ranged between 2 and 40%, with an average of 13% for both groups. Individual genes showing a CV of greater than 30% were normally associated with low signal intensity. The means of sample triplicates were used for statistical analysis. 2.5. Statistical Analysis mRNA levels of each gene, were analyzed by Student’s t-tests for comparisons between Saline/FFPE and Saline/fresh or between TMT/FFPE and TMT/fresh tissue groups. A Benjamini-Hochberg (Benjamini and Hochberg, 1995) procedure was applied separately to the fresh tissue group and to the FFPE tissue group, using a false discovery rate (FDR) of 0.05. Student t-tests were used to determine the influence of tissue preparation with tests run within each dose group for comparisons between fresh and FFPE tissue and a Benjamini-Hochberg procedure applied.
3. RESULTS
3.1. Confirmation of Hippocampal Injury
Prior to molecular analysis, hippocampal damage was confirmed in the TMT-dosed mice. Histological evaluation of the hippocampus confirmed a localization and level of severity of dentate granule cell death in the experimental cohort within 24 h following TMT injection. The injury pattern displayed was consistent with previous reports (Bruccoleri et al., 1998; Harry et al., 2008; McPherson et al., 2011). At this age, no immunoreactivity for active caspase 3 was detected in the vehicle control hippocampus, while a pronounced immunoreactivity was observed in the dentate granule cell layer of mice dosed with TMT (Fig. 1A). The microglia response to TMT was similar to what has been previously described for this model (Bruccoleri et al., 1998; Harry et al., 2008). In the hippocampus of control mice, IB4+ microglia displayed few cell bodies within the granule cell layer (GCL) but showed evidence of fine processes throughout the area (Fig. 1B). With injury induced by TMT, microglia showed a shift in morphology and, at 24 h, the cells displayed either thickened processes with stellate morphology or an amoeboid morphology (Fig. 1B). GFAP+ astrocytes displayed long and thin processes throughout the hippocampus in control animals; while, in TMT-dosed mice, astrocytes displayed signs of hypertrophy with a thickening of cell processes and increased GFAP immunoreactivity evident in the GCL (Fig. 1C).
Figure 1.

Histopathology of the suprapyramidal blade of the hippocampal dentate granule cell layer (GCL) 24 h following a single injection of TMT hydroxide (2.3 mg/kg body wt, ip, 2ml/kg). Consistent with previous reports (Harry et al., 2008, McPherson et al., 2011), the current dosing cohort displayed dentate granule cell death with a concurrent microglia and astrocyte response. (A) Representative image of active caspase 3 immunoreactivity showing dark and collapsed cells (arrow) indicative of apoptosis of dentate granule neurons with TMT and no indication of apoptosis in the control. (B) Representative images of Bandeiraea simplifolia BS-I, IB4+ microglia. Microglia in the saline control showed small cell bodies and elongated distal ramified processes throughout the GCL (arrow). At 24 h post-TMT, microglia displayed an amoeboid morphology (arrow head) indicative of phagocytic activity. (C) Representative image of GFAP+ astrocytes. Cells displayed thin elongated processes (arrow) throughout the GCL in saline control mice. Following TMT, astrocytes showed an increased immunoreactivity of GFAP within the blade. Immunoreactivity was detected with 3,3′-diaminobenzidine (DAB) and sections counterstained with Harris hemotoxylin. Scale bar = 50 μm.
