

Abstract
Atherogenesis, the uncontrolled deposition of modified lipoproteins in inflamed arteries, serves as a focal trigger of cardiovascular disease (CVD). Polymeric biomaterials have been envisioned to counteract atherogenesis based on their ability to repress scavenger mediated uptake of oxidized lipoprotein (oxLDL) in macrophages. Following the conceptualization in our laboratories of a new library of amphiphilic macromolecules (AMs), assembled from sugar backbones, aliphatic chains and poly(-ethylene glycol) tails, a more rational approach is necessary to parse the diverse features such as charge, hydrophobicity, sugar composition and stereochemistry. In this study, we advance a computational biomaterials design approach to screen and elucidate anti-atherogenic biomaterials with high efficacy. AMs were quantified in terms of not only 1D (molecular formula) and 2D (molecular connectivity) descriptors, but also new 3D (molecular geometry) descriptors of AMs modeled by coarse-grained molecular dynamics (MD) followed by all-atom MD simulations. Quantitative structure-activity relationship (QSAR) models for anti-atherogenic activity were then constructed by screening a total of 1164 descriptors against the corresponding, experimentally measured potency of AM inhibition of oxLDL uptake in human monocyte-derived macrophages. Five key descriptors were identified to provide a strong linear correlation between the predicted and observed anti-atherogenic activity values, and were then used to correctly forecast the efficacy of three newly designed AMs. Thus, a new ligand-based drug design framework was successfully adapted to computationally screen and design biomaterials with cardiovascular therapeutic properties.
Keywords: Amphiphilic macromolecules, Macrophages, Atherosclerosis, Molecular modeling, Structure-activity relations
1. Introduction
Atherosclerosis is characterized as an inflammatory disease involving macrophage scavenger receptor (SR) interactions with oxidized low-density lipoproteins (oxLDL) in the vascular intima, leading to plaque initiation and growth [1]. The early stages of atherosclerosis include low-density lipoproteins (LDL) sequestration and oxidation in arterial walls, followed by monocyte recruitment and differentiation into macrophages, which internalize oxLDL via SR-mediated mechanisms [2,3]. This progression results in enhanced inflammatory signaling and lipid-laden foam cell formation [4]. The accumulation of foam cells can lead to a lipid filled necrotic core covered by a fibrous cap, which upon rupture can lead to thrombus formation and the clinical endpoints of myocardial infarction or stroke [5].
Conventional therapies are often limited in terms of their ability to address localized inflammation from pre-existing lipid deposits [6]. As macrophages are crucial to progressing the inflammatory cycle and responsible for the majority of lipid accumulation, they present an ideal target for therapeutics to arrest disease progression [7]. By designing biomaterial nanoassemblies that can simulate the size, amphiphilicity and anionic charge of oxLDL and thus modulate scavenger receptor-mediated oxLDL trafficking, the macrophage pathways that are key to formation of atherosclerotic plaque can be potentially targeted. Studies from our laboratories have revealed a class of amphiphilic macromolecules (AM), which can competitively block oxLDL interaction with scavenger receptors, thereby mitigating downstream consequences [8–15]. Previous work with first generation AM structures qualitatively studied the impact of charge placement and net charge on oxLDL uptake inhibition, but lacked the sophistication to accurately discern the key structural features that govern anti-atherogenic potency [10,11]. More recent studies indicate that AM chemistries with similar chemical composition can elicit markedly different, and somewhat unexpected, interactions with oxLDL when imbued with different stereochemistries [8]. Thus, more rational and computationally guided approaches would aid in elucidating the AM structure-biological activity relations and designing a wider library of AMs with high potency.
Given the large size of the AMs, many current drug design methods of developing “small molecule” therapeutics are not directly applicable to AM structures (due to limits in feature space). High throughput screening could in theory be used to synthesize a large array of AMs but this approach has an inherently high cost barrier and is often is plagued by large false negative and false positive rates [16]. Furthermore, structural optimization of early lead compounds by chemical synthesis of analogs is both time and cost intensive and typically based on heuristic, rather than rational drug design approaches. Computational drug discovery and optimization approaches, based on quantitative structure-activity relationship (QSAR) principles, offer an efficient and economical alternative for drug development when employed in conjunction with synthetic medicinal chemistry and experimental testing of lead compounds [17–20].
Computational (rational) drug design has made significant contributions to the discovery of new and more efficacious therapeutics. The general strategies employed today are broadly divided into ligand-based and structure-based drug design (LBDD and SBDD, respectively) [21–24]. Assuming that drug action operates through the simple mechanism of drug–target interaction, the option to use one or both strategies in a drug discovery campaign depends on the existing knowledge of biologically active ligands for LBDD and the 3D structure of the target protein for SBDD. Modern techniques in LBDD rely heavily on in silico (virtual) screening of often vast chemical libraries, QSAR modeling, and pharmacophore modeling [12,25–29]. Likewise, SBDD studies frequently employ virtual screening procedures using a process commonly known as ligand-receptor docking [8,30–32]. LBDD and SBDD methods remain the subject of intensive research to improve their speed, accuracy, and sophistication [33,34].
We have previously employed several different approaches to construct QSAR models of biological activities of drugs and biomaterials [35–38]. QSAR models associate variations in the chemical structure of the subject materials, as encoded by molecular descriptors, with variations in their corresponding biological activity (e.g., inhibition of oxLDL uptake). QSAR modeling entails two key steps: 1) computing values of an ensemble of molecular descriptors, and 2) creating and validating the regression or classification models by machine learning methods and statistical analysis tools. Molecular descriptors can be categorized as 1D (e.g., molecular weight, number of rings), 2D (e.g., electro-topological/connectivity indices) whose values are conformation invariant, or 3D (e.g., dipole moment, surface area, radius of gyration) whose values are conformation dependent.
The goal of the present study was to propose a LBDD-based approach, using both 2D and 3D descriptors, to identify structure-activity relationships between the AMs and inhibition of oxLDL uptake and foam cell formation in human monocyte-derived macrophages (MDM). One of the specific objectives is to generate QSAR models as means to predict the biological activities of new AMs, thus, guiding the rational design and optimization of AMs. A critical component of the molecular modeling strategy for the current AMs, is the inclusion of 3D descriptors, which uniquely encodes vital information such as stereochemistry of the AMs. We hypothesize that this study will provide a new “in silico” framework to correlate compositional changes in the AM biomaterials with the respective variations in anti-atherogenic potency and thus facilitate the design of new AM structures by predicting biological activity.
