

Abstract
The thienopyrimidinone 5,6-dimethyl-2-(4-nitrophenyl)thieno[2,3-d]pyrimidin-4(3H)-one (DNTP) occupies the interface between the p66 ribonuclease H (RNase H) domain and p51 thumb of human immunodeficiency virus reverse transcriptase (HIV RT), thereby inducing a conformational change incompatible with catalysis. Here, we combined biochemical characterization of 39 DNTP derivatives with antiviral testing of selected compounds. In addition to wild-type HIV-1 RT, derivatives were evaluated with rationally-designed, p66/p51 heterodimers exhibiting high-level DNTP sensitivity or resistance. This strategy identified 3′,4′-dihydroxyphenyl (catechol)-substituted thienopyrimidinones with sub-micromolar in vitro activity against both wild type HIV-1 RT and drug-resistant variants. Thermal shift analysis indicates that, in contrast to active site RNase H inhibitors, these thienopyrimidinones destabilize the enzyme, in some instances reducing the Tm by 5°C. Importantly, catechol-containing thienopyrimidinones also inhibit HIV-1 replication in cells. Our data strengthens the case for allosteric inhibition of HIV RNase H activity, providing a platform for designing improved antagonists for use in combination antiviral therapy.
Introduction
For approximately 15 years, nonnucleoside reverse transcriptase inhibitors (NNRTIs) have constituted a critical component of combination HIV antiretroviral therapy1, acting allosterically by creating and occupying a hydrophobic pocket of the p66 thumb subdomain of the p66/p51 reverse transcriptase (RT) heterodimer2 to compromize enzyme conformational dynamics3 and the chemical step of DNA synthesis4. The possibility of allosteric inhibition of additional HIV enzymes has recently been demonstrated by the report that the 2-(quinolin-3-yl) acetic acid derivatives, LEDGIN-6 and BI-1001 are allosteric inhibitors of HIV-1 integrase5, while Perryman et al. have used a fragment-based approach to highlight proposed non-active site allosteric inhibitors of HIV-1 protease6. With respect to RT-associated ribonuclease H (RNase H) activity, encoded by the C-terminal domain of the p66 subunit and is essential for virus replication7, 8, we demonstrated that α-hydroxytropolones, while effective inhibitors at sub-micromolar concentrations, were efficiently displaced from the RNase active site by nucleic acid9. In light of this limitation, an alternative strategy might therefore be to identify allosteric inhibitors that bind close to the RNase H active site and compromise catalysis by affecting subdomain and/or subunit dynamics.
As a first step in this direction, the vinylogous ureas 2-amino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (NSC727447) and N-[3-(aminocarbonyl)-4,5-dimethyl-2-thienyl]-2-furancarboxamide (NSC727448) were identified as modestly potent HIV-1 RNase H inhibitors. In the absence of crystallographic data, protein footprinting by mass spectrometry implicated p51 thumb residues Cys280 and Lys281 in inhibitor binding10. Since Cys280 - Thr290 of the p51 thumb and Pro537 – Glu546 of the p66 RNase H domain constitute approximately 30% of the buried surface interface11, 12, these compounds, which are equally effective against the enzyme and the enzyme/substrate complex13, 14 represent allosteric inhibitors that affect RNase H activity by destabilizing the subunit interface. A limited structure-activity study subsequently identified the thienopyrimidinone 5,6-dimethyl-2-(4-nitrophenyl)thieno[2,3-d]pyrimidin-4(3H)-one (DNTP), a cyclized derivative of NSC727448, as a lead candidate for further inhibitor optimization. Alanine scanning mutagenesis between p51 residues Lys275 and Thr286 and a vertical scan of Cys28014 identified selectively-mutated RT heterodimers displaying either significantly-increased DNTP sensitivity (Val276Ala, Arg284Ala) or high-level resistance (Cys280Ala, Thr286Ala), strengthening the contention that the p51 thumb as the target site. More importantly this scanning mutagenesis approach provided a unique collection of mutant HIV-1 enzymes that could be exploited to screen for DNTP derivatives with broad-spectrum activity.
In this communication, we report the synthesis, activity, and antiviral properties of 39 novel thienopyrimidinones bearing substitutions on the thiophene or the C2 position of the pyrimidinone ring (Figure 1). Exploiting the panel of selectively-mutated HIV-1 RT mutants allowed us to identify four groups of molecules, based on their resistance/sensitivity profiles. Among these, compounds with a 3′,4′-dihydroxyphenyl (catechol) substitution, displayed activity against both wild type and drug-resistant RT variants at sub-micromolar concentrations and, importantly, inhibited HIV-1 replication in cells. Differential scanning fluorimetry indicated that these compounds, in contrast to α-hydroxytropolone-derived RNase H inhibitors15, destabilized the RT heterodimer, in some instances lowering the Tm by almost 5°C. Collectively, our data provides an important structural platform for the continued development of thienopyrimidinone-based RNase H inhibitors, and highlights the value of genetically-engineered HIV-1 RT variants with altered inhibitor sensitivity profiles for secondary screening.
Figure 1.

Structure of the thienopyrimidinone 5,6-dimethyl-2-(4-nitrophenyl)thieno[2,3-d]pyrimidin-4(3H)-one (DNTP).
Results
Synthesis of Thienopyrimidinones
All thienopyrimidinone derivatives tested were synthesized according to Bakavoli, et al.16. Briefly, 2-amino-4′,5′-disubstituted thiophene-3-carboxamide intermediates were obtained from condensation of the appropriate ketone and α-cyanoacetamide in the presence of elemental sulfur and base17. This intermediate was then treated with the aromatic aldehyde and molecular iodine to produce the corresponding thienopyrimidinone (Scheme 1)16. Additional details are provided in the Supporting Information.
Scheme 1.

General scheme for synthesis of modified thienopyrimidinones.
C2 Aromatic Substitutions of the Thienopyrimidinone
DNTP, identified recently as a submicromolar HIV-1 RNase H inhibitor13, comprises a thiophene-fused pyrimidinone ring containing a C2 4′-nitrophenyl substitution (Figure 1). Previous work13 suggested the 4′-NO2 group was a key element in inhibition, although its role remained to be established. We therefore elected to preserve the pyrimidinone core and introduce substituents on the phenyl ring. Table 1 summarizes the effect of these substitutions on inhibitor potency.