3.2. qNPA
Hippocampus
By 24 h post-TMT, a distinct profile of elevated mRNA levels was demonstrated (Table 1). Prominent and significant changes were observed in genes associated with each of the classifications of inflammation, cell death, glial response, and development. Elevated inflammatory genes included the complement factor, C1qb; TNF related genes (Tnfaip3, Tnfrsfla); the pro-inflammatory cytokine interleukin-1α (Il1a), the cell surface glycoprotein, Cd44; integrin alpha M (Itgam); secreted phosphoprotein 1 (Spp1), as well as the gene for microglia/macrophage marker Iba1 (Aif1-allograft inflammatory factor 1). In the TMT dosed animals, signals were detected for chemokine (C-C motif) ligand (Ccl)2 and Ccl4; Tnf, interferon-γ (Ifng), and the anti-inflammatory cytokine, IL-10 that were not observed in control tissue. Transforming growth factor β1 (Tgfb1) was elevated in TMT-dosed tissue while, Il6 was decreased. Elevated cell death-related genes included those associated with apoptosis (caspase 3), inflammatory mediated apoptosis (Fas and Fasl), ischemia (hypoxia inducible factor 1α [Hif1a]), heat shock (Hspa1b, Hspb1), activating transcription factor 3 (Atf3) and Atf4. No changes were observed in matrix metalloproteinase (Mmp)9 or Mmp12. A number of genes were also elevated that reflected the injury-induced hippocampal neurogenesis that has been reported with TMT injury (Harry et al., 2004; Ogita et al., 2005; McPherson et al., 2011). These included genes for the proliferative protein Ki67 (Mki67) and neurofilament heavy polypeptide (Nefh), with a signal detected for fibroblast growth factor (Fgf) 21 that was not observed in saline control tissue. However, decreases were observed in doublecortin-like kinase 3 (Dclk3), neurotrophin 3 (Ntf3), G protein regulated inducer of neurite outgrowth 2 (Grin2b; glutamate receptor subunit epsilon-2b), and neurotrophic tyrosine kinase, receptor, type 3 (Ntrk3). mRNA levels for the astrocyte structural protein, GFAP were elevated in the TMT dose group. In addition, lipocalin (Lcn2), a protein known to induce an M1 phenotype, was significantly elevated in the TMT dose group. Gene transcript levels for metabotropic glutamate receptor 3 (Grm3) and Ly6/neurotoxin 1 (Lynx1) remained within control levels. Following a Benjamini-Hochberg adjustment (FDR of 0.05) changes in Atf4, Ddit3, Hsp1b, Dclk3, and Nefh were no longer found to reach statistical significance.
Temporal cortex
To evaluate the specificity of the response in the hippocampus, fresh tissue samples of the temporal cortex were examined by qNPA. mRNA levels for immune-related and cell death-related factors remained within normal control levels. Following a Benjamini-Hochberg adjustment (FDR of 0.05), fresh tissue samples of the cortex indicated a significant decrease (less than 1.5-fold) in Lynx1, Grm3, and Grin2b. A significant increase (greater than 1.5-fold) was observed for Gfap (data not shown).
Comparison between FFPE and fresh frozen hippocampus
In the overall evaluation of the array data, the chemiluminescent levels detected for mRNA transcripts in FFPE tissue were slightly higher than those observed in fresh saline control tissue obtained from the same animal. When the FFPE saline control tissue was compared to the matched fresh tissue, 8 transcripts were detected for which there was no detection level in the fresh tissue (Table 1). One gene Hspa1b showed a significantly higher level in the FFPE tissue as compared to fresh tissue from the saline control group. In the TMT-dosed animals, a comparison of the FFPE tissue with fresh tissue indicated that 11 genes were significantly different with 10 higher and 1 lower in tissue that had undergone fixation and embedding (Table 1). While not all genes were found to be significantly higher in the FFPE tissue, the pattern was indicative of an overall higher chemiluminescent signal. However, the most important comparison was whether or not the profile of gene changes observed in fresh tissue as a function of TMT could be recapitulated from FFPE samples. To maximize the comparison, data prior to adjustment for FDR were used to determine the number of possible shared genes (Fig. 2A). Of the genes initially indicated in the fresh hippocampal tissue to be altered by TMT, only 11 were also indicated in the FFPE tissue. The fold-change in the TMT dosed tissue, relative to saline controls, was generally lower in the FFPE tissue than that seen in fresh tissue (Fig. 2B). In addition, data from the FFPE tissue indicated a decrease in 4 genes with TMT exposure that were not found changed in fresh tissue (Fig. 2A).
Figure 2.