2. Materials and methods
2.1. Materials
All chemicals/materials were purchased from Sigma–Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and used as received unless otherwise noted. Deionized (DI) water with a resistivity of 18 MΩ cm is obtained using PicoPure 2 UV Plus (Hydro Service and Supplies–Durham, NC). The following items were purchased from the indicated vendors: RPMI 1640 from ATCC (Manassas, VA), macrophage colony stimulating factor (M-CSF) from PeproTech (Rocky Hill, NJ), 1.077 g/cm3 Ficoll–Paque Premium from GE healthcare (Pittsburgh, PA), FBS and Hoechst 33342 from Life Technologies (Grand Island, NY), 3,3′-dioctadecyloxacarbocyanine (DiO) labeled oxLDL from Kalen Biomedical (Montgomery Village, MD), unlabeled oxidized LDL from Biomedical Technologies Inc. (Stoughton, MA), and human buffy coats from the Blood Center of New Jersey (East Orange, NJ).
2.2. AM synthesis and physicochemical property determination
AMs were synthesized as previously described [10–14,39–41]. The pKa values were estimated based on ionizable functional groups. For example, aliphatic carboxylic acids have a pKa of ∼ 3–5, and primary amines ∼ 35. Attachment length was calculated by counting the number of atoms between the functional group responsible for the charge and the attachment of PEG as shown in Fig. 1. In the current study, 17 different AM structures were examined, varying the overall charge, hydrophobicity, sugar structure (linear vs. cyclic) and stereochemistry. Table 1 summarizes the diverse range of their physicochemical properties.
Fig. 1.
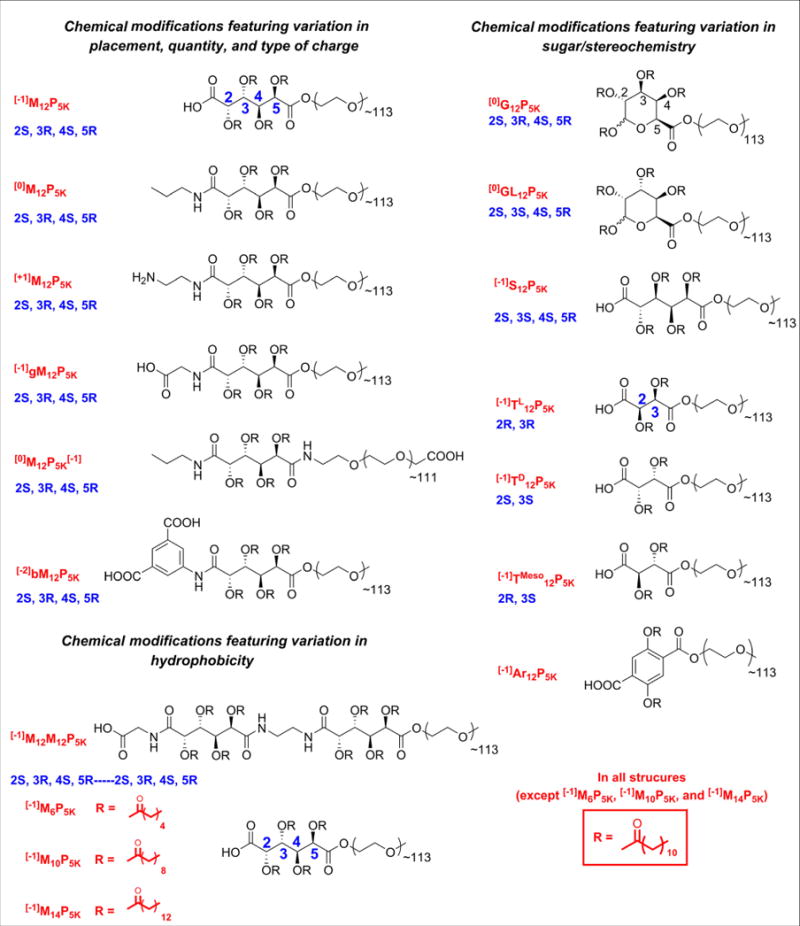
Chemical compositions, stereochemistry, and associated abbreviations for AMs in this study. Nomenclature methodology: [charge]BackboneStereochemistrychain lengthPPEG length. Backbone and functional group abbreviations: M = mucic acid, S = saccharic acid, T = tartaric acid, Ar = 2,5-dihydroxyterephthalic acid, G = d-galacturonic acid, GL = d-glucuronic acid, b = benzene, g = glycine.
Table 1.
Physiochemical properties of AM.
| Mw (kDa) | # hydrophobic chains | Hydrophobic chain length | pKa | Charge source | Charge number | Backbone structure | Attachment length | |
|---|---|---|---|---|---|---|---|---|
| [−1]M12P5K | 5.9 | 4 | 12 | 3–5 | COOH | −1 | Mucic acid | 5 |
| [0]M12P5K | 5.9 | 4 | 12 | – | 0 | Ethylene-mucic acid | – | |
| [+1]M12P5K | 5.9 | 4 | 12 | ∼35 | NH2 | 1 | Ethylene-diamine-mucic acid | 9 |
| [−1]gM12P5K | 5.4 | 4 | 12 | 3–5 | COOH | −1 | Glycine-mucic acid | 8 |
| [0]M12 | 5.9 | 4 | 12 | 3–5 | COOH | −1 (PEG) | Mucic acid | 8 |
| [−2]bM12P5K | 6.1 | 4 | 12 | 3–5 | COOH | −2 | Benzene-mucic acid | 10,10 |
| [0]G12P5K | 5.9 | 4 | 12 | – | 0 | Galacturonic acid | – | |
| [0]GL12P5K | 5.9 | 4 | 12 | – | 0 | Glucuronic acid | – | |
| [−1]S12P5K | 5.9 | 4 | 12 | 3–5 | COOH | −1 | Saccharic acid | 5 |
| [−1]TL 12P5K | 5.4 | 2 | 12 | 3–5 | COOH | −1 | Tartaric acid | 3 |
| [−1]TD 12P5K | 5.4 | 2 | 12 | 3–5 | COOH | −1 | Tartaric acid | 3 |
| [−1]TMeso 12P5K | 5.4 | 2 | 12 | 3–5 | COOH | −1 | Tartaric acid | 3 |
| [−1]Ar12P5K | 5.5 | 2 | 12 | 3–5 | COOH | −1 | 2,4-dihydroxyterephthalic acid | 5 |
| [−1]M12M12P5K | 6.9 | 8 | 12 | 3–5 | COOH | −1 | Glycine-mucic-mucic acid | 18 |
| [−1]M6P5K | 5.6 | 4 | 6 | 3–5 | COOH | −1 | Mucic acid | 5 |
| [−1]M10P5K | 5.8 | 4 | 10 | 3–5 | COOH | −1 | Mucic acid | 5 |
| [−1]M14P5K | 6 | 4 | 14 | 3–5 | COOH | −1 | Mucic acid | 5 |
2.3. Isolation and culture of hMDMs
Peripheral blood mononuclear cells (PBMCs) were isolated from human buffy coats by Ficoll–Paque (1.077 g/cm3) density gradient. Red blood cells were lysed with ACK buffer and platelets were removed by centrifugation at 300 g for 10 m. PBMCs were transferred to flasks containing RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin. Monocytes were selected from PBMCs by adherence after 24 h and then cultured for 7 days in RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin and 50 ng/mL M-CSF for differentiation into macrophages. After the 7 day culture, the macrophages were trypsinized and scraped from flasks, transferred into well plates at 50,000 cells/cm2, and treatments administered after 24 h.