Table 1.
Inhibition of HIV-1 RNase H enzymatic activity by thienopyrimidinones carrying C2 aromatic substitutions. IC50 values represent the average of triplicate analysis.
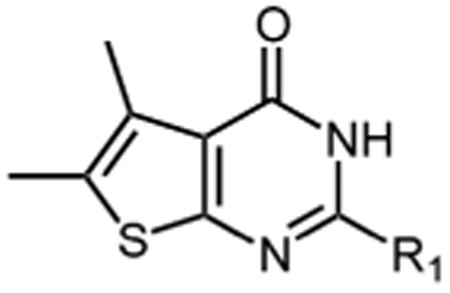
| |||||
|---|---|---|---|---|---|
|
| |||||
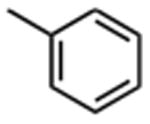
| Compound | R1 | IC50 (μM) | Compound | R1 | IC50 (μM) |
| mono-substituted phenyl | di-substituted phenyl | ||||
| 1 |
|
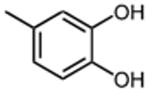
11.3 ±0.6 | 9 |
|
0.26 ±0.01 |
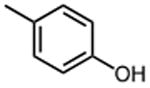
| 2 |
|
0.79 ±0.11 | 10 |
|
12.0 ±0.6 |
| 3 |
|
1.3 ±0.4 | 11 |
|
>50 |
| 4 |
|
1.6 ±0.4 | 12 |
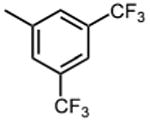
|
>50 |
| 5 |
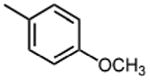
|
9.1 ±2.5 | 13 |
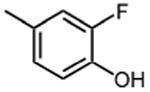
|
>50 |
| 6 |
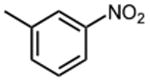
|
1.5 ±0.1 | 14 |
|
12.6 ±0.8 |
| 7 |
|
>50 | 15 |
|
>50 |
| 8 |
|
>50 | |||
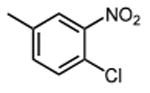
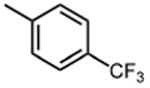
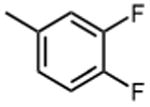
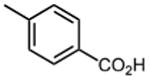
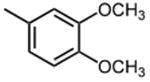
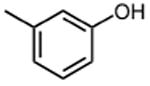
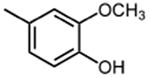
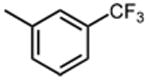
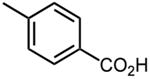
The effect of meta- and para-substituted phenyl rings was first evaluated. The absence of any 50 μM. As previously shown with DNTP, the electron withdrawing effect of the substituent also contributes to inhibitory potency, as compound 3 (-CF3, 1.3 μM), and 4 (-CO2H, 1.6 μM) were 5 - 6 fold more active than compound 5 (-OCH3, 9.1 μM). The exception is compound 2 (-OH, 0.79 μM) which is likely to be involved in hydrogen bonding with residues at the p51-p66 dimer interface. Introducing an additional hydroxyl at the 3′ position of compound 2 (Compound 9) resulted in a 3-fold increase in activity (IC50 0.26 μM), suggesting additional contacts provided by this substitution stabilize inhibitor binding. This notion was strengthened by the observation that replacing the 3′-OH with either –F (Compound 13) or -OCH3 (Compound 15) reduced the IC50 almost 200-fold. Other di-substituted thienopyrimidinones were generally inactive (Compounds 11, and 12) or displayed significantly reduced IC50 values (Compounds 10 and 14).
Table 2 shows that substituting the phenyl ring with other heteroaromatic rings (Compounds 16, 17, and 19) does not significantly affect the activity compared to compound 1. The exception to this was the indole-substituted pyrimidinone, compound 18, for which an IC50 value of 0.48 μM was determined. Despite the apparently favorable properties of compound 18, data of a later section indicates that it failed to inhibit thienopyrimidinone-resistant HIV-1 RT mutants.
Table 2.
Inhibition of HIV-1 RNase H enzymatic activity by thienopyrimidinones carrying C2 heteroaromatic substitutions. IC50 values represent the average of triplicate analysis.
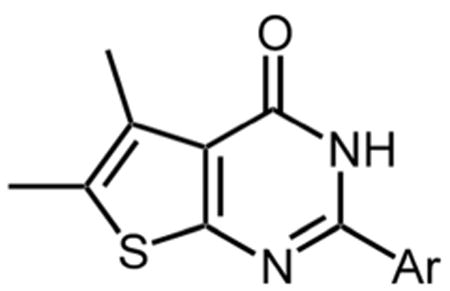
| ||
|---|---|---|
|
| ||
| Compound | Ar | IC50 (μM) |
| 16 |
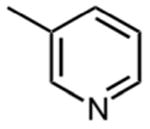
|
8.3 ±0.2 |
| 17 |
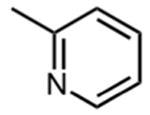
|
4.2 ± 0.05 |
| 18 |
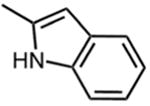
|
0.48 ± 0.06 |
| 19 |
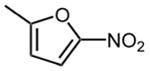
|
16.9 ±1.7 |
C5,C6-Cycloheptene Ring Substitutions
Since previous work with NSC727447 suggested a hydrophobic pocket in the immediate vicinity of the binding site could accommodate its cycloheptene ring, but not other cycloalkenes13, we next evaluated thienopyrimidinones containing a cycloheptene ring (compounds 20 - 23). These compounds displayed IC50 values in the range of 1.9 – 4.1 μM (Table 3), which is slightly higher than derivatives containing a 5,6-dimethyl substitution of the thiophene ring. This result suggests substituting a 5,6-dimethyl with a cycloheptene ring does not significantly affect binding affinity, although we cannot rule out the possibility that the binding mode of these compounds differs from that of 5,6-dimethyl derivatives as was previously suggested13.