Students t-tests were used to identify genes differentially expressed in the hippocampus of mice 24 h post-injection of either saline or trimethyltin hydroxide (TMT; 2.3mg/kg body wt, i.p.). Tissue was generated either as fresh frozen (Fresh) or sections from hippocampus rapidly fixed in formalin and embedding in paraffin (FFPE). (A) Venn diagram representing the distribution of genes either increased (up arrow) or decreased (down arrow) 24 h post-injection unique to and shared between Fresh and FFPE samples (p<0.05; non-adjusted for false detection). (B) Representation of mean (±SEM) fold-change of the shared genes identified in A as compared to saline control samples for each tissue preparation.
4. DISCUSSION
The TMT model of hippocampal damage has been used over the last few decades as a model to examine hippocampal function (Harry and Lefebvre d’Hellencourt, 2003) and has been employed more recently as a model of Alzheimer’s Disease-associated hippocampal dysfunction (Andjus et al., 2009; Gasparova et al., 2012). Early reports on TMT neurotoxicity examined a number of potential neuronal death mechanisms and, in general, excluded the involvement of ischemia/hypoxia and the contribution of excitotoxicity; while, later reports suggested a specific TNFα-receptor-mediated apoptosis of dentate granule cells (Harry et al., 2008). In the current study, genes associated with hypoxia/ischemia or glutamate activation were not elevated and consistent with earlier work, the profile did not support these as primary mechanisms of neuronal death induced by TMT. In agreement with earlier studies demonstrating a TMT-induced pro-inflammatory environment, we now provide data supporting the pro-inflammatory M1 activation state as an early event for neuronal death. The regional specificity of this response was demonstrated by the contrasting molecular pattern obtained from the temporal cortex. As a neuronal projection site from the hippocampus, inflammation was not implicated in the cortex by the gene profile but rather the data suggested activity-related events for down-regulation and remodeling that are possibly related to a M2 activation phenotype.
The characteristic features of neuronal death, astrocyte reactivity, and microglia activation seen with TMT hippocampal injury were reflected by elevations in related gene transcripts on the qNPA. Previous in vivo and in vitro studies established the hypothesis that TMT-induced neuronal death in the mouse was a TNF receptor-mediated event (Bruccoleri et al., 1998; Harry et al., 2008). In the mouse, TMT results in the induction of TNFα production by microglia (Bruccoleri et al., 1998; Figiel and Dzwonek, 2007; Harry et al., 2002) and activation of TNF receptors (Figiel and Dzwonek, 2007; Harry et al., 2008). Further confirmation of the contributory role for TNFα was demonstrated by a diminished neuronal death with the neutralization of TNFα signaling (Harry et al., 2008; Harry et al., 2002). Consistent with these reports, the qNPA demonstrated that elevated gene transcripts were primarily associated with M1 pro-inflammatory processes such as, chemokine and cytokine signaling. The data also suggested that an alternative common neuronal death mechanism such as, excitotoxicity was not initiated. This is in contrast with the microarray gene profile generated for TMT-induced hippocampal injury in the rat (Little et al., 2012). In that case, the localized loss of CA3-4 pyramidal neurons, rather than dentate granule neurons, and the associated microglia activation was not accompanied by an elevation in pro-inflammatory related genes such as TNFα. It was proposed that the difference in an inflammatory response between mice and rats was due to differences in the localization of neuronal damage and microglia heterogeneity (Lawson et al., 1990). However, earlier studies reported that TMT exposure in the rat did result in elevations in mRNA levels for Tnfa (Jahnke et al., 2001; Maier et al., 1995) and TNF elevations have been seen with in vitro studies using rat mixed glia cultures (Figiel and Dzwonek, 2007; Harry et al., 2002; Viviani et al., 2001). While the elevation in TNF related genes seen in the mouse differs from that reported for the rat (Little et al., 2002; Little et al., 2012), we did observe an elevation in mRNA levels for Ccl2 following TMT that is in agreement with elevations observed in the rat (Little et al., 2002). The discrepancy in the pro-inflammatory contribution across species may be a factor of timing, the toxicokinetics of the compound, severity of the damage, or method of detection. However, it does offer an interesting comparison for the future to determine common underlying biomarkers of neurotoxicity and neuroinflammation.
