

Abstract
Among epigenetic “writers”, “readers”, and “erasers”, the lysine methyltransferases G9a and GLP, which catalyze mono- and dimethylation of histone H3 lysine 9 (H3K9me2) and non-histone proteins, have been implicated in a variety of human diseases. A “toolkit” of well-characterized chemical probes will allow biological and disease hypotheses concerning these proteins to be tested in cell-based and animal models with high confidence. We previously discovered potent and selective G9a/GLP inhibitors including the cellular chemical probe UNC0638, which displays an excellent separation of functional potency and cell toxicity. However, this inhibitor is not suitable for animal studies due to its poor pharmacokinetic (PK) properties. Here, we report the discovery of the first G9a and GLP in vivo chemical probe UNC0642, which not only maintains high in vitro and cellular potency, low cell toxicity, and excellent selectivity, but also displays improved in vivo PK properties, making it suitable for animal studies.
Introduction
Protein lysine methylation catalyzed by protein lysine methyltransferases (PKMTs, also known as histone methyltransferases (HMTs)) has been increasingly recognized as a major signaling mechanism in eukaryotic cells.1–5 PKMTs target both histone and non-histone substrates and display significant variation in their ability to catalyze mono-, di-, and/or trimethylation.1, 3, 5–8 In the context of epigenetic gene regulation, the different states of histone lysine methylation encode distinct signals and are recognized by a host of proteins and protein complexes. More than 50 PKMTs have been identified to date and many of them have been implicated in various human diseases.1, 3, 9, 10 During the last several years, the PKMT target class has received considerable attention from the drug discovery and medicinal chemistry community. A number of selective small-molecule inhibitors, which target the PKMT substrate binding groove,11–17 cofactor binding site,18–31 and a PRMT (protein arginine methyltransferase) allosteric binding site,32, 33 have been reported. However, well-characterized chemical probes34–36 of PKMTs that are suitable for cell-based and animal studies are still rare. Such probes are invaluable tools for testing biological and therapeutic hypotheses concerning the PKMTs and for their validation as drug targets.
G9a (also known as KMT1C (lysine methyltransferase 1C) or EHMT2 (euchromatic histone methyltransferase 2)) and GLP (also known as KMT1D (lysine methyltransferase 1D) or EHMT1 (euchromatic histone methyltransferase 1)) are two closely related proteins and were initially identified as H3K9 (histone H3 lysine 9) methyltransferases.37, 38 They share 80% sequence identity in their respective SET (suppressor of variegation 3–9, enhancer of zeste, and trithorax) domains.38 In addition to H3K9, G9a and GLP methylate many non-histone proteins.39, 40 For example, G9a and GLP catalyze dimethylation of the tumor suppressor p53, resulting in inactivation of the transcriptional activity of p53.6 G9a is overexpressed in leukemia,6 prostate carcinoma,6, 41 hepatocellular carcinoma,42 and lung cancer.43 Knockdown of G9a inhibits prostate, lung, and leukemia cancer cell growth.41, 43, 44 Moreover, G9a and/or GLP play a role in cocaine addiction,45, 46 mental retardation,47 maintenance of HIV-1 (human immunodeficiency virus type 1) latency,48 and stem cell function, maintenance, differentiation and reprogramming.49–54 In addition, GLP has been implicated in Kleefstra syndrome,55, 56 a disorder affecting intellectual ability.
BIX01294 (1), the first selective inhibitor of G9a and GLP, was discovered via high throughput screening (Figure 1).11 Optimization of this chemical series based on the cocrystal structure of GLP in complex with inhibitor 157 led to the discovery of potent and selective G9a/GLP inhibitors UNC0224 (2), UNC0321 (3), and E72 (4) (Figure 1).12–14 Further optimization of this quinazoline scaffold resulted in the discovery of the G9a and GLP cellular chemical probe UNC0638 (5), which displays balanced in vitro potency, aqueous solubility, and cell membrane permeability (Figure 1).15, 16 Inhibitor 5 is highly selective for G9a and GLP over a broad range of epigenetic and non-epigenetic targets, and exhibits robust on-target activities in cells and low cell toxicity.15 More recently, BRD9539 (6), a structurally distinct inhibitor of G9a, was reported (Figure 1).20 Although Inhibitor 5 is an excellent chemical probe for cell-based studies,54, 58 it is not suitable for animal studies due to its poor in vivo pharmacokinetic (PK) properties.15 We therefore endeavored to optimize the PK properties of the quinazoline series. Here we report the discovery of UNC0642 (7), the first in vivo chemical probe of G9a and GLP. This inhibitor not only displays high in vitro and cellular potency, low cell toxicity, and excellent selectivity, but also exhibits greatly improved in vivo PK properties as compared to inhibitor 5. We describe: (1) the synthesis of novel compounds aimed at improving PK properties of this series; (2) structure activity relationships (SAR) of these compounds in biochemical and cell-based assays; (3) in vitro and in vivo PK properties of selected inhibitors; and (4) further characterization of inhibitor 7 in a number of biochemical and cell-based studies including mechanism of action, selectivity, and phenotypic effect studies.
Figure 1.

Results and Discussion
Synthesis
We hypothesized that the 2-cyclohexyl group of inhibitor 5 could contribute to its poor metabolic stability due to cytochrome P450 mediated oxidation of the cyclohexyl ring. Because modifications to the 2-substituent of the quinazoline scaffold are well tolerated,14, 16 we extensively explored this region and designed a number of novel analogs aimed at improving metabolic stability while maintaining high in vitro and cellular potency. We also conducted limited exploration of the 4-amino and 7-aminoalkoxy regions.
We previously developed an efficient synthetic route for preparing 2-amino quinazolines, which allows various 2-amino groups to be installed at the last step of the synthetic sequence.16 Using this efficient route, we synthesized the 2-amino quinazolines 7, 13 – 17 from commercially available methyl 4-hydroxy-3-methoxybenzoate (8) in 9 steps in good yields (Scheme 1 and Table 1). Briefly, compound 8 was reacted with 1-chloro-3-iodopropane, followed by nitration, to afford the chloride 9. Substitution of the chloride with pyrrolidine and subsequent reduction of the nitro group yielded the aniline 10. Urea formation and subsequent ring closure afforded the intermediate 11, which was then converted to the 2,4-dichloroquinazoline 12. Two consecutive displacement reactions yielded the desired final products 7, 13 – 17. It is worth noting that 5 g of inhibitor 7 was produced in 14.8% overall yield using this synthetic route. We also synthesized compounds 18 and 19, which contain a cyclopropyl group instead of an isopropyl group as the N-capping of the upper piperidine moiety, following the same synthetic route.
Scheme 1. Synthesis of Compounds 7, 13 – 19a.

a Reagents and conditions: (a) 1-Chloro-3-iodopropane, K2CO3, CH3CN, reflux; (b) HNO3, Ac2O, 0 °C to rt, 75% over 2 steps; (c) pyrrolidine, K2CO3, NaI, cat. tetrabutylammonium iodide, CH3CN, reflux, 81%; (d) Fe dust, NH4OAc, AcOEt, H2O, reflux, 63%; (e) NaOCN, AcOH, H2O, rt; (f) NaOH, H2O, MeOH, reflux; (g) N,N-diethylaniline POCl3, reflux, 50% over 3 steps; (h) 1-isopropyl piperidin-4-amines, DIEA, THF, rt, 94%; (i) 1-cyclopropyl piperidin-4-amines, DIEA, THF, rt, 89%; (j) various amines, i-PrOH, 160 °C, microwave, 74–82%.
Table 1.
SAR of the 2-Amino Moiety.
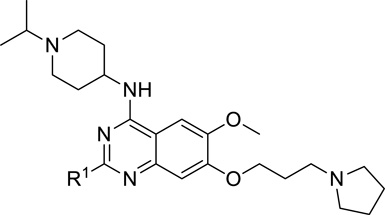 | ||
|---|---|---|
| Compound | R1 | G9a IC50 (nM)a |
| 5 | 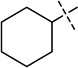 |
< 2.5 |
| 7 (UNC0642) | 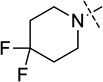 |
< 2.5 |
| 13 (UNC1479) | 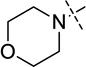 |
< 2.5 |
| 14 | 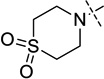 |
14 ± 0.6 |
| 15 | 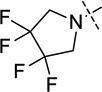 |
26 ± 1 |
| 16 | 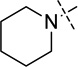 |
< 2.5 |
| 17 | 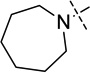 |
< 2.5 |
| 22 | 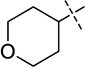 |
9.0 ± 0.1 |
IC50 determination experiments were performed in triplicate.