Table 3.
Inhibition of HIV-1 RNase H enzymatic activity by cycloheptene-containing thienopyrimidinones. IC50 values represent the average of triplicate analysis.
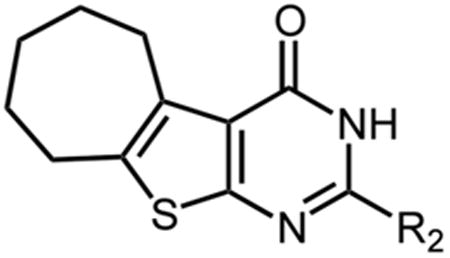
| ||
|---|---|---|
|
| ||
| Compound | R2 | IC50 (μM) |
| 20 |
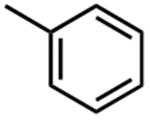
|
4.1 ±0.2 |
| 21 |
|
1.9 ±0.03 |
| 22 |
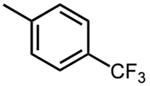
|
3.1 ±0.9 |
| 23 |
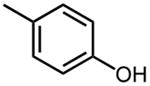
|
3.2 ±0.2 |
Di- or Tri-hydroxyphenyl-Containing Thienopyrimidinone
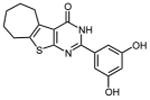
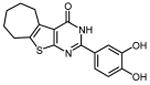
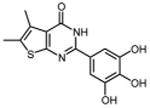
In an effort to improve thienopyrimidinone activity, we focused on compound 9 for further modification. We first investigated how C5 and C6 substitutions affected activity. Table 4 shows that replacing a C5 –CH3 with a bulkier alkyl group, such as n-propyl (Compound 24) and n-butyl (Compound 25) or replacing a C6 -CH3 with ethyl (Compound 26) is tolerated, resulting in IC50 values of 0.49, 0.42, and 0.59 μM respectively. Substituting 5,6-dimethyl with an aliphatic ring also does not affect the activity, regardless of the ring size, suggesting that the bulkier C5 and C6 alkyl groups can be accommodated. This result supports our previous contention that a role of the 3′,4′-dihydroxyphenyl component is to “anchor” thienopyrimidinones at the p51-p66 subunit interface, predicting that substitutions on the thiophene ring would minimally affect the overall binding conformation. However any change in the position of -OH substitution (Compounds 31 – 37) or adding an additional –OH group (Compounds 38 - 39) decreased activity, indicating the necessity of hydroxyl substitutions at 3′ and 4′ positions for optimal binding of these inhibitors.
Table 4.
Inhibition of HIV-1 RNase H enzymatic activity by di- or tri-hydroxyphenyl-containing thienopyrimidinones. IC50 values represent the average of triplicate analysis.
| Compound | IC50 (μM) | Compound | IC50 (μM) | ||
|---|---|---|---|---|---|
| 9 |
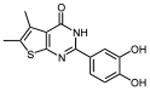
|
0.26 ±0.01 | 32 |
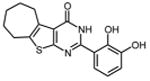
|
1.8 ±0.1 |
| 24 |
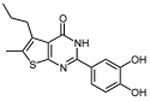
|
0.49 ±0.01 | 33 |
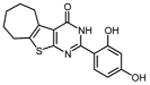
|
9.3 ± 0.3 |
| 25 |
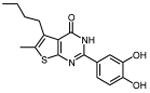
|
0.42 ± 0.02 | 34 |
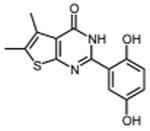
|
9.3 ± 0.3 |
| 26 |
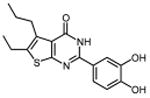
|
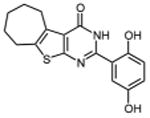
0.59 ±0.04 | 35 |
|
1.7 ±0.04 |
| 27 |
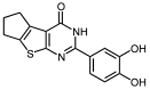
|
0.32 ± 0.02 | 36 |
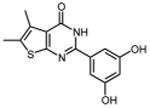
|
5.2 ±0.2 |
| 28 |
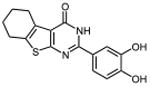
|
0.37 ±0.03 | 37 |
|
2.6 ± 0.02 |
| 29 |
|
0.29 ±0.11 | 38 |
|
0.69 ±0.01 |
| 30 |
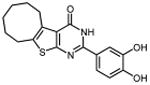
|
0.26 ±0.01 | 39 |
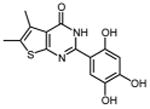
|
1.6 ±0.03 |
| 31 |
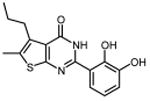
|
1.7 ± 0.1 |
Screening with Engineered HIV-1 RT Mutants Identifies Thienopyrimidinones with Broad Spectrum Activity
Scanning mutagenesis of p51 α-helix I14(Figure 2) demonstrated increased DNTP sensitivity for selectively-mutated HIV-1 RT derivatives p66/p51V276A and p66/p51R284A, in addition to decreased sensitivity for mutants p66/p51C280A and p66/p51T286A. This observation was most pronounced for RT mutant p66/p51C280A, which exhibited ∼50-fold resistance to DNTP14. A high level of resistance was also observed in a vertical scan of p51 residue Cys280, suggesting a critical contribution from this residue to inhibitor binding. We therefore determined IC50 values for all thienopyrimidinones in this study with the panel of engineered RT mutants in an attempt to identify broad-spectrum inhibitors. The results of this analysis are presented in Table 5.
Figure 2.

Model of the interface between the p66 RNase H domain (gold) and p51 thumb subdomain (green) of HIV-1 RT. Active site carboxylates (D443, E478, D498, D549) and the conserved His539 of the RNase H domain are indicated in white. Residues of the p51 thumb subdomain indicated in red were previously shown to confer resistance (C280, T286) or sensitivity (V276, R284) to thienopyrimidinone DNTP when substituted with Ala.
Table 5.