In the mouse hippocampus, the highest level of elevation following TMT was observed in Spp1, secreted phosphoprotein 1. SPP1 is a member of the osteopontin superfamily. It functions as a cytokine that regulates the expression of the M1-associated cytokines, interferon-γ and interleukin-12, in the peripheral immune system (Santamaria and Corral, 2013). It has been associated with microglia upregulation following transient forebrain ischemia (Choi et al., 2007), in a subset of amoeboid microglia in the hippocampus following kainic acid induced injury (Kim et al., 2002), and in peri-arterial areas following TMT (Morita et al., 2008). Its elevation is consistent with the microglia response observed at 24 h following TMT. Also related to the microglia response were elevations in Itgam, one protein subunit of macrophage-1 antigen also known as cluster of differentiation molecule 11b (Cd11b), and Cd44, a receptor for hyaluronic acid that can interact with osteopontin and matrix metalloproteinases and is upregulated in microglia upon focal stroke (Wang et al., 2001). ITGAM is involved in the complement system and has been implicated in several immune processes (Solovjov et al., 2005). The elevation observed in the hippocampus was likely related to the elevation observed in the complement gene C1qb and possibly to synaptic remodeling (Yuzaki, 2010).
Similar to what has been reported with kainic acid-induced hippocampal damage, we observed a significant elevation in lipocalin 2 (Lcn2). Lipocalin is a member of a diverse family of small soluble proteins, many which are secreted extracellularly and implicated in apoptosis and inflammation (Kim et al., 2002; Yang et al., 2009). Recently, secreted Lcn2 was demonstrated to promote microglial polarization to the M1 state in vitro. Upon further examination in vivo, Lcn2 was shown to enhance M1 microglia polarization in the hippocampus following lipopolysaccharide injection (Jang et al., 2013). In response to kainic acid, Lcn2 is elevated in the hippocampus and localized to reactive astrocytes (Chia et al., 2011). In cultured astrocytes, inflammatory stimulation elevates expression and secretion of Lcn2 (Lee et al., 2009). Thus, the combined elevation in Lcn2 and Gfap would provide a molecular indicator of the early astrocyte response or an interaction between microglia and astrocytes occurring with TMT injury (Fig. 1C). Lcn2 has also been identified to modulate the effects of stress on the hippocampus by altering neuronal spine formation (Mucha et al., 2011). Thus, it is likely that the upregulation of Lcn2 mRNA is not only associated with the injury in the hippocampus as a response pro-inflammatory cytokines but also as an effort to protect the hippocampal network from the injury-related stress. Such a mechanism could be linked to the elevation observed in the anti-apoptotic pathway gene, heat shock protein 1b (Hsp1b). Elevations in Ddit3 following TMT were indicated prior to conducting a FDR adjustment and were no longer statistically significant after adjustment however, given the level of fold-change it remains of interest. It is a key regulator of the mammalian target of rapamycin (mTOR) pathway and serves as an important signaling pathway in the hippocampus contributing to translational control and synaptic plasticity (Hoeffer and Klann, 2010; Polman et al., 2012). One could propose that an elevation in Ddit3 might represent the induction of signaling processes necessary for dealing with injury-induced stress and maintaining synaptic integrity.
In considering the specificity of the response in the hippocampus, changes were observed in the cortex of mice exposed to TMT. In general, the response was minimal as compared to the hippocampus. Three genes, Lynx1, Grin2b, and Grm3, were significantly decreased. Both Grin2b and Grm3 are associated with glutamate receptors. Grin2b, or the NR2 subunit, acts as the agonist-binding site for glutamate and Grm3 encodes the metabotropic glutamate receptor 3. Lynx1 is expressed in large projection neurons in the hippocampus and cortex and binding appears to be correlated with the distribution of nicotinic acetylcholine receptors (nAChRs) (Miwa et al., 1999). Lynx1 has been demonstrated to enhance desensitization of nAChRs and modulate their functional properties (Ibanez-Tallon et al., 2002) suggesting that the down regulation of these genes represents an adaptive and protective response of cortical neurons upon stimulation from the injured hippocampus. Such stimulation may also be the basis for the elevation observed in Gfap suggestive of an astrocyte response to early changes in the neuronal projections from the hippocampus.