2.4. OxLDL uptake by hMDMs
To measure AM efficacy at inhibiting oxLDL uptake, hMDMs were incubated with 1 μg/mL of DiO labeled oxLDL and 10−6 m AM in RPMI 1640 for 24 h. Cells were removed from plates by vigorous pipetting in cold PBS with 2 mm EDTA, washed with PBS, centrifuged and fixed in 1% paraformaldehyde. DiO fluorescence (oxLDL uptake) was measured by flow cytometry on a FACScalibur (Beckton Dickenson) in the FL1 channel. A minimum of 15,000 events per sample were collected, and quantified using the geometric mean fluorescence intensity (MFI) of intact hMDMs with FloJo (Treestar). Results are the average of three independent experiments with two technical replicates per experiment. Data is presented as % oxLDL uptake inhibition, which was calculated using the following formula:
2.5. Foam cell formation
To measure the effectiveness at preventing foam cell formation (lipid accumulation), AMs (10−5m) and oxLDL (50 μg/mL) were co-incubated with hMDMs for 24 h. Cells were then washed, fixed, dehydrated with 60% isopropanol, stained with 2 mg/mL Oil Red O in 60% isopropanol for 5 min, washed and the nucleus counterstained with 1 μg/mL Hoechst 33342. Brightfield and epifluorescent images were taken on a Nikon Eclipse TE2000-S and merged using ImageJ. Images shown are representative of two independent experiments with three technical replicates per experiment.
2.6. Statistical analysis
OxLDL uptake results are presented as mean ± standard error of the mean (S.E.M.) and data evaluated by one-way ANOVA and Tukey's test for post-hoc pair-wise comparisons between multiple conditions. A p-value of 0.05 or less was considered statistically significant.
2.7. Molecular modeling
AMs were constructed and minimized in Molecular Operating Environment (MOE), 2011.10 (Chemical Computing Group Inc., Canada). Initial parameterization and charge distribution calculations for AM input structures for MD simulation were performed with the antechamber program from Amber Tools package [42]. Next, a two-tier process was implemented that comprised coarse-grained molecular dynamics (CG MD) simulations on a three-dimensional model system to extract one or more representative (low-energy) structures, followed by all-atom molecular dynamics (AA MD) simulations to yield highly resolved AM conformers from which molecular descriptors were subsequently calculated. Fig. 2 outlines the modeling approach.
Fig. 2.
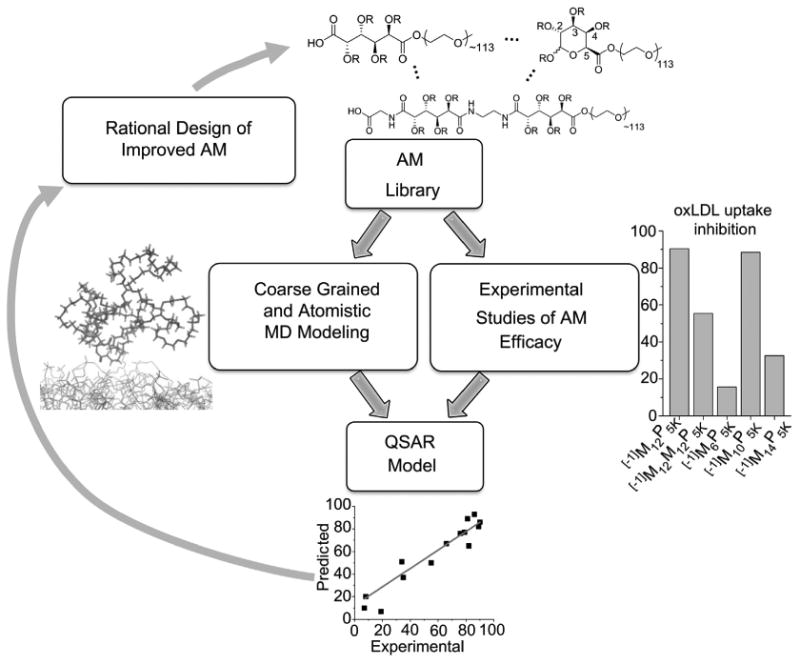
An overview of the QSAR modeling methodology employed for this study. The AM library is evaluated for experimental outcomes (“Degree of oxLDL uptake inhibition”) while 2D and 3D descriptors are identified with coarse grained and atomistic MD modeling. These are correlated to experimental results to develop QSAR and predictive models for new AMs. Optimal AM structures are synthesized and this process repeated.
2.8. Coarse-grained (CG) molecular dynamics simulations
To mimic a cellular environment, polymer simulations were carried out in solution in the presence of a dipalmitoylphosphatidylcholine (DPPC) bilayer. Next, the CG MARTINI force-field, which has been shown successful in the study of numerous systems including lipids, proteins, and polymers, was used [43–45]. Each AM was built by combining already parameterized functional groups such as the PEG tails from Lee et al. and aliphatic chains from Marrink et al. [45,46]. In all simulations, the length of the PEG tail was kept at 46 monomers, roughly correspondingto2000 MW. Lipids, water, and ions were parameterized according to Marrink et al. [46]. Each simulation contained 1 AM, 512 DPPC lipids, 0.1 m NaCl and 20,640 CG water (82,560 water molecules) and was run for 400 ns using the GROMACS software package v.4.5.5 [47].
2.9. Representative structure from CG simulations
Following 400 ns of simulation time, the ensemble of generated AM structures, taken every 1 ns, was clustered with the g_cluster analysis program of GROMACS using the single linkage algorithm. For each AM, the root mean square deviation (RMSD) threshold was chosen as the minimum value such that greater than 50% of structures were members of a single cluster. The representative structure was chosen to be the median structure (in terms of RMSD) of the largest cluster.
2.10. Reverse mapping of CG structures to atomistic AM structures
The reverse transformation technique of Rzepiela et al. was applied to convert the MARTINI CG structures back to all-atom structures [48]. The Antechamber module of the Amber Tools software package was used to generate atomistic topologies for each AM and the program acpype was used to convert Amber topologies into GROMACS format [42,49]. The GROMACS utility g_fg2cg was used to generate an initial approximation of the all-atom structure by placing at random the underlying atoms within the volume of their corresponding CG interaction site. Simulated annealing (SA) was then used to bring the system from 1300 K to 310 K over 100 ps to allow for rearrangement of the atoms and crossing of energetic barriers. During SA, a restraining force is used such that the center of mass (COM) of atoms corresponding to a given CG interaction site align with the COM of the CG site. Following SA, the restraining force is slowly removed over a period of 10 ps. The resultant structure was then subject to energy minimization.