Thienopyrimidinone inhibition of RNase H activity of selectively-mutated p66/p51 HIV-1 RT heterodimers. Alanine substitutions were introduced into the thumb subdomain of the p51 RT subunit. Values reported represent the average of triplicate analysis. Group I, II, III, and IV designations are defined in the text.
| Compound | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| WT | V276A | C280A | R284A | T286A | ||
| I | 2 | 0.79 ±0.11 | 0.14 ±0.04 | >50 | 0.20 ± 0.03 | 3.7 ±0.3 |
| 3 | 1.3 ±0.4 | 0.31 ±0.01 | >50 | 0.23 ±0.01 | 7.5 ±1.0 | |
| 6 | 1.5 ±0.1 | 0.26 ± 0.03 | >50 | 0.34 ±0.05 | 1.5 ±0.2 | |
| 18 | 0.48 ± 0.06 | 0.16 ±0.02 | >50 | 0.12 ±0.001 | 0.96 ±0.02 | |
| 21 | 1.9 ±0.03 | 0.46 ± 0.02 | >50 | 0.95 ± 0.04 | 32.0 ±15.5 | |
|
| ||||||
| II | 20 | 4.1 ±0.2 | 0.96 ±0.01 | 41.4 ±6.5 | 1.3 ±0.06 | 27.9 ± 6.9 |
| 22 | 3.1 ±0.9 | 0.53 ±0.01 | 10.6 ±0.5 | 0.54 ±0.03 | 7.2 ±0.7 | |
| 23 | 3.2 ±0.2 | 0.64 ± 0.03 | 15.8 ±2.0 | 0.65 ± 0.02 | 6.6 ±0.3 | |
|
| ||||||
| III | 31 | 1.7 ±0.1 | 4.1 ±0.1 | 2.9 ±0.1 | 3.5 ±0.1 | 5.7 ±0.2 |
| 32 | 1.8 ±0.1 | 2.8 ±0.2 | 1.6 ±0.03 | 2.9 ±0.1 | 3.6 ±0.2 | |
|
| ||||||
| IV | 9 | 0.26 ±0.01 | 0.39 ±0.04 | 0.32 ±0.01 | 0.30 ±0.01 | 0.28 ± 0.04 |
| 24 | 0.49 ±0.01 | 0.56 ±0.02 | 0.37 ±0.001 | 0.80 ±0.01 | 0.57 ± 0.02 | |
| 25 | 0.42 ± 0.02 | 0.91 ±0.02 | 0.49 ± 0.005 | 1.2 ±0.04 | 1.1 ±0.01 | |
| 26 | 0.59 ± 0.04 | 0.76 ± 0.03 | 0.50 ±0.01 | 1.4 ±0.04 | 0.82 ± 0.02 | |
| 27 | 0.32 ± 0.02 | 0.21 ±0.01 | 0.26 ±0.01 | 0.47 ±0.01 | 0.37 ±0.01 | |
| 28 | 0.37 ±0.03 | 0.41 ±0.01 | 0.29 ±0.01 | 0.92 ± 0.04 | 0.46 ±0.01 | |
| 29 | 0.29 ±0.11 | 0.37 ±0.01 | 0.26 ± 0.02 | 0.55 ± 0.004 | 0.36 ±0.02 | |
| 30 | 0.26 ±0.01 | 0.11 ±0.01 | 0.23 ± 0.004 | 0.28 ±0.01 | 0.29 ±0.01 | |
In general, thienopyrimidinones could be classed into four groups, depending on their activity profiles. Group I inhibitors, comprising compounds 2, 3, 6, 18, and 21, while active against wild type RT at concentrations ranging from 0.48 – 1.9 μM, uniformly failed to inhibit p66/p51C280A RT at a concentration of 50 μM. At the same time, these compounds showed enhanced activity against mutant p66/p51V276A (e.g. Compound 2: IC50WT = 0.79 μM vs IC50Mut = 0.14 μM) and reduced activity against mutant p66/p51T286A (e.g. Compound 21: IC50WT = 1.9 μM vs IC50Mut = 32.0 μM). Group II inhibitors, exemplified by compounds 20, 22, and 23 were slightly less active than the parent thienopyrimidinone, DNTP, against wild type RT with IC50 values varying from 3.1 - 4.1 μM. However, these inhibitors compromised RNase H activity of RT mutant p66/p51C280A, albeit at IC50s ranging from 10.6 - 41.4 μM. Group II compounds also displayed a similar trend with respect to mutants p66/p51V276A (increased sensitivity), p66/p51R284A (increased sensitivity) and p66/p51T286A (decreased sensitivity).
Group III and IV inhibitors were dramatically different. Interestingly, both groups contain a catechol moiety (2′,3′-dihydroxy for Group III and 3′,4′-dihydroxy phenyl for Group IV) and, in contrast to Group I and II inhibitors, were effective against RT mutant p66/p51C280A. Group IV inhibitors (IC50 = 0.26 – 0.59 μM) were in general 4-8 fold more potent than those of Group III (IC50 = 1.7 – 1.8 μM). More importantly, this broad-spectrum activity was also extended to drug-sensitive mutants p66/p51V276A, p66/p51R284A and drug-resistant mutant p66/p51T286A. Supplementary Table S1 provides IC50 values for compound 9 across the entire panel of selectively-mutated p51 thumb α-helix I variants (i.e. Lys275 – Arg286), indicating that within experimental error, they are uniformly sensitive to this thienopyrimidinone. Although such an observation cannot exclude the possibility that Group III and IV inhibitors might interact with p51 RT at a site slightly removed from that previously proposed10, 13, data shown below suggests this is unlikely. In summary, although carrying a variety of substituents on the thiophene ring, the catechol moiety common to Group III and IV inhibitors appears to play a critical role in inhibitory potency.