Inflammation as a mechanism for initiating neuronal death or exacerbating neuronal injury and degeneration has gained a significant level of interest and enthusiasm; however, understanding the nature of any such response is necessary for interpretation of any biological impact. In the current study, we utilized a specific set of probes that were designed to detect changes in genes associated with pro-inflammatory events, neuronal death processes, and repair mechanisms. Using this approach, we have now identified a number of gene transcripts that may be utilized as specific markers of an M1 microglia state or M1 environment. While there are numerous methods that would suffice for analyzing fresh tissue, we were also interested in determining if we could obtain a reliable molecular profile from tissue that had been processed for histological analysis. Quite often, the initial assessments of neurotoxicity rely heavily on an anatomical demonstration of neuronal death or other forms of neuropathology. If such lesions could then be evaluated at a molecular level an evaluation of region specific changes could be conducted to characterize the neurotoxicity by identifying changes preceding or maintained after neuronal death (O’Callaghan and Sriram, 2005; Spradling et al., 2011; 2013; Yin et al., 2011; Woodcock and Morganti-Kossmann, 2013). Reports in the literature provided promising data regarding the utility of the qNPA platform for evaluating FFPE tissue (Roberts et al., 2007; Rimsza et al., 2008; Rimsza et al., 2011). However, so far, success has been limited to defining specific tumors and the method has not been evaluated for assessing multiple cellular processes in a tissue specific injury or in a direct comparison with fresh tissue. When we compared fresh and FFPE tissue, mRNA levels were generally higher in the FFPE tissue and in some cases a signal was detected that was not present in the fresh tissue. This raised concerns that the process of fixation and embedding of the tissue introduced artifacts that could lead to a misinterpretation of data. In comparison to fresh tissue, only approximately one-third of transcripts elevated by TMT were elevated in the FFPE tissue. Whether this was due to an increased non-specific background level of chemiluminescence that hindered the ability to detect subtle changes or if the data obtained from FFPE tissue was not valid are not known. These differences were observed in tissue that represented somewhat of a best-case scenario with the tissue being immediately fixed and processed for paraffin embedding and assays completed within 1-month. Whether the limited concordance would decrease with additional sample to assay time interval or increase with a shorter interval is not known. Remaining cautious, we considered the types of genes reflected in this one-third subset and concluded that, while the qNPA platform has been used for tumor characterization, the use of FFPE brain tissue would be limited and, at a minimum, would require very careful probe selection and data validation with fresh tissue.
Acknowledgements
The authors thank the NIEHS Microarray Core for their expert assistant with the initial microarray analysis used for selection of qNPA probes, Dr. Grace Kissling for statistical expertise, and Drs. Stephanie L. Smith-Roe and Chad Blystone of NIEHS for reviewing the final manuscript. This study was funded by the Division of Intramural Research and the Division National Toxicology Program, NIEHS/NIH (1Z01ES101623). The qNPA was conducted at HTG under an NTP contract with technical expertise from Dr John Luecke. The statements, opinions or conclusions contained within this manuscript do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the United States government.
Footnotes
Conflict of interest statement.
The authors declare no conflict of interest associated with this study.