2.11. Atomistic MD simulations
Followed the long-range CG MD simulation for 400 ns, the average structures of the AMs were subjected to refinement with AA MD in aqueous solution over the surface of the membrane bilayer, constructed to mimic the macrophage cellular surface. Each reverse mapped AM was placed over the surface of the constructed membrane comprising phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylserine (PS) (courtesy of Dr. William Moyle, Department of OBGYN, RWJMS, Rutgers University), neutralized with sodium ions and solvated with tleap subroutine from Amber 12 software package [42]. High level AA MD simulation totaling 10 ns for each of the 17 AMs contained 1 AM, 812 lipids and 82,560 water molecules in a periodic box was performed on the specifically built and dedicated high performance GPU Linux cluster.
2.12. 3D molecular descriptors and QSAR analysis
The representative low energy 3D structures emerging from the AA MD simulations were subjected to QSAR analysis to find correlations between polymer structural features and their effect on oxLDL uptake. Each polymer was encoded with 1664 distinct molecular descriptors using Dragon v.5.4 software program. Sets of descriptors were limited to 3D descriptor blocks as defined in Dragon, namely Randic molecular profiles, geometrical descriptors, RDF descriptors, 3d-MoRSE, Weighted Holistic Invariant Molecular (WHIM), GEometry, Topology, and Atom-Weights AssemblY (GETAWAY) and charge descriptors (total of 735). After removing highly correlated pairs and descriptors with standard deviation below the program default threshold, the final set of descriptors was reduced to 115. Partial least squares (PLS) regression method implemented in MOE was used to model the experimental data.
3. Results
3.1. oxLDL uptake in hMDMs
The different AM structures have demonstrated varying abilities to inhibit oxLDL uptake in MDMs. As shown in Fig. 3A–C, [−1]M12P5K, [0]M12P5K, [0]G12P5K, [−1]TMeso12P5K, [−1]gM12P5K, and [−1]M10P5K display the most significant reduction in oxLDL accumulation. In contrast, AM structures, [−1]S12P5K, [−1]Ar12P5K, and [−1]M6P5K, did not show a statistically significant decrease in oxLDL uptake as compared to the oxLDL control.
Fig. 3.
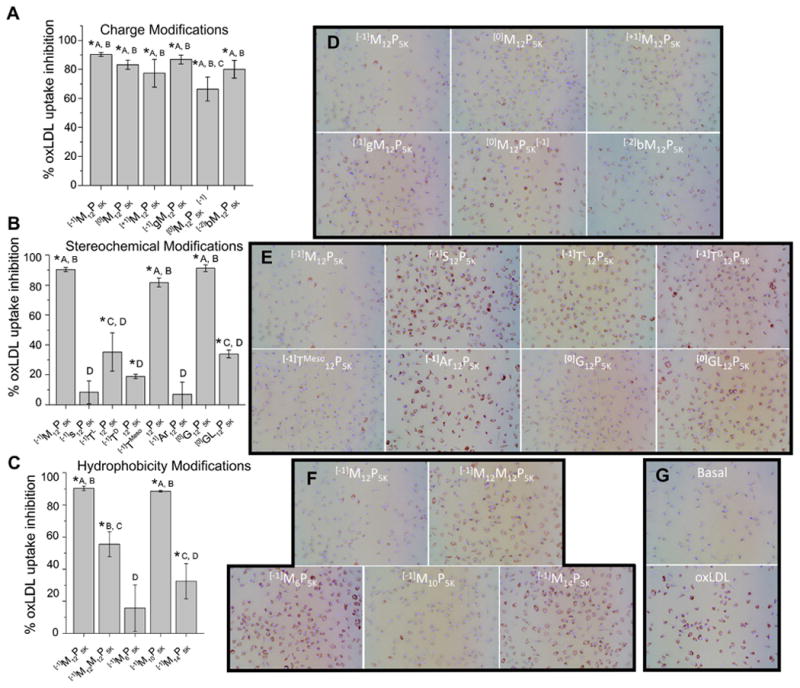
The AM library shows graded efficacy of anti-atherogenesis in hMDMs A–C) The percentage of inhibition of oxLDL uptake was plotted vs. nature of AM treatment in hMDMs. D–G) Representative micrographs showing modulation of lipid uptake and foam cell phenotype. AM are grouped to show effects of charge (A/D), stereochemical (B/E) and hydrophobic modifications (C/F). Treatments with the same letter are not statistically significant from one another and the asterisk (*) indicates statistical significance (p < .05) from the control (no AM, oxLDL only).
3.2. Foam cell formation in hMDMs
Treatment of hMDMs with oxLDL generated the foam cell phenotype as evidenced by intracellular lipid droplet accumulation (Fig. 3G). The foam cell formation results qualitatively parallel the oxLDL uptake results, and show that the AM library has differential ability to reduce lipid accumulation (Fig. 3D–F). These results demonstrate the ability of AMs to mitigate actual atherosclerotic-relevant endpoints.
3.3. CGMD in conjunction with atomistic MD
The two-tiered approach used in this study provided detailed conformers for all AM structures. A schematic of an AM along with its CG representation and atomistic transformation is shown in Fig. 4. Snapshots of selected AM are presented in Fig. 5.
Fig. 4.
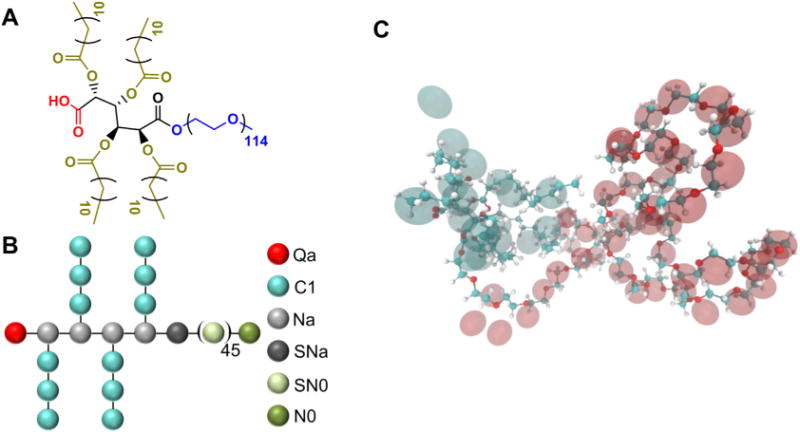
A) The molecular and B) coarse grained structure of [−1]M12P5K shown in a ball-and-stick representation. C) The conformation of [−1]M12P5K along with its CG representation in the MARTINI force field where CG beads are shown as transparent spheres.
Fig. 5.