Thienopyrimidinone Inhibitors Destabilize HIV-1 RT in the Absence and Presence of Substrate
Differential scanning fluorimetry (ThermoFluor18) is a simple, rapid and inexpensive means of determining protein stability in the presence of small molecule ligands19, 20, an example of which is the demonstration by Su et al. that naphthyridinone-based RNase H active site inhibitors increased the Tm of HIV-1 RT by 3 – 4 °C21. More recently, we have also shown that allosteric HIV-1 integrase inhibitors (ALLINIs) stabilize the integrase tetramer and increase its melting temperature5. Since computer-assisted modeling studies located the parent thienopyrimidinone, DNTP, at the interface of the p51 and p66 RT subunits, we investigated whether binding of the thienopyrimidinone compounds affected the stability of the p66/p51 RT heterodimer. The results of ThermoFluor analysis are provided in Figure 3. For comparative purposes, we first examined alterations to the thermal stability of wild type HIV-1 RT induced by the RNase H active site hydroxytropolone inhibitor, β-thujaplicinol15. In accordance with studies of Su et al.21, we observed a Tm increase of ∼2.0 °C in the presence of Mg2+ and the active site inhibitor. In contrast, all thienopyrimidinones tested reduced the Tm by 0.5 - 5.5 °C (Figure 3) although there was no linear correlation between IC50 and ΔTm. The ability of compounds 9 and 29 to destabilize the binary enzyme/substrate complex was also examined. For these experiments, we chose an RNA/DNA hybrid containing the polypurine tract (PPT) primer of HIV-1 (+) strand DNA synthesis. In the absence of inhibitor, binding of the PPT-containing duplex increased the Tm by 9.3 °C, indicating significant stabilization of HIV-1 RT (Supplementary Figure S1). However, compounds 9 and 29 retained their destabilizing property, reducing the Tm of the enzyme/substrate complex by 5.8 and 5.9 °C, respectively.
Figure 3.

Effect of thienopyrimidinone RNase H inhibitors on the thermal stability of p66/p51 HIV-1 RT. β-TP, RNase H active site inhibitor β-thujaplicinol. Tm values are the average of triplicate analysis.
These results collectively support the notion that thienopyrimidinones inhibit RNase H activity allosterically by binding at the interface of two subunits in the presence of the nucleic acid substrate and destabilizing those interactions. However, such changes in thermal stability cannot discriminate between denaturation of individual RT subunits, dissociation of the p66/p51 heterodimer or a combination of both processes.
Inhibition of DNA Polymerase and Integrase Activities by 3′,4′-Dydroxyphenyl-containing Thienopyrimidinones
In order to examine the specificity of our thienopyrimidinones, we determined their effect on DNA-dependent DNA polymerase and integrase activities. Figure 4a shows that compounds 25-28, and 30 inhibited > 60% of DNA-dependent DNA polymerase activity at 50 μM concentration. These results is not unexpected since these compounds most likely bind at the dimer interface and induce alterations of overall RT-substrate interactions, which could affect both RNase H and polymerase activities. These compounds are equally active against NNRTI resistant mutants (K103N, Y181C) (data not shown), suggesting that they are less likely to bind to the NNRTI binding pocket.
Figure 4.

Evaluation of inhibitory activity of 3′,4′-dihydroxyphenyl-containing thienopyrimidinones against HIV-1 DNA polymerase activities (a) and HIV-1 integrase (b) in the presence of 50 μM 3′,4′-dihydroxyphenyl-containing thienopyrimidinones.
It has been reported that compounds possessing catechol moieties were active against purified HIV-1 integrase (IN)22. However, the results of our analysis presented in Figure 4b, indicate these thienopyrimidinones have minimal effect on 3′-processing of integrase, suggesting specificity for RT.
Antiviral Activity of 3′,4′-Dihydroxyphenyl-Containing Thienopyrimidinones
Based on their favorable properties against the panel of selectively-mutated p51 RT mutants, the ability of compounds 9, 24 – 26, 29, and 31 - 32 to inhibit virus replication was assessed. Data of Table 6 indicates that dihydroxyphenyl-containing thienopyrimidinones inhibited HIV-1 replication in the low micromolar range, with selectivity ratios ranging from 2.3 (Compound 9, EC50 3.8 μM: CC50 8.9 μM) to ∼6 (Compound 24, EC50 1.9 μM: CC50 11.2 μM). The exception to this was compound 28. Although active in vitro at low micromolar concentrations, this compound failed to elicit protection from HIV infection.
Table 6.
Antiviral activity of catechol-containing thienopyrimidinones. sr, selectivity ratio, i.e., CC50/EC50.
| Compound | EC50 (μM) | CC50 (μM) | sr |
|---|---|---|---|
| 9 | 3.8 | 8.9 | 2.3 |
| 24 | 1.9 | 11.2 | 5.9 |
| 25 | 7.9 | 25.4 | 3.2 |
| 26 | 9.2 | 33.7 | 3.7 |
| 29 | 1.6 | 6.8 | 4.3 |
| 31 | 3.1 | 12.4 | 4.0 |
| 32 | 2.7 | 9.8 | 3.6 |
Discussion and Conclusions
The demonstration that NNRTIs interrupt HIV-1 DNA synthesis by influencing enzyme conformational dynamics3, 23 has provided a novel and important platform for identification of small molecules that impose allosteric control of critical HIV enzymes, a notion that has been extended to HIV-1 integrase5 and proposed for HIV-1 protease6. It is therefore not unreasonable to consider allosteric inhibition of HIV-1 RT-associated RNase H activity, especially in light of observations that interactions involving p51 thumb residues Cys280 - Thr290 and Pro537 - Glu546 of the p66 RNase H domain constitute ∼33% of the buried surface at the subunit interface24. Since the p66 thumb does not participate in an equivalent inter-subunit interaction, vinylogous ureas previously described by Wendeler et al. 10 most likely interfere with RNase H activity by perturbing the dimer interface, inducing alterations to active site geometry that are incompatible with catalysis10, 13, 14.