References
- Andjus PR, Bataveljic D, Vanhoutte G, Mitrecic D, Pizzolante F, Djogo N, Nicaise C, Gankam Kengne F, Gangitano C, Michetti F, van der Linden A, Pochet R, Bacic G. In vivo morphological changes in animal models of amyotrophic lateral sclerosis and Alzheimer’s-like disease: MRI approach. Anat Rec (Hoboken) 2009;292:1882–1892. doi: 10.1002/ar.20995. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Royal Stat. Soc. Ser. B (Methodological) 1995;57:289–300. [Google Scholar]
- Bruccoleri A, Brown H, Harry GJ. Cellular localization and temporal elevation of tumor necrosis factor-alpha, interleukin-1 alpha, and transforming growth factor-beta 1 mRNA in hippocampal injury response induced by trimethyltin. J Neurochem. 1998;71:1577–1587. doi: 10.1046/j.1471-4159.1998.71041577.x. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Harry GJ. Chemical-induced hippocampal neurodegeneration and elevations in TNFalpha, TNFbeta, IL-1alpha, IP-10, and MCP-1 mRNA in osteopetrotic (op/op) mice. J Neurosci Res. 2000;62:146–155. doi: 10.1002/1097-4547(20001001)62:1<146::AID-JNR15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, Degos V, Jacotot E, Hagberg H, Savman K, Mallard C, Gressens P, Fleiss B. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013 doi: 10.1016/j.bbi.2013.02.005. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia WJ, Dawe GS, Ong WY. Expression and localization of the iron-siderophore binding protein lipocalin 2 in the normal rat brain and after kainate-induced excitotoxicity. Neurochem Int. 2011;59:591–599. doi: 10.1016/j.neuint.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim HY, Cha JH, Choi JY, Lee MY. Transient microglial and prolonged astroglial upregulation of osteopontin following transient forebrain ischemia in rats. Brain Res. 2007;1151:195–202. doi: 10.1016/j.brainres.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Figiel I, Dzwonek K. TNFalpha and TNF receptor 1 expression in the mixed neuronal-glial cultures of hippocampal dentate gyrus exposed to glutamate or trimethyltin. Brain Res. 2007;1131:17–28. doi: 10.1016/j.brainres.2006.10.095. [DOI] [PubMed] [Google Scholar]
- Funk JA, Gohlke J, Kraft AD, McPherson CA, Collins JB, Jean Harry G. Voluntary exercise protects hippocampal neurons from trimethyltin injury: Possible role of interleukin-6 to modulate tumor necrosis factor receptor-mediated neurotoxicity. Brain Behav Immun. 2011;6:1063–1077. doi: 10.1016/j.bbi.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparova Z, Janega P, Stara V, Ujhazy E. Early and late stage of neurodegeneration induced by trimethyltin in hippocampus and cortex of male Wistar rats. Neuro Endocrinol Lett. 2012;33:689–696. [PubMed] [Google Scholar]
- Geloso MC, Vercelli A, Corvino V, Repici M, Boca M, Haglid K, Zelano G, Michetti F. Cyclooxygenase-2 and caspase 3 expression in trimethyltin-induced apoptosis in the mouse hippocampus. Exp Neurol. 2002;175:152–160. doi: 10.1006/exnr.2002.7866. [DOI] [PubMed] [Google Scholar]
- Graeber MB. Changing face of microglia. Science. 2010;330:783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Lefebvre d’Hellencourt C. Dentate gyrus: alterations that occur with hippocampal injury. Neurotoxicology. 2003;24:343–356. doi: 10.1016/S0161-813X(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Lefebvre d’Hellencourt C, McPherson CA, Funk JA, Aoyama M, Wine RN. Tumor necrosis factor p55 and p75 receptors are involved in chemical-induced apoptosis of dentate granule neurons. J Neurochem. 2008;106:281–298. doi: 10.1111/j.1471-4159.2008.05382.x. [DOI] [PubMed] [Google Scholar]