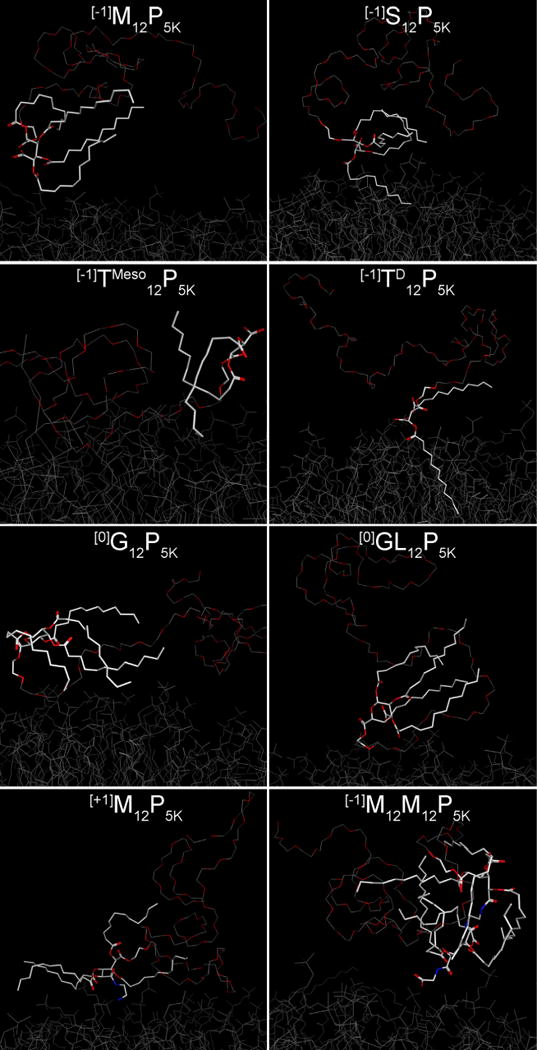
AMs exhibiting high to moderate efficiency in reduction of oxLDL uptake (left column) have their aliphatic arms in an extended conformation while less effective polymers (right column) form more compact globular structures with aliphatic arms pointing in the direction opposite to the cell membrane. These snapshots of AMs were obtained after 400 ns of CG MD simulation and additional 2 ns of AA MD simulation over the surface of membrane bilayer. For simplicity, hydrogen atoms are omitted. The hydrophobic heads of the AMs are highlighted as white sticks and the PEG tail is shown as a trace attached to the AM “head”. For visual comparison, the top three rows show stereo pairs of polymers that have distinctive behavior in reduction of oxLDL uptake: [−1]M12P5K/[−1]S12P5K, [−1]TMeso12P5K/[−1]TD12P5K, and [0]G12P5K/[0]GL12P5K.
3.4. Simulations of AM conformations
The MD results suggest that the most effective AM at inhibiting oxLDL ([−1]M12P5K, [−1]gM12P5K, [+1]M12P5K) tend to stay in the extended conformation during the entire MD simulation, while their less active counterparts ([−1]S12P5K, [0]GL12P5K, [−1]M12M12P5K) generally form more compact globular structures with lauryl arms pointing in the direction opposite of the cell membrane (Fig. 5).
3.5. Establishment of a QSAR model
Molecular descriptors were generated to encode various physicochemical properties of the AM including spatial organization, chemical composition and stereochemistry. Following filtering and prioritization of descriptors based on their information content, QSAR models were constructed using PLS regression to predict the AM efficacy (i.e., inhibition of oxLDL uptake). Several QSAR models were built and key descriptors that explain oxLDL uptake-related behavior of AM stereo pairs were identified. A statistically strong correlation between predicted and observed results was achieved with only five descriptors. Employing these descriptors, a QSAR model was established which exhibited a strong linear correlation (r2 = 0.91, ) between predicted and observed values of oxLDL uptake (Table 2).
Table 2.
QSAR equation related oxLDL uptake inhibition and descriptors of polymers, statistical analysis of the model fit and relative influence of descriptors on the QSAR model. oxLDL uptake inhibition = −617.97111 + 6528.05803*G3p + 187.62572*HOMA + 608:22915*Ds − 391.41561*R5u + 1179.44106*G1u.
| Relative influence of descriptors | ||
|---|---|---|
| G3p | 1.000000 | 3rd component symmetry directional WHIM index/weighted by atomic polarizabilities |
| HOMA | 0.329363 | Harmonic oscillator model of aromaticity index |
| Ds | 0.382432 | D total accessibility index/weighted by atomic electrotopological states |
| R5u | 0.314173 | R autocorrelation of lag 5/unweighted |
| G1u | 0.245292 | 1st component symmetry directional WHIM index/unweighted. |
| QSAR fit | ||
|
| ||
| Root mean square error (RMSE) | 8.65491 | |
| Correlation coefficient (R2) | 0.91 | |
| Cross-validated RMSE | 14.65 | |
| Cross-validated R2 | 0.77 | |
WHIM descriptors (G3p, Ds, G1u): (Weighted Holistic Invariant Molecular descriptors), which are geometrical descriptors based on statistical indices calculated on the projections of the atoms along principal axes.
Geometrical descriptors (HOMA): different kinds of conformationally dependent descriptors based on the molecular geometry.
GETAWAY descriptors: descriptors calculated from the leverage matrix obtained by the centered atomic coordinates (molecular influence matrix, MIM).
3.6. QSAR predictive capability
The final model was applied to predict oxLDL uptake inhibition of new AMs, namely, [−1]M12P5K analogs with variable aliphatic arms ([−1]M6P5K, [−1]M10P5K, and [−1]M14P5K) (Fig. 6). The predicted and experimental values for the test set, comprising three newly designed AM with varying hydrophobic moieties were correctly ranked from low to high values. This points to the prediction ability of the final QSAR model and its utility for optimizing AM structures that mitigate adverse athero-relevant endpoints.
Fig. 6.
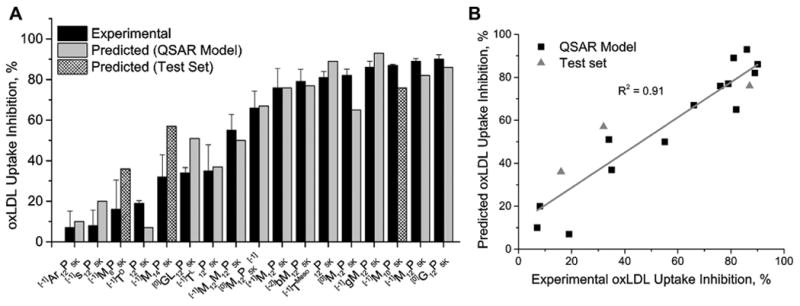
The final QSAR model was successful at predicting the bioactivity (oxLDL uptake inhibition) of the new, three member AM test set. A) The QSAR fit showing that the residual from simulation is often within the S.E.M. from the experimental data. B) The linear fit of experimental vs. predicted outcomes with R2 = 0.91.
4. Discussion
Inhibition of the athero-inflammatory phenotype of macrophages is considered to be as a major strategic target for the management of atherosclerosis underlying cardiovascular disease [50–53]. While a range of recently advanced AMs show continued promise in inhibiting atherogenesis in inflamed macrophages, a rational framework to designing improved AMs is currently lacking [8,10–12,14,54]. In this work, a multiscale modeling approach was advanced by employing molecular descriptors of the AM chemical structures and geometric parameters based on conformational changes of the AMs on model lipid membranes. By correlating biological efficacy data from hMDMs with a wide range of AM descriptors, a QSAR model was derived, which affords new insights to optimize and predict the efficacy of new AM structures.