This study evaluated 39 related and novel thienopyrimidinones as HIV-1 RNase H inhibitors and, more importantly, combined chemical synthesis with a multi-enzyme counter-screen using unique genetically manipulated HIV-1 RT mutants that display enhanced resistance or sensitivity to the parent molecule, DNTP. This strategy identified several compounds containing a catechol-substituted pyrimidinone ring that inhibited both wild-type and drug-resistant HIV-1 RT in vitro, and HIV-1 replication in culture. Since the catechol component of these compounds has been shown to possess metal chelating properties25, the observation that they are equally active against our panel of engineered enzymes might be indicative of active site inhibitors that chelate the catalytically-critical Mg2+ ions. However, thermal denaturation studies of Su et al.21, together with our data shown in Figure 3, demonstrate that binding of metalchelating RNase H inhibitors stabilizes the HIV-1 RT heterodimer, increasing the Tm by ∼2.0°C. In contrast, compounds 9, and 24 - 32 decrease the Tm, in some instances by more than 5°C. In addition, Yonetani-Theorell analysis26 shows that compounds 9 and 29 are mutually-exclusive with respect to the active site inhibitor, β-thujaplicinol (Figure 5). Taken together, these observations strengthen our postulate that compounds 9 and 24 - 30 compromise dynamics at the dimer interface, and as a consequence interrupt catalysis by altering active site geometry 10. This notion is supported by our recent success in solving the structure of p66/p51 HIV-1 RT complexed with an NNRTI and a non-polypurine tract-containing RNA/DNA hybrid. Among several significant conformational changes nucleic acid binding promotes an “expansion” of the enzyme. In this model, an ∼2.5 Å outward movement of the p66 fingers subdomain and RNase H domain is required to accommodate the hybrid and, as a consequence, the p66 thumb tilts toward the duplex, while the p51 thumb moves in concert with the RNase H domain27. A small molecule antagonist interacting at the p51 thumb/p66 RNase H interface could therefore inhibit or alter subdomain movement and modify nucleic acid trajectory or active site geometry. Although high-resolution structural data is presently unavailable, preliminary X-ray crystallographic analysis of HIV-1 RT containing an RNA/DNA hybrid and compound 9 shows electron density corresponding to the inhibitor in the immediate vicinity of p51 α-helix H (W. Yang, personal communication).
Figure 5.

Yonetani-Theorell plot for inhibition of HIV-1 RT-RNase H activity in the presence of β-thujaplicinol and (a) Compound 9 or (b) Compound 29. The inhibitory activity (defined as V0/Vi) for each concentration of β-thujaplicinol (β-TP: 0, 0.125, 0.25, 0.5, and 1 μM) is plotted as a function of the concentrations of the thienopyrimidinone inhibitors (0, 0.625, 1.25, 2.5, and 5 μM). The assay was performed at fixed concentrations of substrate and enzyme (250 nM and 17 nM, respectively).
Since a catechol-substituted pyrimidinone is common to compounds 9 and 24-30, their activity differences should reflect the consequences of their thiophene ring substitutions. However, increasing length of C5 (methyl, propyl and butyl, respectively) or C6 alkyl substitution (ethyl) does not significantly affect activity. Moreover, introducing an aliphatic ring also does not change the IC50, regardless of the ring size, which is in contrast to our previous data with the 2-amino-thiophene-3-carboxamide derivatives, where only the cycloheptene containing compound (NSC727447) was active13. The favorable properties of these compounds are intriguing, since their cycloalkene substituent introduces considerable bulk and rigidity which may drive a favorable binding conformation. One explanation may be that the 3′,4′-dihydroxyphenyl substitution “anchors” the compounds at the p51-p66 subunit interface deeper than the other thienopyrimidinones, which would strengthen their binding affinities and provide more room to accommodate bulky substituents on the thiophene ring without affecting overall binding. Any change in the position of -OH substitution or adding an additional –OH group appears to lower this anchoring effect of the inhibitors, thus decrease inhibitory activity. Improving the selectivity ratio of this class of RNase H inhibitor will permit selection of drug-resistant variants to verify that the dimer interface is the bona fide target. Efforts are presently underway to obtain a co-crystal of HIV-1 RT and compound 29, and compare this with data for compound 9, as well as study the effect of structural modifications of the thiophene-pyrimidinone core in order to understand its role in binding. For example, since the thiophene ring is susceptible to hepatic oxidation by cytochrome P450 and sulfur itself can also undergo oxidation, it would be beneficial to find alternative aromatic rings which may possess better pharmacological properties in the future. Our preliminary data have already shown that substituting the thiophene with a benzene ring, a well-known bioisostere for a thiophene ring, retains its inhibitory activity against our engineered drug-resistant enzymes (Data not shown). This would open a new door to the development of next generation inhibitors by taking advantage of a highly diverse library of substituted benzenes.
Experimental Section
General Experimental
All reagents were purchased from commercial sources and used without further purification. Reaction progress was monitored by TLC using Aldrich silica gel 60 F254 (0.25 mm) with detection by UV. 1H and 13C NMR spectra were recorded on a Varian Unity Inova 500 MHz spectrometer. IR spectra were recorded in KBr on a Perkin-Elmer 1310 spectrophotometer. Melting points were determined on a Stuart Scientific SMP 11 melting point apparatus. HPLC/APCI-MS analysis: a Varian tandem mass spectrometer (Palo Alto, CA, USA) 1200 L triple quadrupole mass spectrometer was used with an APCI source. Varian MS workstation version 6.7 software was used for data acquisition and processing. APCI was operated in both positive and negative ion modes. The capillary potential was 75 V, the APCI torch 450°C, and the shield was 600 V. Nitrogen at 48 mTorr was set at 400°C. Full scan spectra were obtained in the range of 100-1000 amu, scan time 0.75 amu, scan width 0.70 amu, detector at 1450V. For APCI the atmospheric pressure ionization (API) housing was kept at 50°C. Parent compounds were dissolved in a mixture consisting of (A) acetonitrile 90% (v/v) and (B) double distilled water 10% (v/v) and infused in the source at the rate of 0.05 mL/min.
Preparation and purification of wild type p66/p51 HIV-1 RT and reconstituted p51 thumb mutants has been previously reported14. All enzymes were stored at -20°C in a 50% glycerol-containing buffer.
General Procedure of Syntheses of 2-Amino-4,5-disubstituted-thiophene-3-carboxamides (40 -47)
The appropriate ketone (14 mmol), cyanoacetamide (14 mmol), sulfur (14 mmol) and diethylamine (15.4 mmol) in 15 mL of dry ethanol were stirred overnight at room temperature. The reaction was quenched with 100 mL of water and extracted several times with ethyl acetate:ethanol (3:1). The organic layer was dried over Na2SO4 and, after filtration, the solvent was evaporated under vacuum to give a brown oil or solid. The crude product was then crystallized from ethanol to give compounds 40 - 47. Characterization of all thienopyrimidinones used in the study is provided in Supporting Information.