Compound 22 which contains a tetrahydropyran-4-yl group at the 2-position was prepared according to the synthetic route15 developed for synthesis of inhibitor 5 (Scheme 2). In brief, the aniline 10 was reacted with tetrahydro-2H-pyran-4-carbonitrile to yield the cyclization product 20, which was then converted to the 4-chloroquinazoline 21. Subsequent displacement of the chloride with 1-isopropylpiperidin-4-amine afforded the desired product 22.
Scheme 2. Synthesis of Compound 22a.

a Reagents and conditions: (a) 4 N HCl, dioxane, tetrahydro-2H–pyran-4-carbonitrile, 100 °C; (b) N,N-diethylaniline, POCl3, reflux, 48% over 2 steps; (c) 1-isopropylpiperidin-4-amine, K2CO3, DMF, 60 °C, 78%.
Finally, compound 26, which was designed to explore the 7-aminoalkoxy moiety, was synthesized according to Scheme 3. Substitution of the chloride 9 with 4,4-difluoropiperidine and subsequent reduction of the nitro group afforded the aniline 23, which was reacted with cyclohexanecarbonitrile or tetrahydro-2H-pyran-4-carbonitrile to yield the quinazoline 24. Similarly to synthesis of compound 22, the intermediate 24 was converted to the 4-chloroquinazoline 25, which was subsequently converted to the desired product 26.
Scheme 3. Synthesis of Compounds 26a and 26ba.

a Reagents and conditions: (a) 4,4-difluoropiperidine, K2CO3, NaI, cat. tetrabutylammonium iodide, CH3CN, reflux, 81%; (b) Fe dust, NH4OAc, AcOEt-H2O, reflux, 60%; (c) 4 N HCl, dioxane, cyclohexanecarbonitrile or tetrahydro-2H–pyran-4-carbonitrile, 100 °C; (d) N,N-diethylaniline, POCl3, reflux, 49–53% over 2 steps; (e) 1-isopropylpiperidin-4-amines, K2CO3, DMF, 60 °C, 75–77%.
SAR in a G9a Biochemical Assay
The synthesized compounds were evaluated in a radioactive biochemical assay which measures the transfer of the tritiated methyl group from the cofactor 3H – S-adenosyl methionine (SAM) to a peptide substrate catalyzed by G9a. IC50 values of these compounds in this biochemical assay are summarized in Tables 1 – 3.
Table 3.
SAR of the 7-Aminoalkoxy Moiety.
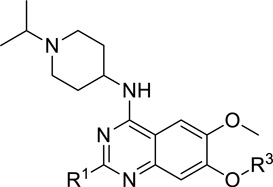 | |||
|---|---|---|---|
| Compound | R1 | R3 | G9a IC50 (nM)a |
| 5 | 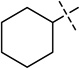 |
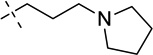 |
< 2.5 |
| 26a | 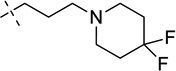 |
> 26,000 | |
| 22 | 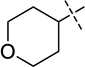 |
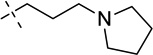 |
9.0 ± 0.1 |
| 26b | 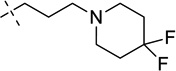 |
> 50,000 | |
IC50 determination experiments were performed in triplicate.
We were pleased to find that the 2-cyclohexyl group (5) can be replaced by a variety of 2-substituents without a significant loss of potency (Table 1). In particular, compounds possessing a 2-(4,4-difluoropiperidin-1-yl) (7), 2-(morpholin-4-yl) (13), 2-(piperidin-1-yl) (16) or 2(azepan-1-yl) (17) maintained high in vitro potency (IC50 < 2.5 nM). Although compounds 14, 15, and 22, which contain a 2-(1,1-dioxidethiomorpholin-4-yl), 2-(3,3,4,4-tetrafluoropyrrolin-1yl) or 2-(tetrahydropyran-4-yl) group, were not as potent as compound 5, the in vitro potency of these inhibitors is still quite good (IC50 < 30 nM). These SAR findings are consistent with our previous results,14, 16 further demonstrating that modifications to the 2-amino region of the quinazoline scaffold are well tolerated.
For the N-capping of the upper piperidine moiety, we found that the isopropyl group can be replaced by a cyclopropyl group (7 versus 18, and 14 versus 19) without a significant change in potency (Table 2). Although we previously showed that a large N-capping group such as cyclohexylmethyl is tolerated in this region,16 we thought that such a large lipophilic group might increase metabolic liability, and therefore, did not explore this region further. In addition, we attempted to replace the 7-(3-(pyrrolidin-1-yl)propoxy) group with the 7-(3-(4,4-difluoropiperidin-1-yl)propoxy) group (Table 3). To our surprise, this replacement resulted in a complete loss of potency (5 versus 26a, and 22 versus 26b). Because a basic nitrogen in this region is required for maintaining high in vitro potency and few modifications are tolerated,14, 16 we did not further investigate this region.
Table 2.
SAR of the N capping of the Upper Piperidine Moiety.
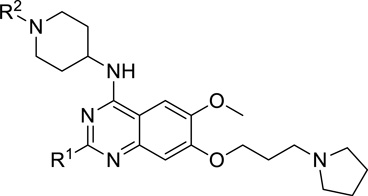 | |||
|---|---|---|---|
| Compound | R1 | R2 | G9a IC50 (nM)a |
| 7 | 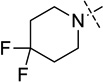 |
i-propyl | < 2.5 |
| 18 | c-propyl | 3.0 ± 0.1 | |
| 14 | 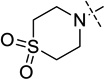 |
i-propyl | 14 ± 0.6 |
| 19 | c-propyl | 4.0 ± 0.2 | |
IC50 determination experiments were performed in triplicate.
Assessment of Functional Potency and Cell Toxicity
The inhibitors which displayed IC50 < 10 nM in the biochemical assay were next evaluated in an H3K9me2 cell immunofluorescence in-cell western (ICW) assay for assessing their cellular potency, and a standard resazurin (Alamar Blue) reduction assay for assessing their cell toxicity. MDA-MB-231 cells were used in this study because this cell line possesses robust H3K9me2 levels.
We were pleased to find that all 8 inhibitors exhibited good potency at reducing cellular levels of H3K9me2 (IC50 = 100 – 600 nM), low cell toxicity (EC50 > 5,000 nM), and a good separation of functional potency and cell toxicity with a ratio of toxicity to functional potency (tox/function ratio, which is determined by dividing the EC50 value of the observed toxicity by the IC50 value of the functional potency) > 45 (Table 4). In particular, inhibitors 7, 13, and 22 displayed high cellular potency and an excellent tox/function ratio (> 130), similar to that of our G9a/GLP cellular chemical probe 5.
Table 4.
Functional Potency and Cell Toxicity of Selected Inhibitors in MDA-MB-231 Cellsa
| Compound | H3K9me2 IC50 (nM) |
Cell Toxicity EC50 (nM) |
Tox/function Ratio |
|---|---|---|---|
| 5 | 81 | 11,000 | 136 |
| 7 | 110 | 16,700 | 152 |
| 13 | 180 | 23,700 | 132 |
| 16 | 150 | 7,000 | 47 |
| 17 | 100 | 5,700 | 57 |
| 18 | 310 | 14,700 | 47 |
| 19 | 590 | > 50,000 | > 85 |
| 22 | 310 | > 50,000 | > 161 |
IC50 or EC50 values are the average of experimental triplicates with standard deviation (SD) values that are about 3-fold less than the average.