Developing QSAR and principal component analysis (PCA) models to isolate critical molecular features is a versatile approach applied to a wide array of molecular feature spaces [55,56]. Even a small library size of molecules such as the AMs show a high variability in biological efficacy, as slight changes in AM structure produce markedly different levels of efficacy at inhibiting oxLDL uptake and foam cell formation (Fig. 3). While this result is quite common for small molecule therapeutics, this effect was somewhat intriguing for the current AM library given the large MW and the conformational flexibility of the AMs, thus prompting further mechanistic modeling efforts.
Although previous studies found AM efficacy differences dependent on charge type and charge placement [10,11], this work has identified that the largest differences in AM efficacy arise from variations in stereochemical and hydrophobic modifications (Fig. 3A–C). For instance, [0]M12P5K and [0]G12P5K show relatively high levels of bioactivity despite having an overall neutral charge that should make them less effective at binding to the positive charged SR binding domains, which mediate oxLDL binding and uptake, and similarly AM uptake. This result may indicate that the folded conformation of the AM and the presentation of hydrophobic moieties can contribute to improved binding. Additionally, the mucic acid sugar backbone seems to provide increased efficacy as [−1]M12P5K, [0]M12P5K, , [−2]bM12P5K, [−1]gM12P5K and [+1]M12P5K all show higher levels of bioactivity (inhibition of oxLDL uptake) than tartaric and saccharic acid backbone AMs. However, changes in stereochemistry resulted in the most drastic differences in inhibition efficiency, for example, with the substitution of saccharic acid in [−1]S12P5K for mucic acid in [−1]M12P5K where the only structural difference is one stereocenter. Thus, [−1]M12P5K was the second most effective AM at inhibiting oxLDL uptake, whereas its stereo-counterpart, [−1]S12P5K consistently showed little inhibition.
While measurements of oxLDL uptake provide early insights into variations in lipid influx, they do not capture the longer-term effects of lipid accumulation, metabolism and secretion [57]. Transition to the foam cell phenotype defined by large lipid droplets developing within the cell, is an alternative, physiologically relevant marker of atherogenesis as it marks the first stage in lesion development in vivo and is often accompanied by increased inflammatory cytokine secretion. Our results indicate that the AM library shows similar effects on inhibition of the foam cell phenotype as that on oxLDL uptake in vitro. As these results mimic those of oxLDL uptake, they show that AM could be effective at managing atherosclerotic disease even in areas of high oxLDL concentration.
The molecular dynamics simulations shed light into potential mechanisms underlying how the AMs with different alkyl arms exhibit differential bioactivity. For example, the MD snapshots indicate that the extended conformation of alkyl arms is required for effective bioactivity and conversely increasingly globular AMs were found to be less effective. This divergence may arise from stronger interactions occurring between the extended lauryl arms with the membrane bilayer surface during simulation. As a result, these interactions would generate deeper and more rapid penetration of the AM into the cell membrane, suggesting that tighter AM binding leads to more effective inhibition of oxLDL uptake and foam cell formation.
The advantage of using combined coarse grained and atomistic MD modeling is that the resultant descriptors could also be used to establish QSARmodels for predicting AM efficacy. One of the notable results from this study is that a statistically significant model was established between predicted and experimental outcomes using merely five AM descriptors. The descriptor set, although not easily transferable into simple chemical terms, encompasses a wide range of AM descriptors such as aromatic rings and unsaturated bonds (HOMA descriptor), molecular geometry, size, shape and stereochemistry (Table 2). Among the prominent 3D descriptors are the WHIM descriptors, which are geometrical descriptors based on statistical indices calculated on the projections of the atoms along principal axes and the GETAWAY descriptors [58–61]. WHIM descriptors encode 3D information on molecular size, shape, symmetry and atom distribution with respect to invariant reference frames, while the GETAWAY descriptors encode atomic properties such as atomic mass, atomic polarizability, atomic electronegativity, van der Waals atomic volume, and the unit weight. Overall, this QSAR model was successful at predicting the efficacy of [−1]M12P5K analogs with variable hydrophobicity ([−1]M6P5K, [−1]M10P5K, and [−1]M14P5K).
Despite the success of the proposed QSAR formalism, further modeling improvements will be forthcoming as the AM library continues to evolve, to include, for example, newer and more complex carbohydrate-derived core structures, aliphatic chains, and PEG chains. Moreover, the QSAR models will gain in predictive performance and statistical robustness as more experimental results are obtained for model building. Analogous QSAR relations could also be sought in the future between the AM chemistry and downstream activity such as membrane binding/partitioning, or modulation of inflammatory activity. For example, we hypothesize that certain AMs could attenuate the downstream pro-inflammatory signaling cascade either directly through the scavenger receptor-mediated uptake processes or indirectly through other unrelated mechanisms. The modeling approach could potentially help to tease apart the role of different members of the AM library that could trigger one or more such pathways in silico. Improved molecular level binding data of how the AMs bind to scavenger receptors is necessary to further validate the relevance of the AM conformers on the bioactivity. In the absence of such information, the current modeling QSAR framework that ties together chemical and molecular level inputs from biomaterial structures with downstream biological outputs is particularly valuable. In summary, the implementation of the validated QSAR model, together with insights into structure-activity relationships gained from analyzing the leading molecular descriptors, can guide the functional design of AM biomaterials as therapeutics for cardiovascular disease.
5. Conclusion
A modeling framework was developed for the design and predictive enhancements to biomaterials that can inhibit the uptake of oxidized lipoproteins in inflammatory macrophages, a process called atherogenesis. An expanded biomaterial library of amphiphilic macromolecules (AM) modulating key molecular features was created that displayed high variability in the reduction of oxLDL uptake and foam cell formation. As 2D molecular parameters cannot adequately describe a large 3D structure, MD simulations were used to generate 3D structures in solution. These 2D and 3D molecular models yielded several descriptors, which were correlated to biological activity using QSAR. This model based upon five key descriptors was then implemented to successfully predict the efficacy of new AM structures. This work demonstrates the unique interplay of 2D and 3D computational approaches combined with powerful bioactivity prediction models that can be used for the rational design and optimization of macromolecular therapeutics.
Supplementary Material
Acknowledgments
The authors thank the National Heart, Lung and Blood Institute (R01HL107913, R21HL93753–PVM, KEU), the Coulter Foundation for Biomedical Engineering Translational Research Award (PVM), National Institute of Biomedical Imaging and Bioengineering (T32 EB005583–DRL), for financial support.
Footnotes
Appendix A. Supplementary data: Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2013.07.011.