- Harry GJ, McPherson CA, Wine RN, Atkinson K, Lefebvre d’Hellencourt C. Trimethyltin-induced neurogenesis in the murine hippocampus. Neurotox Res. 2004;5:623–627. doi: 10.1007/BF03033182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ, Tyler K, d’Hellencourt CL, Tilson HA, Maier WE. Morphological alterations and elevations in tumor necrosis factor-alpha, interleukin (IL)-1alpha, and IL-6 in mixed glia cultures following exposure to trimethyltin: modulation by proinflammatory cytokine recombinant proteins and neutralizing antibodies. Toxicol Appl Pharmacol. 2002;180:205–218. doi: 10.1006/taap.2002.9390. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Miwa JM, Wang HL, Adams NC, Crabtree GW, Sine SM, Heintz N. Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron. 2002;33:893–903. doi: 10.1016/s0896-6273(02)00632-3. [DOI] [PubMed] [Google Scholar]
- Jahnke GD, Brunssen S, Maier WE, Harry GJ. Neurotoxicant-induced elevation of adrenomedullin expression in hippocampus and glia cultures. J Neurosci Res. 2001;66:464–474. doi: 10.1002/jnr.1237. [DOI] [PubMed] [Google Scholar]
- Jang E, Lee S, Kim JH, Seo JW, Lee WH, Mori K, Nakao K, Suk K. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 2013;27:1176–1190. doi: 10.1096/fj.12-222257. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kim SY, Choi YS, Choi JS, Cha JH, Kim ON, Lee SB, Chung JW, Chun MH, Lee MY. Osteopontin in kainic acid-induced microglial reactions in the rat brain. Mol Cells. 2002;13:429–435. [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29:234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little AR, Benkovic SA, Miller DB, O’Callaghan JP. Chemically induced neuronal damage and gliosis: enhanced expression of the proinflammatory chemokine, monocyte chemoattractant protein (MCP)-1, without a corresponding increase in proinflammatory cytokines(1) Neuroscience. 2002;115:307–320. doi: 10.1016/s0306-4522(02)00359-7. [DOI] [PubMed] [Google Scholar]
- Little AR, Miller DB, Li S, Kashon ML, O’Callaghan JP. Trimethyltin-induced neurotoxicity: gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol Teratol. 2012;34:72–82. doi: 10.1016/j.ntt.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Maier WE, Brown HW, Tilson HA, Luster MI, Harry GJ. Trimethyltin increases interleukin (IL)-1 alpha, IL-6 and tumor necrosis factor alpha mRNA levels in rat hippocampus. J Neuroimmunol. 1995;59:65–75. doi: 10.1016/0165-5728(95)00026-x. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- McPherson CA, Aoyama M, Harry GJ. Interleukin (IL)-1 and IL-6 regulation of neural progenitor cell proliferation with hippocampal injury: Differential regulatory pathways in the subgranular zone (SGZ) of the adolescent and mature mouse brain. Brain Behav Immun. 2011;25:850–862. doi: 10.1016/j.bbi.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, Heintz N. 0lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–114. doi: 10.1016/s0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- Morita M, Imai H, Liu Y, Xu X, Sadamatsu M, Nakagami R, Shirakawa T, Nakano K, Kita Y, Yoshida K, Tsunashima K, Kato N. FK506-protective effects against trimethyltin neurotoxicity in rats: hippocampal expression analyses reveal the involvement of periarterial osteopontin. Neuroscience. 2008;153:1135–1145. doi: 10.1016/j.neuroscience.2008.01.078. [DOI] [PubMed] [Google Scholar]
- Mucha M, Skrzypiec AE, Schiavon E, Attwood BK, Kucerova E, Pawlak R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc Natl Acad Sci U S A. 2011;108:18436–18441. doi: 10.1073/pnas.1107936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf. 2005;4:433–442. doi: 10.1517/14740338.4.3.433. [DOI] [PubMed] [Google Scholar]
- Ogita K, Nishiyama N, Sugiyama C, Higuchi K, Yoneyama M, Yoneda Y. Regeneration of granule neurons after lesioning of hippocampal dentate gyrus: evaluation using adult mice treated with trimethyltin chloride as a model. J Neurosci Res. 2005;82:609–621. doi: 10.1002/jnr.20678. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia. 2005;50:287–298. doi: 10.1002/glia.20181. [DOI] [PubMed] [Google Scholar]