Assessment of in vitro and in vivo PK Properties
We next evaluated in vitro metabolic stability of selected inhibitors using mouse liver microsomes. As shown in Table 5, inhibitors 7 and 13 displayed improved intrinsic clearance (CLint) and half-life (T1/2) compared with inhibitor 5. Interestingly, inhibitor 15, which possesses a 2-(3,3,4,4-tetrafluoropyrrolin-1yl) group, exhibited the best in vitro metabolic stability although this inhibitor was significantly less potent than inhibitors 5, 7 and 13 (see Table 1). On the other hand, compounds 22 and 26, which contain a 2-(tetrahydropyran-4-yl) group, displayed a significant decrease in metabolic stability. Surprisingly, inhibitor 26 which has a 7-(3-(4,4-difluoropiperidin-1-yl)propoxy) group was not only less potent than compound 22 which contains 7-(3-(pyrrolidin-1-yl)propoxy) group (see Table 3), but also metabolically less stable compared with inhibitor 22. Taken together, these results indicate that modifications aimed at preventing CYP450 mediated oxidation of the 2-substituent of the quinazoline scaffold can lead to improved in vitro metabolic stability.
Table 5.
In vitro Metabolic Stability of Selected Inhibitors.
| Compound | CLint (mL/min/g liver) |
T1/2 (min) |
|---|---|---|
| 5 | 1.0 | 73 |
| 7 | 0.5 | > 90 |
| 13 | 0.7 | > 90 |
| 15 | < 0.5 | > 90 |
| 22 | 4.7 | 16 |
| 26 | 7.4 | 10 |
On the basis of high in vitro and cellular potencies, low cell toxicity, and good in vitro metabolic stability, we selected inhibitors 7 and 13 for evaluation of their in vivo PK properties in male Swiss Albino mice. A single intraperitoneal (IP) injection (5 mg/kg) of inhibitor 7 resulted in a plasma Cmax (maximum concentration) of 947 ng/mL, which is more than 10-fold higher than that of inhibitor 5, and an AUC (area under the curve) of 1265 hr*ng/mL, which is also much higher than that of inhibitor 5 (Table 6). Similarly, a single 5 mg/kg IP injection of inhibitor 13 gave a much improved Cmax and AUC in plasma compared with inhibitor 5. In addition, while inhibitor 7 displayed modest brain penetration with a brain/plasma ratio of 0.33, inhibitor 13 exhibited increased CNS (central nervous system) penetration with a brain/plasma ratio of 0.68. These encouraging PK results suggest that inhibitors 7 and 13 could be used for animal studies and 13 might be suitable for studies to explore the role of G9a/GLP in the CNS. These results also indicate that appropriate modifications to the 2-substituent of the quinazoline scaffold can improve in vivo PK properties, thus validating our probe design hypothesis.
Table 6.
In vivo PK Properties of Selected Inhibitorsa
| Compound | Matrix | Cmax (ng/mL) |
AUC0–24h (hr*ng/mL) |
Brain/Plasma ratio |
|---|---|---|---|---|
| 5 | Plasma | 66 | 404 | — |
| 7 | Brainb | 68 | 412 | 0.33 |
| Plasma | 947 | 1265 | ||
| 13 | Brainb | 99 | 721 | 0.68 |
| Plasma | 731 | 1061 |
Plasma and brain concentrations were measured at 8 time points with 3 animals per time point. Clearance, half-life, and volume distribution values are not reported because only IP administration was performed.
The density of brain homogenate was considered as 1 which is equivalent to plasma density (1); Brain concentrations and exposures are expressed as ng/g and hr*ng/g, respectively.
Further Characterization of Inhibitor 7
We next carried out a number of probe characterization studies. First, we studied the MOA (mechanism of action) of inhibitor 7 by determining Michaelis-Menten kinetic parameters associated with both the peptide substrate and cofactor SAM. As shown in Figure 2, the apparent Km of the peptide (Kmapp) increased linearly with inhibitor concentration, whereas the Kmapp of SAM remained constant in the presence of increasing concentrations of the inhibitor. These results indicate that inhibitor 7 is competitive with the peptide substrate and non-competitive with the cofactor SAM, the same MOA as inhibitor 5.15 The Ki of inhibitor 7 was determined to be 3.7 ± 1 nM (n = 3).
Figure 2.

MOA studies of inhibitor 7. (A) and (B) The competition of inhibitor 7 and the H3K9 peptide indicates that 7 is competitive with the peptide substrate. The Kmapp of the peptide increased linearly with compound concentration. (C) and (D) The competition with the cofactor SAM reveals that inhibitor 7 is non-competitive with the cofactor. The Kmapp of SAM were not affected by inhibitor concentration.
We next determined the selectivity of inhibitor 7 versus 15 methyltransferases and a broad range of non-epigenetic targets. We expected this inhibitor to be highly potent for GLP on the basis of the high sequence identity between G9a and GLP.10 Indeed, inhibitor 7 displayed high in vitro potency for GLP (IC50 < 2.5 nM), similar to G9a (Figure 3A, Table 1). This result is consistent with the selectivity profile of our cellular chemical probe 5.15 Importantly, inhibitor 7 was more than 20,000-fold selective for G9a and GLP over 13 other methyltransferases (IC50 > 50,000 nM) and more than 2,000-fold selective over PRC2 – EZH2 (polycomb repressive complex 2 – enhancer of zeste homolog 2, IC50 > 5,000 nM) (Figure 3B). In addition, inhibitor 7 showed no appreciable inhibition (less than 20% inhibition at 10,000 nM) against a panel of 50 representative kinases (Table S1). We also tested inhibitor 7 against 44 GPCRs (G protein-coupled receptors), transporters, and ion channels in the National Institute of Mental Health – Psychoactive Drug Screen Program Selectivity Panel. This inhibitor was found to show less than 50% inhibition at 1,000 nM against 39 targets and > 50% inhibition at 1,000 nM against 5 targets in the panel (Table S2). Ki values for each of the 5 interacting targets were then determined in radioligand binding assays. Inhibitor 7 had Ki values of 4,500 nM, > 10,000 nM, 45 nM, > 10,000 nM, and 900 nM for α1D, α2C, histamine H3, µ opioid, and σ2 receptors, respectively (Table S3). Therefore, with the exception of the histamine H3 receptor, inhibitor 7 was more than 300-fold selective for G9a and GLP over a broad range of kinases, GPCRs, transporters, and ion channels.
Figure 3.

Selectivity of inhibitor 7 versus 15 other methyltransferases.
In addition to MDA-MB-231 cells, we assessed functional potency and cell toxicity of inhibitor 7 in several other cell lines (Table 7). Inhibitor 7 displayed high potency (IC50 < 150 nM) in reducing cellular levels of H3K9me2, low cell toxicity (EC50 > 3,000 nM), resulting in a good separation of functional potency and cell toxicity with a tox/function ratio of > 45 in U2OS, PC3, and PANC-1 cells. In particular, this inhibitor exhibited excellent potency (IC50 = 40 nM) in PANC-1 cells with a good tox/function ratio of 88.
Table 7.
Functional Potency and Cell Toxicity of Inhibitor 7 in Various Cell linesa
| Cell line | H3K9me2 IC50 (nM) |
Cell Toxicity EC50 (nM) |
Tox/function Ratio |
|---|---|---|---|
| MDA-MB-231 | 110 | 16,700 | 152 |
| U2OS | 130 | 6,000 | 46 |
| PC3 | 130 | 8,900 | 68 |
| PANC-1 | 40 | 3,500 | 88 |
IC50 or EC50 values are the average of experimental triplicates with SD values that are about 3-fold less than the average.
Lastly, we evaluated effects of inhibitor 7 on clonogenicity in PANC-1 and MDA-MB-231 cells. As shown in Figure 4, inhibitor 7 reduced clonogenicity in PANC-1 cells in a concentration-dependent manner while it had no effect on clonogenicity in MDA-MB-231 cells. These results are consistent with the previous report that PANC-1 cells are highly sensitive to G9a inhibitors20 and our previous finding that MDA-MB-231 cells are insensitive to G9a/GLP inhibitors.15 These observations suggest that pharmacological inhibition of G9a and GLP can have differential phenotypic effects depending on the cell type and/or disease setting.
Figure 4.