References
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Sahoo D, Drover V. The role of scavenger receptors in signaling, inflammation and atherosclerosis. In: Cheema S, editor. Biochemistry of atherosclerosis. US: Springer; 2006. pp. 70–91. [Google Scholar]
- 3.Fuhrman B, Partoush A, Volkova N, Aviram M. Ox-LDL induces monocyte-to-macrophage differentiation in vivo: possible role for the macrophage colony stimulating factor receptor (M-CSF-R) Atherosclerosis. 2008;196(2):598–607. doi: 10.1016/j.atherosclerosis.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 4.Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med. 1996;20(5):707–27. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- 5.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005;3:63–8. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 6.Gotto AM. Antioxidants, statins, and atherosclerosis. J Am Coll Cardiol. 2003;41(7):1205–10. doi: 10.1016/s0735-1097(03)00082-2. [DOI] [PubMed] [Google Scholar]
- 7.Saha P, Modarai B, Humphries J, Mattock K, Waltham M, Burnand KG, et al. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol. 2009;9(2):109–18. doi: 10.1016/j.coph.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Hehir S, Plourde NM, Gu L, Poree DE, Welsh WJ, Moghe PV, et al. Carbohydrate composition of amphiphilic macromolecules influences physicochemical properties and binding to atherogenic scavenger receptor A. Acta Biomater. 2012;8(11):3956–62. doi: 10.1016/j.actbio.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iverson NM, Plourde NM, Sparks SM, Wang J, Patel EN, Shah PS, et al. Dual use of amphiphilic macromolecules as cholesterol efflux triggers and inhibitors of macrophage athero-inflammation. Biomaterials. 2011;32(32):8319–27. doi: 10.1016/j.biomaterials.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iverson NM, Sparks SM, Demirdirek B, Uhrich KE, Moghe PV. Controllable inhibition of cellular uptake of oxidized low-density lipoprotein: structure-function relationships for nanoscale amphiphilic polymers. Acta Biomater. 2010;6(8):3081–91. doi: 10.1016/j.actbio.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Plourde NM, Iverson N, Moghe P, Uhrich KE. Nanoscale amphiphilic macromolecules as lipoprotein inhibitors: the role of charge and architecture. Int J Nanomedicine. 2007;2(4):697–705. [PMC free article] [PubMed] [Google Scholar]
- 12.Plourde NM, Kortagere S, Welsh W, Moghe PV. Structure-activity relations of nanolipoblockers with the atherogenic domain of human macrophage scavenger receptor A. Biomacromolecules. 2009;10(6):1381–91. doi: 10.1021/bm8014522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chnari E, Nikitczuk JS, Wang J, Uhrich KE, Moghe PV. Engineered polymeric nanoparticles for receptor-targeted blockage of oxidized low density lipoprotein uptake and atherogenesis in macrophages. Biomacromolecules. 2006;7(6):1796–805. doi: 10.1021/bm0600872. [DOI] [PubMed] [Google Scholar]
- 14.Chnari E, Nikitczuk JS, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules can inhibit cellular uptake of differentially oxidized LDL. Biomacromolecules. 2006;7(2):597–603. doi: 10.1021/bm0506905. [DOI] [PubMed] [Google Scholar]
- 15.Chnari E, Lari HB, Tian L, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules for selective retention of low-density lipoproteins. Biomaterials. 2005;26(17):3749–58. doi: 10.1016/j.biomaterials.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 16.Mayr LM, Bojanic D. Novel trends in high-throughput screening. Curr Opin Pharmacol. 2009;9(5):580–8. doi: 10.1016/j.coph.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Wang CY, Ai N, Arora S, Nagarajan K, Zauhar R, Young D, et al. Identification of previously unrecognized antiestrogenic chemicals using a novel virtual screening approach. Chem Res Toxicol. 2006;19(12):1595–601. doi: 10.1021/tx060218k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chekmarev D, Kholodovych V, Kortagere S, Welsh W, Ekins S. Predicting inhibitors of acetylcholinesterase by regression and classification machine learning approaches with combinations of molecular descriptors. Pharm Res. 2009;26(9):2216–24. doi: 10.1007/s11095-009-9937-8. [DOI] [PubMed] [Google Scholar]
- 19.Paranjpe PV, Chen Y, Kholodovych V, Welsh W, Stein S, Sinko PJ. Tumor-targeted bioconjugate based delivery of camptothecin: design, synthesis and in vitro evaluation. J Control Release. 2004;100(2):275–92. doi: 10.1016/j.jconrel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Passic SR, Ferguson ML, Catalone BJ, Kish-Catalone T, Kholodovych V, Zhu W, et al. Structure-activity relationships of polybiguanides with activity against human immunodeficiency virus type 1. Biomed Pharmacother. 2010;64(10):723–32. doi: 10.1016/j.biopha.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kortagere S, Welsh W. Development and application of hybrid structure based method for efficient screening of ligands binding to g-protein coupled receptors. J Comput Aided Mol Des. 2006;20(12):789–802. doi: 10.1007/s10822-006-9077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aparoy P, Kumar Reddy K, Reddanna P. Structure and ligand based drug design strategies in the development of novel 5-lox inhibitors. Curr Med Chem. 2012;19(22):3763–78. doi: 10.2174/092986712801661112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh JH, Yin S, Wang XS, Liu S, Dokholyan NV, Tropsha A. Cheminformatics meets molecular mechanics: a combined application of knowledge-based pose scoring and physical force field-based hit scoring functions improves the accuracy of structure-based virtual screening. J Chem Inf Model. 2011;52(1):16–28. doi: 10.1021/ci2002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mize CD, Abbott AM, Gacasan SB, Parrill AL, Baker DL. Ligand-based autotaxin pharmacophore models reflect structure-based docking results. J Mol Graph Model. 2011;31(0):76–86. doi: 10.1016/j.jmgm.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatt HG, Patel PK. Pharmacophore modeling, virtual screening and 3D-QSAR studies of 5-tetrahydroquinolinylidine aminoguanidine derivatives as sodium hydrogen exchanger inhibitors. Bioorg Med Chem Lett. 2012;22(11):3758–65. doi: 10.1016/j.bmcl.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Sushko I, Novotarskyi S, Körner R, Pandey A, Rupp M, Teetz W, et al. Online chemical modeling environment (ochem): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des. 2011;25(6):533–54. doi: 10.1007/s10822-011-9440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dube D, Periwal V, Kumar M, Sharma S, Singh T, Kaur P. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model. 2012;18(5):1701–11. doi: 10.1007/s00894-011-1187-0. [DOI] [PubMed] [Google Scholar]
- 28.Dong XO, Ebalunode J, Yang SY, Zheng W. Receptor-based pharmacophore and pharmacophore key descriptors for virtual screening and QSAR modeling. Curr Comput Aided Drug Des. 2011;7(3):181–9. doi: 10.2174/157340911796504332. [DOI] [PubMed] [Google Scholar]