General Procedure of Syntheses of Thieno[2,3-d]pyrimidin-4(3H)-ones (1 - 39)
A mixture of 2-amino-4,5-disubstituted-thiophene-3-carboxamide (40 - 47) (1.5 mmol), molecular iodine (1.95 mmol) and the appropriate aromatic aldehyde (1.8 mmol) in the presence of acetonitrile as solvent (15 mL) was stirred at room temperature for the period indicated (TLC) (1-5 h). The reaction was quenched with 35 mL of 5% Na2S2O3 solution and the resulted precipitate was filtered off. The crude product was washed with petroleum ether:ethyl acetate (1:1) and re-crystallized from ethanol to give compounds 1 - 39. Characterization of all thienopyrimidinones used in the study is provided in Supporting Information. The purity of synthetic compounds 1 - 39 was assessed to be ≥95% by HPLC analysis (Agilent HP-1100).
RNase H Inhibitor Analysis
IC50 values were determined as previously reported13, using an 18-nt 3′-fluorescein-labeled RNA annealed to a complementary 18-nt 5′-dabsyl-labeled DNA. To a 96-well plate was added 1 μL of each inhibitor (in DMSO), followed by 10 μL of the appropriate RT (15 - 80 ng/mL) in reaction buffer. Hydrolysis was initiated by adding 10 μL of RNA/DNA hybrid (2.5 μM). Final assay conditions were 50 mM Tris·HCl, pH 8.0, 60 mM KCl, 10 mM MgCl2, 1% DMSO, 150 - 800 ng RT, 250 nM substrate, and increasing concentrations of inhibitor. Wells containing only DMSO was used as negative control. Plates were incubated at 37°C in a Spectramax Gemini EM fluorescence spectrometer for 10 minutes, and fluorescence (lex = 485 nm; lem = 520 nm) was measured at 1-minute intervals such that linear initial rates could be measured in the presence (vi) and absence (vo) of inhibitor. Percent inhibition was calculated as 100 × (vo – vi)/vo, and plotted against log[I]. IC50 values were determined using Prism5 (GraphPad Software). All assays were performed in triplicate.
Yonetani-Theorell Analysis
To determine whether the binding site for thienopyrimidinones overlaps with that of the active site RNase H inhibitor β-thujaplicinol26. RNase H activity was measured in the presence of varying concentrations of compounds 9 and 29 at fixed concentrations of β-thujaplicinol. The assay was conducted at fixed concentrations of RNA/DNA substrate (250 nM) and enzyme (4 nM). Hydrolysis was initiated by adding RNA/DNA hybrid, and monitored for 60 minutes. Data analysis was performed with SigmaPlot (SPSS Inc). The following equation was used for data analysis:
where nij = enzyme velocity in the presence of both compounds at concentrations (I) and (J) and g is the interaction term that defines the degree to which binding of one compound perturbs binding of the other.
Differential Scanning Fluorimetry (ThermoFluor)
Thermal stability assays were performed according to Nettleship et al.28. To a LightCycler®480 96-well plate (Roche) was added 1 μL of 500 μM inhibitor in DMSO, followed by 49 μL of 300 nM HIV-1 RT in reaction buffer containing 20 mM HEPES, pH 7.5, 10 mM MgCl2, 100 mM NaCl, and a 1:1000 dilution of Sypro® Orange dye (Invitrogen). The mixture was heated from 30 to 90°C in increments of 0.2 °C. Fluorescence intensity was measured using excitation/emission wavelengths of 483 nm and 568 nm, respectively. Changes in protein thermal stability (ΔTm) upon inhibitor binding were analyzed by using LightCycler® 480 Software. All assays were performed in triplicate.
HIV-1 IN assay
The evaluate 3′-end processing activity assay of HIV-1 IN, a duplex DNA substrate was prepared by annealing 20-nt 32P-labeled DNA (5′-32P-TGTGGAAAATCTCTAGCAGT-3′) with its complementary strand (5′-ACTGCTAGAGATTTTCCACA-3′). Recombinant HIV-1 IN (750 nM) was preincubated with inhibitor (in DMSO) for 10 min at room temperature. 3′-processing was initiated by adding DNA substrate, and then incubated for an hour at 37°C. Final assay conditions were 50 mM MOPS, pH 7.2, 10 mM MgCl2, 50 mM NaCl, 2 mM 2-mercaptoethanol, 10% DMSO, 50 nM substrate, and 50 μM inhibitor. The reaction was stopped by adding loading buffer containing 89 mM Tris-borate pH 8.3, 6 M urea, 25 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol and analyzed on a 15% denaturing polyacrylamide gel. The fraction of substrate converted to the specific 3′-processed product was determined by PhosphorImager analysis (Typhoon Trio+; GE Healthcare).
DNA-dependent DNA polymerase assay
DNA-dependent DNA synthesis was measured on a fluorescently labeled duplex DNA generated by annealing a 42-nucleoptide (nt) template (5′-TAC ATA CCC ATA CAT AAA TCC TAA CCT TGA AGA ACT CGT CAC-3′) to the 5′ Cy5-labeled primer 5′-ATG TAT GGG TAT GTA TTT AGG-3′. Polymerization was initiated by adding 1 μL of 2 mM deoxynucleoside triphosphates (dNTPs) to 9 μL of a mixture containing 15 ng RT, 200 nM substrate, 10 mM Tris-HCl (pH 7.8), 80 mM KCl, 10 mM MgCl2, 10% DMSO, and 50 μM inhibitor at 37°C and was terminated after 15 min by adding an equal volume of 8M urea. Reaction products were analyzed by 15% denaturing polyacrylamide gel electrophoresis and fluorescent imaging (Typhoon Trio+; GE Healthcare).
Antiviral Activity
The antiviral activity of selected thienopyrimidinones was determined via the 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-5-[(phenylamino)carbonyl]-2H tetrazolium hydroxide (XTT)-based cell viability assay of Weislow et al. 29 using the HIV-1RF isolate and human T-cell line CEM-SS.