Inhibitor 7 affects clonogenicity of PANC-1 cells but not MDA-MB-231 cells. (A) PANC-1 cell colonies after 2 weeks of growth in the presence of inhibitor 7 at 0, 0.5 or 1 µM. (B) Graphical representation of data in (A). (C) MDA-MB-231 cell colonies after 2 weeks of growth in the presence of inhibitor 7 at 0, 0.5 or 1 µM. (D) Graphical representation of data in (C).
Conclusions
We designed, synthesized, and biologically evaluated a set of novel compounds aimed at improving the in vivo PK properties of our previously reported G9a/GLP cellular chemical probe 5. From these studies, we discovered inhibitor 7, which is the first in vivo chemical probe of G9a and GLP. This inhibitor: (1) displayed high in vitro potency for G9a and GLP (IC50 < 2.5 nM); (2) was > 2,000-fold selective for G9a and GLP over PRC2 – EZH2 and > 20,000-fold selective over 13 other methyltransferases; (3) was > 300-fold selective for G9a and GLP over a broad range of kinases, GPCRs, ion channels, and transporters with the exception of the histamine H3 receptor; (4) was competitive with the peptide substrate and non-competitive with the cofactor SAM; (5) exhibited high potency at reducing the H3K9me2 mark, low cell toxicity, and good separation of functional potency and cell toxicity in a number of cell lines; (6) reduced clonogenicity in PANC-1 cells, a pancreatic carcinoma cell line; and (7) importantly, displayed improved in vitro and in vivo PK properties. We also discovered inhibitor 13, which had better brain penetration than inhibitor 7 and might be suitable for CNS studies. These two inhibitors are valuable additions to our G9a/GLP inhibitor toolbox, and are freely available to the research community for investigating the role of G9a and GLP in health and disease.
Experimental Section
Chemistry general procedures
HPLC spectra for all compounds were acquired using an Agilent 6110 Series system with UV detector set to 254 nm. Samples were injected (5 µL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 µM, C18 column at room temperature. A linear gradient from 10% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min was followed by pumping 100% B for another 2 minutes with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. High-resolution (positive ion) mass spectrum (HRMS) for compound 7 and 13 were acquired using a Thermo LTqFT mass spectrometer under FT control at 100,000 resolution. Nuclear Magnetic Resonance (NMR) spectra were recorded at Varian Mercury spectrometer with 400 MHz for proton (1H NMR), 100 MHz for carbon (13C NMR) and 376 MHz for Fluorine (19F NMR). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 220 nm. Samples were injected onto a Phenomenex Luna 75 × 30 mm, 5 µM, C18 column at room temperature. The flow rate was 30 mL/min. A linear gradient was used with 10% of MeOH (A) in 0.1 % TFA in H2O (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All compounds had > 95% purity using the HPLC methods described above.
Synthesis of compound 5 was reported previously.15
2-(4,4-Difluoropiperidin-1-yl)-N-(1-isopropylpiperidin-4-yl)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-4-amine (7)
A mixture of compound 12 (0.72 g, 2.02 mmol) (prepared from commercially available methyl 3-methoxyl-4-hydroxy benzoate (8) according to the procedures described previously15), 1-isopropylpiperidin-4-amine·2TFA salt (2.3 g, 6.06 mmol) (this amine and other non-commercially available amines were prepared according to previously reported procedures59), and DIEA (0.67 mL, 4.04 mmol) in THF (10 mL) was stirred overnight at rt. After concentration in vacuo, the crude product was purified by silica gel chromatography (0 – 20% MeOH (1% NH3)/CH2Cl2) to afford the desired product 2-chloro-N-(1-isopropylpiperidin-4-yl)-6-methoxyl-7-(3-(pyrrolidin-1-yl)propoxyl) quinazolin-4-amine as a yellow solid (0.88 g, 94% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (s, 1H), 6.78 (s, 1H), 5.36 (d, J = 7.7 Hz, 1H), 4.31 – 4.20 (m, 1H), 4.17 (t, J = 6.7 Hz, 2H), 3.96 (s, 3H), 2.90 (d, J = 11.9 Hz, 2H), 2.84 – 2.73 (m, 1H), 2.67 – 2.59 (m, 2H), 2.57 – 2.46 (m, 4H), 2.45 – 2.34 (m, 2H), 2.22 – 2.05 (m, 4H), 1.84 – 1.74 (m, 4H), 1.07 (d, J = 6.6 Hz, 6H). MS (ESI): 462 [M + H]+. A mixture of this intermediate (55 mg, 0.12 mmol), 4,4-difluoropiperidine hydrochloride salt (37 mg, 0.24 mmol), and TFA (55 mg, 0.48 mmol) in i-PrOH (0.25 mL) was heated by microwave irradiation at 160 °C for 15 min in a sealed tube. After concentration in vacuo the crude product was purified by preparative HPLC with a gradient from 10% MeOH (A) in 0.1% TFA in H2O (B) to 100% of MeOH (A). The resulting product was basified with saturated aq. NaHCO3 and extracted with CH2Cl2 to afford the title compound 7 as a white solid (52 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 6.91 (s, 1H), 6.70 (s, 1H), 4.97 (d, J = 7.2 Hz, 1H), 4.18 (t, J = 6.8 Hz, 2H), 4.13 – 4.03 (m, 1H), 4.02 – 3.94 (m, 4H), 3.92 (s, 3H), 2.97 – 2.86 (m, 2H), 2.83 – 2.72 (m, 1H), 2.63 (t, J = 7.2 Hz, 2H), 2.58 – 2.46 (m, 4H), 2.34 (td, J = 11.6, 2.2 Hz, 2H), 2.21 – 2.14 (m, 2H), 2.14 – 2.07 (m, 2H), 2.07 – 1.94 (m, 4H), 1.82 – 1.74 (m, 4H), 1.58 (ddd, J = 14.9, 11.8, 3.7 Hz, 2H), 1.08 (d, J = 6.5 Hz, 6H). 19F NMR (376 MHz, CDCl3) δ −96.10 – −97.04 (m, 2F). 13C NMR (100 MHz, CDCl3) δ 158.49, 158.32, 154.23, 149.33, 146.00, 122.95 (t, J = 241.4 Hz), 107.18, 103.18, 101.34, 67.47, 56.69, 54.59, 54.28 (two carbons), 53.01 (two carbons), 48.77, 47.85, 41.25 (t, J = 4.9 Hz) (two carbons), 34.00 (t, J = 22.4 Hz) (two carbons), 32.72 (two carbons), 28.62 (two carbons), 23.57, 18.58 (two carbons). HPLC: 100%; tR 2.37 min. MS (ESI): 547 [M + H]+. HRMS (ESI) calcd for C29H44F2N6O2Na [M + Na]+: 569.3392. Found: 569.3384.
N-(1-Isopropylpiperidin-4-yl)-6-methoxy-2-morpholino-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-4-amine (13)
The procedure used for preparation of compound 7 was followed for synthesis of compound 13. The title compound 13 was obtained as a white solid (55 mg, 78% yield). 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 6.72 (s, 1H), 5.04 (d, J = 7.3 Hz, 1H), 4.15 (t, J = 6.8 Hz, 2H), 4.12 – 4.02 (m, 1H), 3.89 (s, 3H), 3.85 – 3.72 (m, 8H), 2.95 – 2.85 (m, 2H), 2.81 – 2.71 (m, 1H), 2.61 (t, J = 7.2 Hz, 2H), 2.55 – 2.44 (m, 4H), 2.32 (td, J = 11.6, 2.1 Hz, 2H), 2.21 – 2.12 (m, 2H), 2.12 – 2.02 (m, 2H), 1.82 – 1.71 (m, 4H), 1.57 (ddd, J = 14.9, 11.8, 3.7 Hz, 2H), 1.06 (d, J = 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 159.06, 158.35, 154.21, 149.23, 145.96, 107.19, 103.38, 101.40, 67.47, 67.23 (two carbons), 56.71, 54.63, 54.29 (two carbons), 53.04 (two carbons), 48.68, 47.90, 44.80 (two carbons), 32.71 (two carbons), 28.63 (two carbons), 23.59, 18.58 (two carbons). HPLC: 100%; tR 2.19 min. MS (ESI): 513 [M + H]+. HRMS (ESI) calcd for C28H45N6O3 [M + H]+: 513.3553. Found: 513.3550.