- 29.Ebalunode J, Zheng W, Tropsha A. Application of QSAR and shape pharma-cophore modeling approaches for targeted chemical library design. In: Zhou JZ, editor. Chemical library design Methods in molecular biology. Vol. 685. Humana Press; 2011. pp. 111–33. [DOI] [PubMed] [Google Scholar]
- 30.Kooistra AJ, Roumen L, Leurs R, de Esch IJ, de Graaf C. From heptahelical bundle to hits from the haystack: structure-based virtual screening for GPCR ligands. Meth Enzymol. 2013;522:279–336. doi: 10.1016/B978-0-12-407865-9.00015-7. [DOI] [PubMed] [Google Scholar]
- 31.Kamal A, Kashi Reddy M, Viswanath A. The design and development of imidazothiazole–chalcone derivatives as potential anticancer drugs. Expert Opin Drug Discov. 2013;8(3):289–304. doi: 10.1517/17460441.2013.758630. [DOI] [PubMed] [Google Scholar]
- 32.Krasowski MD, Hopfinger AJ. The discovery of new anesthetics by targeting GABAA receptors. Expert Opin Drug Discov. 2011;6(11):1187–201. doi: 10.1517/17460441.2011.627324. [DOI] [PubMed] [Google Scholar]
- 33.Martin TM, Harten P, Young DM, Muratov EN, Golbraikh A, Zhu H, et al. Does rational selection of training and test sets improve the outcome of QSAR modeling? J Chem Inf Model. 2012;52(10):2570–8. doi: 10.1021/ci300338w. [DOI] [PubMed] [Google Scholar]
- 34.Ertl P, Lewis R. Iade: a system for intelligent automatic design of bioisosteric analogs. J Comput Aided Mol Des. 2012;26(11):1207–15. doi: 10.1007/s10822-012-9609-3. [DOI] [PubMed] [Google Scholar]
- 35.Gubskaya AV, Kholodovych V, Knight D, Kohn J, Welsh WJ. Prediction of fibrinogen adsorption for biodegradable polymers: integration of molecular dynamics and surrogate modeling. Polymer. 2007;48(19):5788–801. doi: 10.1016/j.polymer.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith JR, Knight D, Kohn J, Rasheed K, Weber N, Kholodovych V, et al. Using surrogate modeling in the prediction of fibrinogen adsorption onto polymer surfaces. J Chem Inf Comput Sci. 2004;44(3):1088–97. doi: 10.1021/ci0499774. [DOI] [PubMed] [Google Scholar]
- 37.Duan P, Li S, Ai N, Hu L, Welsh WJ, You G. Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: discovery and molecular characterization. Mol Pharmacol. 2012;9(11):3340–6. doi: 10.1021/mp300365t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fomovska A, Wood RD, Mui E, Dubey JP, Ferreira LR, Hickman MR, et al. Salicylanilide inhibitors of toxoplasma gondii. J Med Chem. 2012;55(19):8375–91. doi: 10.1021/jm3007596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian L, Yam L, Zhou N, Tat H, Uhrich KE. Amphiphilic scorpion-like macro-molecules: design, synthesis, and characterization. Macromolecules. 2004;37(2):538–43. [Google Scholar]
- 40.Djordjevic J, del Rosario LS, Wang J, Uhrich KE. Amphiphilic scorpion-like macromolecules as micellar nanocarriers. J Bioact Compat Polym. 2008;2008(23):532–51. [Google Scholar]
- 41.Wang J, del Rosario LS, Demirdirek B, Bae A, Uhrich KE. Comparison of peg chain length and density on amphiphilic macromolecular nanocarriers: self-assembled and unimolecular micelles. Acta Biomater. 2009;5:883–92. doi: 10.1016/j.actbio.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 42.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, et al. Amber 12. San Francisco: University of California; 2012. [Google Scholar]
- 43.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111(27):7812–24. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 44.Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput. 2008;4(5):819–34. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 45.Lee H, de Vries AH, Marrink SJ, Pastor RW. A coarse-grained model for polyethylene oxide and polyethylene glycol: conformation and hydrodynamics. J Phys Chem B. 2009;113(40):13186–94. doi: 10.1021/jp9058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marrink SJ, de Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B. 2003;108(2):750–60. [Google Scholar]
- 47.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–47. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 48.Rzepiela AJ, Schäfer LV, Goga N, Risselada HJ, De Vries AH, Marrink SJ. Reconstruction of atomistic details from coarse-grained structures. J Comput Chem. 2010;31(6):1333–43. doi: 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- 49.Sousa da Silva A, Vranken W. ACPYPE–AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5(1):367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis DR, Kamisoglu K, York AW, Moghe PV. Polymer-based therapeutics: nanoassemblies and nanoparticles for management of atherosclerosis. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3(4):400–20. doi: 10.1002/wnan.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iverson N, Plourde N, Chnari E, Nackman GB, Moghe PV. Convergence of nanotechnology and cardiovascular medicine. Biodrugs. 2008;22(1):1–10. doi: 10.2165/00063030-200822010-00001. [DOI] [PubMed] [Google Scholar]
- 52.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29(10):1419–23. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 53.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. 2007;37(1):14–6. doi: 10.1002/eji.200636910. [DOI] [PubMed] [Google Scholar]
- 54.York AW, Zablocki KR, Lewis DR, Gu L, Uhrich KE, Prud'homme RK, et al. Kinetically assembled nanoparticles of bioactive macromolecules exhibit enhanced stability and cell-targeted biological efficacy. Adv Mater. 2012;24(6):733–9. doi: 10.1002/adma.201103348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kou PM, Pallassana N, Bowden R, Cunningham B, Joy A, Kohn J, et al. Predicting biomaterial property-dendritic cell phenotype relationships from the multivariate analysis of responses to polymethacrylates. Biomaterials. 2012;33(6):1699–713. doi: 10.1016/j.biomaterials.2011.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen LK, Ramer-Tait AE, Broderick SR, Kong CS, Ulery BD, Rajan K, et al. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32(28):6815–22. doi: 10.1016/j.biomaterials.2011.05.063. [DOI] [PubMed] [Google Scholar]
- 57.Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306(5701):1558–61. doi: 10.1126/science.1101909. [DOI] [PubMed] [Google Scholar]
- 58.Todeschini R, Lasagni M, Marengo E. New molecular descriptors for 2D and 3D structures. Theory J Chemom. 1994;8(4):263–72. [Google Scholar]
- 59.Consonni V, Todeschini R, Pavan M. Structure/response correlations and similarity/diversity analysis by getaway descriptors. 1. Theory of the novel 3D molecular descriptors. J Chem Inf Comput Sci. 2002;42(3):682–92. doi: 10.1021/ci015504a. [DOI] [PubMed] [Google Scholar]
- 60.Consonni V, Todeschini R, Pavan M, Gramatica P. Structure/response correlations and similarity/diversity analysis by getaway descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. J Chem Inf Comput Sci. 2002;42(3):693–705. doi: 10.1021/ci0155053. [DOI] [PubMed] [Google Scholar]
- 61.Kubinyi H, Folkers G, Martin YC, editors. 3D QSAR in drug design. Dordrecht (The Netherlands): Kluwer/ESCOM; 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.