Supplementary Material
Acknowledgments
We would like to thank Dr Wei Yang, NIH, for communication of crystallographic data on HIV-1 RT containing compound 9 prior to publication.
Funding Sources: T. M., S. C., M. W., H. T. -S., J. R., J. A. B. and S. Le G. were supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. G.T. was supported by Ministero dell'Università, dell'Istruzione e della Ricerca MIUR- PRIN 2008 and Fondazione Banco di Sardegna.
Abbreviations Used
- DNTP
5,6-dimethyl-2-(4-nitrophenyl)thieno[2,3-d]pyrimidin-4(3H)-one
- RNase H
ribonuclease H
- NNRTI
nonnucleoside reverse transcriptase inhibitors
- RT
reverse transcriptase
Footnotes
Supporting Information: Spectroscopic characterization of the compounds in this paper and details related to in vitro studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Luzuriaga K, Bryson Y, Krogstad P, Robinson J, Stechenberg B, Lamson M, Cort S, Sullivan JL. Combination treatment with zidovudine, didanosine, and nevirapine in infants with human immunodeficiency virus type 1 infection. N Engl J Med. 1997;336:1343–1349. doi: 10.1056/NEJM199705083361902. [DOI] [PubMed] [Google Scholar]
- 2.Das K, Lewi PJ, Hughes SH, Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Abbondanzieri EA, Bokinsky G, Rausch JW, Zhang JX, Le Grice SF, Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptas. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, Fuchs JR, Kvaratskhelia M. A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem. 2012;287:16801–16811. doi: 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perryman AL, Zhang Q, Soutter HH, Rosenfeld R, McRee DE, Olson AJ, Elder JE, Stout CD. Fragment-based screen against HIV protease. Chem Biol Drug Des. 2010;75:257–268. doi: 10.1111/j.1747-0285.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schatz O, Cromme F, Naas T, Lindemann D, Gruninger Leitch F, Mous J, Le Grice SFJ. Inactivation of the RNase H domain of HIV 1 reverse transcriptase blocks viral infectivity. Portfolio Publishing Company; Houston, Texas: 1990. pp. 293–303. [Google Scholar]
- 8.Tisdale M, Schulze T, Larder BA, Moelling K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J Gen Virol. 1991;72(Pt 1):59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- 9.Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol. 2009;388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wendeler M, Lee HF, Bermingham A, Miller JT, Chertov O, Bona MK, Baichoo NS, Ehteshami M, Beutler J, O'Keefe BR, Gotte M, Kvaratskhelia M, Le Grice S. Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem Biol. 2008;3:635–644. doi: 10.1021/cb8001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava S, Sluis-Cremer N, Tachedjian G. Dimerization of human immunodeficiency virus type 1 reverse transcriptase as an antiviral target. Curr Pharm Des. 2006;12:1879–1894. doi: 10.2174/138161206776873590. [DOI] [PubMed] [Google Scholar]
- 12.Tachedjian G, Aronson HE, de los Santos M, Seehra J, McCoy JM, Goff SP. Role of residues in the tryptophan repeat motif for HIV-1 reverse transcriptase dimerization. J Mol Biol. 2003;326:381–396. doi: 10.1016/s0022-2836(02)01433-x. [DOI] [PubMed] [Google Scholar]
- 13.Chung S, Wendeler M, Rausch JW, Beilhartz G, Gotte M, O'Keefe BR, Bermingham A, Beutler JA, Liu S, Zhuang X, Le Grice SF. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob Agents Chemother. 2010;54:3913–3921. doi: 10.1128/AAC.00434-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung S, Miller JT, Johnson BC, Hughes SH, Le Grice SF. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J Biol Chem. 2011;287:4066–4075. doi: 10.1074/jbc.M111.314781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakavoli M, Bagherzadeh G, Vaseghifar M, Shiri A, Pordeli P. Iodine catalysed synthesis and antibacterial evaluation of thieno[2,3-d] pyrimidine derivatives. Journal of Chemical Research-S. 2009:653–655. [Google Scholar]
- 17.Jamieson C, Basten S, Campbell RA, Cumming IA, Gillen KJ, Gillespie J, Kazemier B, Kiczun M, Lamont Y, Lyons AJ, Maclean JK, Moir EM, Morrow JA, Papakosta M, Rankovic Z, Smith L. A novel series of positive modulators of the AMPA receptor: discovery and structure based hit-to-lead studies. Bioorg Med Chem Lett. 20:5753–5756. doi: 10.1016/j.bmcl.2010.07.138. [DOI] [PubMed] [Google Scholar]
- 18.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 19.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 20.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol. 2010;84:7625–33. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubey S, Satyanarayana YD, Lavania H. Development of integrase inhibitors for treatment of AIDS: an overview. Eur J Med Chem. 2007;42:1159–68. doi: 10.1016/j.ejmech.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322:1092–7. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Smerdon SJ, Jager J, Kohlstaedt LA, Rice PA, Friedman JM, Steitz TA. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc Natl Acad Sci U S A. 1994;91:7242–7246. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tchertanov L, Mouscadet JF. Target recognition by catechols and beta-ketoenols: potential contribution of hydrogen bonding and Mn/Mg chelation to HIV-1 integrase inhibition. J Med Chem. 2007;50:1133–1145. doi: 10.1021/jm061375j. [DOI] [PubMed] [Google Scholar]
- 26.Yonetani T. The Yonetani-Theorell graphical method for examining overlapping subsites of enzyme active centers. Methods Enzymol. 1982;87:500–509. doi: 10.1016/s0076-6879(82)87028-6. [DOI] [PubMed] [Google Scholar]
- 27.Lapkouski M, Tian L, Miller JT, Le Grice SF, Yang W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat Struct Mol Biol. 2013;20:230–236. doi: 10.1038/nsmb.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nettleship JE, Brown J, Groves MR, Geerlof A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 2008;426:299–318. doi: 10.1007/978-1-60327-058-8_19. [DOI] [PubMed] [Google Scholar]
- 29.Weislow OS, Kiser R, Fine DL, Bader J, Shoemaker RH, Boyd MR. New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst. 1989;81:577–586. doi: 10.1093/jnci/81.8.577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.