4-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-2-yl)thiomorpholine 1,1-dioxide (14)
The procedure used for preparation of compound 7 was followed for synthesis of compound 14. The title compound 14 was obtained as a white solid (47 mg, 81% yield). 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.79 (s, 1H), 5.32 (d, J = 7.3 Hz, 1H), 4.42 – 4.24 (m, 4H), 4.14 (t, J = 6.8 Hz, 2H), 4.07 – 3.94 (m, 1H), 3.88 (s, 3H), 3.09 – 2.96 (m, 4H), 2.93 – 2.82 (m, 2H), 2.79 – 2.68 (m, 1H), 2.59 (t, J = 7.3 Hz, 2H), 2.55 – 2.41 (m, 4H), 2.34 – 2.22 (m, 2H), 2.14 – 2.01 (m, 4H), 1.81 – 1.68 (m, 4H), 1.58 (ddd, J = 14.9, 12.0, 3.7 Hz, 2H), 1.03 (d, J = 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 158.69, 157.01, 154.25, 148.86, 146.47, 107.18, 103.50, 101.25, 67.40, 56.61, 54.56, 54.19 (two carbons), 52.89 (two carbons), 51.55 (two carbons), 48.80, 47.74, 42.96 (two carbons), 32.46 (two carbons), 28.46 (two carbons), 23.49, 18.44 (two carbons). HPLC: 98%; tR 1.97 min. MS (ESI): 561 [M + H]+.
N-(1-Isopropylpiperidin-4-yl)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-2-(3,3,4,4-tetrafluoropyrrolidin-1-yl)quinazolin-4-amine (15)
The procedure used for preparation of compound 7 was followed for synthesis of compound 15. The title compound 15 was obtained as a white solid (46 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 6.94 (s, 1H), 6.73 (s, 1H), 5.12 (d, J = 7.3 Hz, 1H), 4.25 – 4.01 (m, 7H), 3.91 (s, 3H), 2.98 – 2.85 (m, 2H), 2.83 – 2.71 (m, 1H), 2.66 – 2.58 (m, 2H), 2.56 – 2.45 (m, 4H), 2.34 (td, J = 11.6, 2.1 Hz, 2H), 2.20 – 2.04 (m, 4H), 1.84 – 1.71 (m, 4H), 1.58 (qd, J = 11.8, 3.7 Hz, 2H), 1.07 (d, J = 6.6 Hz, 6H). 19F NMR (376 MHz, CDCl3) δ −77.18. 13C NMR (100 MHz, CDCl3) δ 158.49, 156.76, 154.46, 148.92, 146.37, 118.61 (m, two carbons), 107.17, 103.61, 101.22, 67.59, 56.70, 54.65, 54.32 (two carbons), 53.02 (two carbons), 50.76 (t, J = 28.8 Hz) (two carbons), 48.90, 47.87, 32.74 (two carbons), 28.62 (two carbons), 23.61, 18.57 (two carbons). HPLC: 99%; tR 2.49 min. MS (ESI): 569 [M + H]+ .
Compounds 16 and 17 were synthesized according to previously reported procedures.16
N-(1-Cyclopropylpiperidin-4-yl)-2-(4,4-difluoropiperidin-1-yl)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-4-amine (18)
The procedure used for preparation of compound 7 was followed for synthesis of compound 18. The title compound 18 was obtained as a yellowish solid (47 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 6.73 (s, 1H), 5.08 (d, J = 7.2 Hz, 1H), 4.21 – 4.05 (m, 3H), 4.02 – 3.93 (m, 4H), 3.88 (s, 3H), 3.08 – 2.98 (m, 2H), 2.68 – 2.57 (m, 2H), 2.57 – 2.46 (m, 4H), 2.38 (td, J = 11.7, 2.2 Hz, 2H), 2.16 – 2.06 (m, 4H), 2.06 – 1.92 (m, 4H), 1.82 – 1.72 (m, 4H), 1.65 – 1.59 (m, 1H), 1.52 (ddd, J = 14.9, 12.2, 3.8 Hz, 2H), 0.49 – 0.43 (m, 2H), 0.43 – 0.37 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 158.49, 158.29, 154.18, 149.30, 145.97, 122.93 (t, J = 4.8 Hz), 107.17, 103.19, 101.34, 67.39, 56.66, 54.24 (two carbons), 52.99 (two carbons), 52.81, 48.54, 41.24 (t, J = 241.4 Hz) (two carbons), 38.61, 33.99 (t, J = 22.4 Hz) (two carbons), 32.21 (two carbons), 28.50 (two carbons), 23.56, 6.15 (two carbons). HPLC: 99%; tR 2.45 min. MS (ESI): 545 [M + H]+.
4-(4-((1-Cyclopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-2-yl)thiomorpholine 1,1-dioxide (19)
The procedure used for preparation of compound 7 was followed for synthesis of compound 19. The title compound 19 was obtained as a yellowish solid (42 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 6.76 (s, 1H), 5.21 (d, J = 7.3 Hz, 1H), 4.44 – 4.26 (m, 4H), 4.16 (t, J = 6.8 Hz, 2H), 4.13 – 4.01 (m, 1H), 3.90 (s, 3H), 3.10 – 2.96 (m, 6H), 2.66 – 2.58 (m, 2H), 2.57 – 2.46 (m, 4H), 2.43 – 2.29 (m, 2H), 2.15 – 2.01 (m, 4H), 1.83 – 1.69 (m, 4H), 1.66 – 1.48 (m, 3H), 0.49 – 0.43 (m, 2H), 0.42 – 0.36 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 158.72, 157.06, 154.35, 148.94, 146.57, 107.28, 103.50, 101.17, 67.45, 56.68, 54.25 (two carbons), 52.96 (two carbons), 52.73, 51.61 (two carbons), 48.67, 43.01 (two carbons), 38.57, 32.17 (two carbons), 28.49 (two carbons), 23.55, 6.17 (two carbons). HPLC: 98%; tR 1.97 min. MS (ESI): 559 [M + H]+.
4-Chloro-6-methoxy-2-(phosphinan-4-yl)-7-(3-(pyrrolidin-1-yl)propoxy)quinazoline (21)
In a sealed tube, a mixture of compound 10 (0.61 g, 2.0 mmol) (prepared according to the procedures described previously15), tetrahydro-2H-pyran-4-carbonitrile (2.2 g, 19.8 mmol) and HCl (4 N solution in dioxane, 8 mL, 32 mmol) was stirred overnight at 100 °C. The reaction mixture was poured into water and neutralized with NaHCO3. The resulting precipitate was collected and dried to provide the desired crude product 20 (0.58 g, 1.5 mmol). A mixture of this crude compound 20 (0.58 g, 1.5 mmol) and N,N–diethylaniline (0.24 mL, 1.5 mmol) in POCl3 (10 mL) was heated at reflux for 4h. The reaction mixture was concentrated in vacuo and saturated aq. NaHCO3 (15 mL) was added. The resulting mixture was extracted with chloroform (15 mL × 3). The combined organic layers were dried, concentrated and purified by flash column chromatography on silica gel (0 – 10% MeOH / CH2Cl2) to afford the title compound 21 as a yellow solid (0.38 g, 48% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 7.23 (s, 1H), 4.22 (t, J = 6.4 Hz, 2H), 4.07–4.02 (m, 2H), 3.96 (s, 3H), 3.50 (td, J = 11.2, 2.8 Hz, 2H), 3.12–3.06 (m, 1H), 2.65 (t, J = 7.2 Hz, 2H), 2.55–2.53 (m, 4H), 2.15–2.09 (m, 2H), 2.05–1.92 (m, 4H), 1.77–1.74 (m, 4H).