- Pechhold S, Stouffer M, Walker G, Martel R, Seligmann B, Hang Y, Stein R, Harlan DM, Pechhold K. Transcriptional analysis of intracytoplasmically stained, FACS-purified cells by high-throughput, quantitative nuclease protection. Nat Biotechnol. 2009;27:1038–1042. doi: 10.1038/nbt.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman JA, Hunter RG, Speksnijder N, van den Oever JM, Korobko OB, McEwen BS, de Kloet ER, Datson NA. Glucocorticoids modulate the mTOR pathway in the hippocampus: differential effects depending on stress history. Endocrinology. 2012;153:4317–4327. doi: 10.1210/en.2012-1255. [DOI] [PubMed] [Google Scholar]
- Reuhl KR, Cranmer JM. Developmental neuropathology of organotin compounds. Neurotoxicology. 1984;5:187–204. [PubMed] [Google Scholar]
- Rimsza LM, Leblanc ML, Unger JM, Miller TP, Grogan TM, Persky DO, Martel RR, Sabalos CM, Seligmann B, Braziel RM, Campo E, Rosenwald A, Connors JM, Sehn LH, Johnson N, Gascoyne RD. Gene expression predicts overall survival in paraffin-embedded tissues of diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2008;112:3425–3433. doi: 10.1182/blood-2008-02-137372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimsza LM, Wright G, Schwartz M, Chan WC, Jaffe ES, Gascoyne RD, Campo E, Rosenwald A, Ott G, Cook JR, Tubbs RR, Braziel RM, Delabie J, Miller TP, Staudt LM. Accurate classification of diffuse large B-cell lymphoma into germinal center and activated B-cell subtypes using a nuclease protection assay on formalin-fixed, paraffin-embedded tissues. Clin Cancer Res. 2011;17:3727–3732. doi: 10.1158/1078-0432.CCR-10-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria MH, Corral RS. Osteopontin-dependent regulation of Th1 and Th17 cytokine responses in Trypanosoma cruzi-infected C57BL/6 mice. Cytokine. 2013;61:491–498. doi: 10.1016/j.cyto.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovjov DA, Pluskota E, Plow EF. Distinct roles for the alpha and beta subunits in the functions of integrin alphaMbeta2. J Biol Chem. 2005;280:1336–1345. doi: 10.1074/jbc.M406968200. [DOI] [PubMed] [Google Scholar]
- Spradling KD, Lumley LA, Robison CL, Meyerhoff JL, Dillman JF. Transcriptional responses of the nerve agent-sensitive brain regions amygdala, hippocampus, piriform cortex, septum, and thalamus following exposure to the organophosphonate anticholinesterase sarin. J Neuroinflammation. 2011;8:84. doi: 10.1186/1742-2094-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J Histochem Cytochem. 1990;38:1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL. Role of microglia in the central nervous system’s immune response. Neurol Res. 2005;27:685–691. doi: 10.1179/016164105X49463a. [DOI] [PubMed] [Google Scholar]
- Viviani B, Corsini E, Pesenti M, Galli CL, Marinovich M. Trimethyltin-activated cyclooxygenase stimulates tumor necrosis factor-alpha release from glial cells through reactive oxygen species. Toxicol Appl Pharmacol. 2001;172:93–97. doi: 10.1006/taap.2001.9136. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhan Y, Xu L, Feuerstein GZ, Wang X. Use of suppression subtractive hybridization for differential gene expression in stroke: discovery of CD44 gene expression and localization in permanent focal stroke in rats. Stroke. 2001;32:1020–1027. doi: 10.1161/01.str.32.4.1020. [DOI] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, Strong RK, Zurakowski D, Moses MA. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci U S A. 2009;106:3913–3918. doi: 10.1073/pnas.0810617106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Lu L, Prasad K, Richfield EK, Unger EL, Xu J, Jones BC. Genetic-based, differential susceptibility to paraquat neurotoxicity in mice. Neurotoxicol Teratol. 2011;33:415–421. doi: 10.1016/j.ntt.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. Synapse formation and maintenance by C1q family proteins: a new class of secreted synapse organizers. Eur J Neurosci. 2010;32:191–197. doi: 10.1111/j.1460-9568.2010.07346.x. [DOI] [PubMed] [Google Scholar]
- Zhang D, Hu X, Qian L, O’Callaghan JP, Hong JS. Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol. 2010;41:232–241. doi: 10.1007/s12035-010-8098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]