N-(1-Isopropylpiperidin-4-yl)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-2-(tetrahydro-2H-pyran-4-yl)quinazolin-4-amine (22)
A suspension of compound 21 (95 mg, 0.23 mmol), 1-isopropylpiperidin-4-amine 2TFA salt (0.24 g, 0.72 mmol), and K2CO3 (0.33 g, 2.4 mmol) in DMF (3.0 mL) was stirred overnight at 60 °C. The reaction mixture was cooled to rt, water (10 mL) was added and the mixture was extracted with CH2Cl2. The combined organic layers were concentrated and purified by preparative HPLC. The resulting product was basified with saturated aq. NaHCO3 and extracted with CH2Cl2 to afford the desired product 22 as a white solid (91 mg, 78% yield). 1H NMR (400 MHz, CDCl3) δ 7.14 (s, 1H), 6.81 (s, 1H), 5.20 (d, J = 7.2 Hz, 1H), 4.26 – 4.12 (m, 3H), 4.12 – 4.02 (m, 2H), 3.91 (s, 3H), 3.54 (td, J = 11.9, 1.9 Hz, 2H), 3.00 – 2.85 (m, 3H), 2.83 – 2.71 (m, 1H), 2.65 – 2.55 (m, 2H), 2.55 – 2.42 (m, 4H), 2.42 – 2.31 (m, 2H), 2.25 – 2.14 (m, 2H), 2.13 – 1.97 (m, 4H), 1.93 – 1.83 (m, 2H), 1.81 – 1.68 (m, 4H), 1.56 (ddd, J = 23.4, 11.9, 3.7 Hz, 2H), 1.06 (d, J = 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 167.45, 158.00, 153.85, 148.68, 147.34, 108.53, 106.83, 100.04, 68.19 (two carbons), 67.64, 56.50, 54.63, 54.28 (two carbons), 52.95 (two carbons), 48.74, 47.85, 44.82, 32.76 (two carbons), 31.63 (two carbons), 28.52 (two carbons), 23.58, 18.56 (two carbons). HPLC: 98%; tR 2.31 min. MS (ESI): 512 [M + H]+.
Methyl 2-amino-4-(3-(4,4-difluoropiperidin-1-yl)propoxy)-5-methoxybenzoate (23)
The procedure used for preparation of compound 10 was followed for synthesis of compound 23. The title compound 23 was obtained as a yellow oil (2.1 g, 49% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 1H), 6.17 (s, 1H), 5.58 (br, 2H), 4.10 (t, J = 6.4 Hz, 2H), 3.85 (s, 3H), 3.79 (s, 3H), 3.17–2.81 (m, 6H), 2.48–2.19 (m, 6H).
4-Chloro-2-cyclohexyl-7-(3-(4,4-difluoropiperidin-1-yl)propoxy)-6-methoxyquinazoline (25a)
In a sealed tube, a mixture of compound 23 (1.1 g, 3.0 mmol), cyclohexanecarbonitrile (3.3 g, 30.0 mmol) and HCl (4 N solution in dioxane, 15 mL, 60 mmol) was stirred overnight at 100 °C. The reaction mixture was poured into water and neutralized with NaHCO3. The resulting precipitate was collected and dried to provide the intermediate 24a (1.0 g, 2.3 mmol). A mixture of the crude compound 24a (1.0 g, 2.3 mmol) and N,N–diethylaniline (0.37 mL, 2.3 mmol) in POCl3 (20 mL) was heated at reflux for 4 h. The reaction mixture was concentrated in vacuo and saturated aq. NaHCO3 (30 mL) was added. The resulting mixture was extracted with chloroform (30 mL × 3). The combined organic layers were dried, concentrated and purified by flash column chromatography on silica gel (0 – 10% MeOH / CH2Cl2) to afford the 25a as a yellowish solid (0.72 g, 53% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.35 (s, 1H), 7.28 (s, 1H), 4.28 (t, J = 6 Hz, 2H), 4.02 (s, 3H), 2.93 (tt, J = 11.6, 3.6 Hz, 1H), 2.83–2.41 (m, 6H), 2.27–1.96 (m, 6H), 1.93–1.84 (m, 4H), 1.81–1.64 (m, 3H), 1.51–1.29 (m, 3H).
7-(3-(4,4-Difluoropiperidin-1-yl)propoxy)-N-(1-isopropylpiperidin-4-yl)-6-methoxy-2-cyclohexyl quinazolin-4-amine (26a)
The procedure used for preparation of compound 5 was then followed for synthesis of compound 26a. The title compound 26a was obtained as a yellowish solid from intermediate 25a (46 mg, 77% yield). 1H NMR (400 MHz, CDCl3) δ 7.15 (s, 1H), 6.81 (s, 1H), 5.18 (d, J = 7.2 Hz, 1H), 4.24 – 4.13 (m, 3H), 4.13 – 4.02 (m, 2H), 3.90 (s, 3H), 2.90 (d, J = 12 Hz, 2H), 2.80–2.66 (m, 2H), 2.55–2.50 (m, 6H), 2.36 (td, J = 11.6, 2 Hz, 2H), 2.22–2.19 (m, 2H), 2.05–1.90 (m, 8H), 1.85–1.81 (m, 2H), 1.73–1.51 (m, 5H), 1.45–1.27 (m, 3H), 1.06 (d, J = 6.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 169.42, 157.82, 153.44, 148.25, 147.26, 122.06 (t, J = 240 Hz), 108.44, 106.68, 99.89, 67.11, 56.28, 54.48, 53.94 (two carbons), 49.99 (t, J = 5 Hz) (two carbons), 48.56, 47.88, 47.74, 33.99 (t, J = 23 Hz) (two carbons), 33.76, 32.67 (two carbons), 26.61 (two carbons), 26.39 (two carbons), 26.21, 18.49 (two carbons). HPLC: 97%; tR 2.74 min. MS (ESI): 560 [M + H]+.
4-Chloro-7-(3-(4,4-difluoropiperidin-1-yl)propoxy)-6-methoxy-2-(piperidin-1-yl)quinazoline (25b)
The procedure used for preparation of compound 25a was then followed for synthesis of compound 25b. The title compound 25b was obtained as a yellowish solid (0.39 g, 49% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.29 (s, 1H), 4.29 (t, J = 6 Hz, 2H), 4.13–4.09 (m, 2H), 4.03 (s, 3H), 3.57 (td, J = 11.6, 2.4 Hz, 2H), 3.21–3.14 (m, 1H), 2.74–2.43 (m, 6H), 2.16–1.96 (m, 10H).
7-(3-(4,4-Difluoropiperidin-1-yl)propoxy)-N-(1-isopropylpiperidin-4-yl)-6-methoxy-2-(tetrahydro-2H-pyran-4-yl)quinazolin-4-amine (26b)
The procedure used for preparation of compound 26a was followed for synthesis of compound 26b. The title compound 26b was obtained as a yellowish solid from compound 25b (48 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 7.15 (s, 1H), 6.80 (s, 1H), 5.16 (d, J = 7.2 Hz, 1H), 4.28 – 4.13 (m, 3H), 4.13 – 4.02 (m, 2H), 3.93 (s, 3H), 3.55 (td, J = 11.9, 2.0 Hz, 2H), 3.01 – 2.85 (m, 3H), 2.83 – 2.71 (m, 1H), 2.63 – 2.47 (m, 6H), 2.36 (td, J = 11.6, 2.2 Hz, 2H), 2.26 – 2.14 (m, 2H), 2.14 – 1.82 (m, 10H), 1.56 (qd, J = 11.8, 3.7 Hz, 2H), 1.07 (d, J = 6.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 167.56, 158.00, 153.75, 148.65, 147.32, 122.17 (t, J = 241.4 Hz), 108.53, 106.88, 99.95, 68.20 (two carbons), 67.29, 56.47, 54.63, 54.08 (two carbons), 50.15 (t, J = 5.4 Hz) (two carbons), 48.78, 47.86, 44.86, 34.13 (t, J = 22.9 Hz) (two carbons), 32.82 (two carbons), 31.64 (two carbons), 26.75, 18.59 (two carbons). HPLC: 97%; tR 1.63 min. MS (ESI): 562 [M + H]+ .
Methyltransferase activity assays
Methyltransferase activity assays were performed by monitoring the incorporation of tritium-labeled methyl group to biotinylated peptide substrates using Scintillation Proximity Assay (SPA) for G9a, GLP, PRMT3, SETD7, SETDB1, SETD8, SUV420H1, SUV420H2, SUV39H2, PRC2 trimeric complex (EZH2:EED:SUZ12), MLL1 tetrameric complex (MLL:WDR5:RbBP5:ASH2L), PRMT5-MEP50 complex and SMYD2. Assay components for all assays are summarized in Table S4. The reaction buffer for SMYD2 and SMYD3 was 50 mM Tris pH 9.0, 5 mM DTT, 0.01%TritonX-100; for G9a, GLP and SUV39H2 was 25 mM potassium phosphate pH 8.0, 1 mM EDTA, 2 mM MgCl2 and 0.01% Triton X-100; and for other HMTs 20 mM Tris pH 8.0, 5 mM DTT, 0.01% TritonX-100. To stop the enzymatic reactions, 10 µL of 7.5 M guanidine hydrochloride was added, followed by 180 µL of buffer, mixed and transferred to a 96-well FlashPlate (Cat.# SMP103; Perkin Elmer; www.perkinelmer.com). After mixing, the reaction mixtures were incubated and the CPM counts were measured using Topcount plate reader (Perkin Elmer, www.perkinelmer.com). The CPM counts in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM counts in each data set were defined as background (0%). IC50 values were determined using compound concentrations ranging from 100 nM to 100 µM. The IC50 values were determined using SigmaPlot software.
For DNMT1, the assay was performed as described above using hemimethylated dsDNA as a substrate. The dsDNA substrate was prepared by annealing two complementary strands (biotintlated forward strand: B-GAGCCCGTAAGCCCGTTCAGGTCG and reverse strand: CGACCTGAACGGGCTTACGGGCTC), synthesized by Eurofins MWG Operon. Reaction buffer was 20 mM Tris-HCl, pH 8.0, 5mM DTT, 0.01% Triton X-100.
Methyltransferase activity assays for DOT1L and SMYD3 were performed using Filter-plates (Millipore; cat.# MSFBN6B10; www.millipore.com). Reaction mixtures in 20 mM Tris-HCl, pH 8.0, 5 mM DTT, 2 mM MgCl2 and 0.01% Triton X-100 were incubated at room temperature for 1h, 100 µL 10% TCA was added, mixed and transferred to filter-plate. Plates were centrifuged at 2000 rpm for 2 min followed by 2 additional 10% TCA wash and one ethanol wash (180 µL) followed by centrifugation. Plates were dried and 100 µL MicroO was added and centrifuged. 70 µL MicroO was added and CPM were measured using Topcount plate reader.
Ki determination for inhibitor 7
A competition between inhibitor 7 and the peptide was measured using microfluidic capillary electrophoresis to monitor the methylation status of peptide substrates. Reactions (15 µL) were set up in Nunc polypropylene shallow low volume 384-well microplates containing a 5 µL spot of the compound in 1.5% DMSO and 1X Assay Buffer (20 mM Tris-HCl pH = 8, 25 mM NaCl, 0.05% Tween 20 and 2 mM DTT) and a 5 µL spot of the peptide substrate60 in 1X Assay Buffer arrayed in an 6 × 8 grid pattern. Inhibitor 7 was titrated from 20 nM to 1.76 nM using 1.5-fold dilution and peptide was titrated from 50 µM to 1.6 µM using 2-fold dilution scheme. A total of 4 grids for each assay time point were set up. 5 µL of G9a and SAM cocktail were added to initiate the reaction (to final concentration of 5 nM and 200 µM, respectively). The reactions were allowed to proceed for 20, 30, 40 and 60 min at 25 °C, and 10 µL of Endo-LysC (40 pg/µL) and inhibitor 5 (10 µM) mix were added to stop the reaction and digest remaining unmethylated peptide. After 1 h, peptide concentrations over 10 µM were diluted to 10 µM in 1X reaction buffer (to avoid saturation of the optics) and the plate was read on a Caliper Life Sciences EZR II, using upstream voltage = −500 V, downstream voltage = −1200 V and pressure = 1.5 psi. Pre-digested peptide was used as the marker. The steady-state velocity was analyzed by linear regression of the peptide methylation versus time and plotted to determine Michaelis-Menten kinetics (GraphPad Prism 5.0). Km and kcat values were plotted to determine the relationship of the peptide and the inhibitor on enzyme kinetics. The Ki value is an average of 3 replicates ± SD.
Competition of inhibitor 7 with SAM
This assay was performed as described previously.15
Kinase selectivity assays
Selectivity of inhibitor 7 against a panel of 50 kinases was conducted using a standard off-chip mobility shift assay technology. The full list of the 50 kinases is included in Table S1.
GPCRs, ion channels, and transporters selectivity assays
Selectivity of inhibitor 7 against 44 GPCRs, ion channels, and transporters was performed in standard radioligand binding assays. The full list of the 44 targets is included in Table S2.
Cellular Assays
MDA-MB-231, PC3, and U2OS cells were cultured in RPMI with 10% FBS, PANC-1 cells in DMEM with 10% FBS. Cells were treated with inhibitors for 48 h. Cell viability assays were performed by incubating cells with 0.1 mg/mL of resazurin (Sigma) for 3 – 4 h. Resazurin reduction was monitored with 544 nm excitation, measuring fluorescence at 590 nm. In-cell western assay was performed as described previously.15
For clonogenicity assays, PANC-1 and MDA-MB-231 cells were cultured in the presence of the inhibitor for 2 days and seeded in 12 well plates at the density of 200 – 300 cells per well in triplicates. The cells were cultured for 2 weeks until colonies were visible. Cell media and the inhibitor were changed every 3 – 4 days. Colonies were stained in 1% methylene blue in 50% methanol and counted. The experiments repeated twice with consistent results. Colonies were scored using Clono-Counter.
In vitro metabolic stability studies
Standard in vitro metabolic stability studies were conducted. Compound concentrations were measured after 0, 5, 15, 30, and 45 min incubation of test compounds with mouse liver microsomes.
Mouse PK studies
Standard PK studies were performed using male Swiss albino mice. Plasma and brain concentrations were measured at 0.08, 0.25, 0.5, 1, 2, 4, 8, and 24 h following a single IP injection of inhibitor 7 or 13 at 5 mg/kg. The compound concentration at each time point in plasma or brain is the average value from 3 test animals.
Supplementary Material
Acknowledgement
The research described here was supported by the grant R01GM103893 from the National Institute of General Medical Sciences of the National Institutes of Health, the University Cancer Research Fund and Carolina Partnership from University of North Carolina at Chapel Hill, the V Foundation for Cancer Research, and the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canada Foundation for Innovation, Eli Lilly Canada, GlaxoSmithKline, the Ontario Ministry of Economic Development and Innovation, the Novartis Research Foundation, Pfizer, AbbVie, Takeda, Janssen, Boehringer Ingelheim and the Wellcome Trust.
ABBREVIATIONS USED
- H3K9me2
dimethylation of histone H3 lysine 9
- PK
pharmacokinetic
- PKMTs
protein lysine methyltransferases
- HMTs
histone methyltransferases
- PRMT
protein arginine methyltransferase
- KMT1C
lysine methyltransferase 1C
- EHMT2
euchromatic histone methyltransferase 2
- KMT1D
lysine methyltransferase 1D
- EHMT1
euchromatic histone methyltransferase 1
- H3K9
histone H3 lysine 9
- SET
suppressor of variegation 3–9, enhancer of zeste, and trithorax
- HIV-1
human immunodeficiency virus type 1
- SAR
structure activity relationships
- CYP450
cytochrome P450
- SAM
S-adenosyl-L-methionine
- ICW
in-cell western
- tox/function ratio
ratio of toxicity to functional potency
- CLint
intrinsic clearance
- T1/2
half-life
- IP
intraperitoneal
- Cmax
maximum concentration
- AUC
area under the curve
- MOA
mechanism of action
- Kmapp
apparent Km
- PRC2 – EZH2
polycomb repressive complex 2 – enhancer of zeste homolog 2
- GPCRs
G protein-coupled receptors
Footnotes
Supporting Information Available. Kinases, GPCRs, ion channels, and transporters selectivity assay results of inhibitor 7. Methyltransferase assay components and conditions. 1H and 13C NMR spectra of compounds 7 and 13. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 3.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Dorsey J, Chuikov S, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9A and GLP methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 8.Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campagna-Slater V, Mok MW, Nguyen KT, Feher M, Najmanovich R, Schapira M. Structural chemistry of the histone methyltransferases cofactor binding site. Journal of chemical information and modeling. 2011;51:612–623. doi: 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, Plotnikov AN, Schapira M. Structural biology of human H3K9 methyltransferases. PLoS ONE. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, Zhang X, Bedford MT, Shinkai Y, Snyder JP, Cheng X. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong A, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin J. Protein Lysine Methyltransferase G9a Inhibitors: Design, Synthesis, and Structure Activity Relationships of 2,4-Diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, DiMaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang S-C, Chen X, Chau I, Mangano T, Huang X-P, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase Activity in Cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He Y, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of Cellular Activity of G9a Inhibitors 7-Aminoalkoxy-quinazolines. J Med Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, Cheung T, Garcia-Arenas R, Cowen S, Wu J, Godin R, Chen H, Keen N. Structural Basis of Substrate Methylation and Inhibition of SMYD2. Structure. 2011;19:1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 18.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BVV, Song Y. Selective Inhibitors of Histone Methyltransferase DOT1L: Design, Synthesis and Crystallographic Studies. J Am Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, Shamji AF, Wagner BK, Schreiber SL. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–1157. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 22.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Iii AD, Diaz E, Lafrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 23.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GSV, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng W, Ibáñez G, Wu H, Blum G, Zeng H, Dong A, Li F, Hajian T, Allali-Hassani A, Amaya MF, Siarheyeva A, Yu W, Brown PJ, Schapira M, Vedadi M, Min J, Luo M. Sinefungin Derivatives as Inhibitors and Structure Probes of Protein Lysine Methyltransferase SETD2. J Am Chem Soc. 2012;134:18004–18014. doi: 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang J-H, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, Marcellus R, Iacob RE, Engen JR, Griffin C, Aman A, Wienholds E, Li F, Pineda J, Estiu G, Shatseva T, Hajian T, Al-awar R, Dick JE, Vedadi M, Brown PJ, Arrowsmith CH, Bradner JE, Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 27.Williams DE, Dalisay DS, Li F, Amphlett J, Maneerat W, Chavez MAG, Wang YA, Matainaho T, Yu W, Brown PJ, Arrowsmith CH, Vedadi M, Andersen RJ. Nahuoic Acid A Produced by a Streptomyces sp. Isolated From a Marine Sediment Is a Selective SAM-Competitive Inhibitor of the Histone Methyltransferase SETD8. Org Lett. 2013;15:414–417. doi: 10.1021/ol303416k. [DOI] [PubMed] [Google Scholar]
- 28.Anglin JL, Deng L, Yao Y, Cai G, Liu Z, Jiang H, Cheng G, Chen P, Dong S, Song Y. Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J Med Chem. 2012;55:8066–8074. doi: 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chemical Biology. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Shaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel-Wahab O, Licht JD, Elemento O, Melnick AM. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney Gregory A, Chau I, Marcellus R, Hajian T, Liu F, Korboukh I, Smil D, Bolshan Y, Min J, Wu H, Zeng H, Loppnau P, Poda G, Griffin C, Aman A, Brown PJ, Jin J, Al-awar R, Arrowsmith CH, Schapira M, Vedadi M. An Allosteric Inhibitor of Protein Arginine Methyltransferase 3. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Li F, Ma A, Dobrovetsky E, Dong A, Gao C, Korboukh I, Liu J, Smil D, Brown PJ, Frye SV, Arrowsmith CH, Schapira M, Vedadi M, Jin J. Exploiting an Allosteric Binding Site of PRMT3 Yields Potent and Selective Inhibitors. J Med Chem. 2013;56:2110–2124. doi: 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 35.Workman P, Collins I. Probing the probes: fitness factors for small molecule tools. Chem Biol. 2010;17:561–577. doi: 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bunnage ME, Chekler EL, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 37.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore KE, Carlson SM, Camp ND, Cheung P, James RG, Chua KF, Wolf-Yadlin A, Gozani O. A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol Cell. 2013;50:444–456. doi: 10.1016/j.molcel.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Islam K, Bothwell I, Chen Y, Sengelaub C, Wang R, Deng H, Luo M. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J Am Chem Soc. 2012;134:5909–5915. doi: 10.1021/ja2118333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JP. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, Kato H, Mizuno Y, Yokoe M, Sugauchi F, Hirashima N, Orito E, Osada H, Ueda R, Guo Y, Chen X, Issa JP, Sekido Y. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res. 2007;37:974–983. doi: 10.1111/j.1872-034X.2007.00141.x. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S, Naoki K, Ishizaka A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008;8:15. doi: 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goyama S, Nitta E, Yoshino T, Kako S, Watanabe-Okochi N, Shimabe M, Imai Y, Takahashi K, Kurokawa M. EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia. 2010;24:81–88. doi: 10.1038/leu.2009.202. [DOI] [PubMed] [Google Scholar]
- 45.Maze I, Covington HE, 3rd., Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Covington HE, 3rd., Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, Dietz DM, Lobo MK, Ghose S, Mouzon E, Neve RL, Tamminga CA, Nestler EJ. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P. Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron. 2009;64:678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imai K, Togami H, Okamoto T. Involvement of histone H3 Lysine 9 (H3K9) methyl transferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Link PA, Gangisetty O, James SR, Woloszynska-Read A, Tachibana M, Shinkai Y, Karpf AR. Distinct roles for histone methyltransferases G9a and GLP in cancer germ-line antigen gene regulation in human cancer cells and murine embryonic stem cells. Mol Cancer Res. 2009;7:851–862. doi: 10.1158/1541-7786.MCR-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008;27:2681–2690. doi: 10.1038/emboj.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, Yang HW, Lam LL, Mager DL, Schubeler D, Tachibana M, Shinkai Y, Lorincz MC. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008;27:2691–2701. doi: 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Shi Y, Do JT, Desponts C, Hahm HS, Scholer HR, Ding S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;2:525–528. doi: 10.1016/j.stem.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Skutt-Kakaria K, Davison J, Ou YL, Choi E, Malik P, Loeb K, Wood B, Georges G, Torok-Storb B, Paddison PJ. G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev. 2012;26:2499–2511. doi: 10.1101/gad.200329.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Genevieve D, Cormier-Daire V, van Esch H, Fryns JP, Hamel BC, Sistermans EA, de Vries BB, van Bokhoven H. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. American journal of human genetics. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, van Dooren M, Willemsen MH, Pfundt R, Turner A, Wilson M, McGaughran J, Rauch A, Zenker M, Adam MP, Innes M, Davies C, Lopez AG, Casalone R, Weber A, Brueton LA, Navarro AD, Bralo MP, Venselaar H, Stegmann SP, Yntema HG, van Bokhoven H, Brunner HG. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. Journal of medical genetics. 2009;46:598–606. doi: 10.1136/jmg.2008.062950. [DOI] [PubMed] [Google Scholar]
- 57.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a–like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauser AT, Jung M. Chemical probes: sharpen your epigenetic tools. Nat Chem Biol. 2011;7:499–500. doi: 10.1038/nchembio.615. [DOI] [PubMed] [Google Scholar]
- 59.Nazare M, Will DW, Matter H, Schreuder H, Ritter K, Urmann M, Essrich M, Bauer A, Wagner M, Czech J, Lorenz M, Laux V, Wehner V. Probing the subpockets of factor Xa reveals two binding modes for inhibitors based on a 2-carboxyindole scaffold: a study combining structure-activity relationship and X-ray crystallography. J Med Chem. 2005;48:4511–4525. doi: 10.1021/jm0490540. [DOI] [PubMed] [Google Scholar]
- 60.Wigle TJ, Provencher LM, Norris JL, Jin J, Brown PJ, Frye SV, Janzen WP. Accessing Protein Methyltransferase and Demethylase Enzymology Using Microfluidic Capillary Electrophoresis. Chemistry & Biology. 2010;17:695–704. doi: 10.1016/j.chembiol.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.