

Abstract
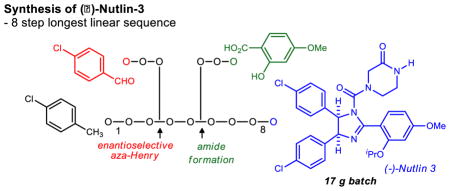
Chiral nonracemic cis-4,5-bis(aryl) imidazolines have emerged as a powerful platform for the development of cancer chemotherapeutics, stimulated by the Hoffmann-La Roche discovery that Nutlin-3 can restore apoptosis in cells with wild-type p53. The lack of efficient methods for the enantioselective synthesis of cis-imidazolines, however, has limited their more general use. Our disclosure of the first enantioselective synthesis of (−)-Nutlin-3 provided a basis to prepare larger amounts of this tool used widely in cancer biology. Key to the decagram-scale synthesis described here was the discovery of a novel bis(amidine) organocatalyst that provides high enantioselectivity at warmer reaction temperature (−20 °C) and low catalyst loadings. Further refinements to the procedure led to the synthesis of (−)-Nutlin-3 in a 17 gram batch, and elimination of all but three chromatographic purifications.
INTRODUCTION
The development of small molecule inhibitors of p53/MDM2 has grown rapidly 1 in the intervening years since the 2004 discovery by scientists at Hoffmann-La Roche (HLR) that a new class of cis-4,5-disubstituted imidazolines were effective inhibitors of this protein-protein interaction.2 Among these heterocycles, Nutlin-3 was the most potent derivative and the levorotatory enantiomer (1) is now known to be the more potent antipode.2,3,4 The HLR discovery fueled countless studies in basic cancer biology, stimulated the discovery of similar small molecule inhibitors by others,1,5 and provided optimism in cancer drug development.6 HLR recently advanced one member of this class to Phase I clinical trials.7
Studies that use (−)-Nutlin 3 as a tool to elucidate basic pathways in cancer cell biology must ultimately be validated in vivo. A typical experiment in a mouse model requires 500 mg of (−)-Nutlin-3, an amount that would require approximately US $15,000 if provided by a commercial source.8 Although this cost represents a discount of 50% since our report in 2011, it remains a significant financial obstacle to most biology laboratories. Alternatively, the HLR synthesis reported in patents4 and two reports in late 2012,9 can be used to prepare Nutlin-3 in racemic form, but the need for supercritical fluid chromatography to obtain the more potent enantiomer10 diminishes its general access as well.
We developed a highly diastereo- and enantioselective addition of aryl nitromethanes to imines and applied the reaction to the first synthesis selective for (−)-Nutlin-3.3 A key advantage of this approach is the access it provides to desymmetrized cis-stilbene diamines in two steps through a carbon-carbon bond forming reaction. Seidel’s desymmetrization of cis-stilbene diamines using an enantioselective acylation is a promising alternative strategy and may ultimately provide an enantioselective route from the cis-stilbene diamine common to the HLR synthesis.11
Although the aza-Henry approach appeared to be advantageous by both strategic and practical criteria, we sought to examine several factors critical to validation of the route’s suitability for large scale implementation, including: 1) optimization of the key enantioselective step (catalyst loading & temperature), and 2) minimization or elimination of chromatography steps. The result of this study is a batch procedure to produce up to 17 grams of (−)-Nutlin 3 and a new catalyst that allows the key enantioselective step to operate at −20 °C with high enantioselectivity.12
Hoffmann-La Roche patents outline their use of meso-diamine 2 (synthesized in 5 steps from benzil, Scheme 1). Compound 2 could be cyclized with amidate 313 or methyl ester 4a in the presence of AlMe314 to provide meso-imidazoline 5a/5b. Treatment of 5a/5b with phosgene gave a mixture of acid chloride enantiomers which were separated by chiral HPLC before or after substitution of the chloride with the amine to give (−)-Nutlin-3 and derivatives. Details related to HLR’s process development of Nutlin-3 have emerged recently,9 describing a boric acid catalyzed cyclization to form imidazoline intermediate 5b. While this is a significant improvement, the approach provides Nutlin-3 in racemic form and relies upon supercritical fluid chromatography to separate the desired enantiomer in the final step.10
Scheme 1.

Hoffmann-La Roche Synthesis of rac-Nutlin-3
RESULTS AND DISCUSSION
Our approach to Nutlin-3 is based upon four key building blocks as illustrated in Figure 1. In order to scale this synthesis, each building block needed to be accessible in significant quantity.
Figure 1.
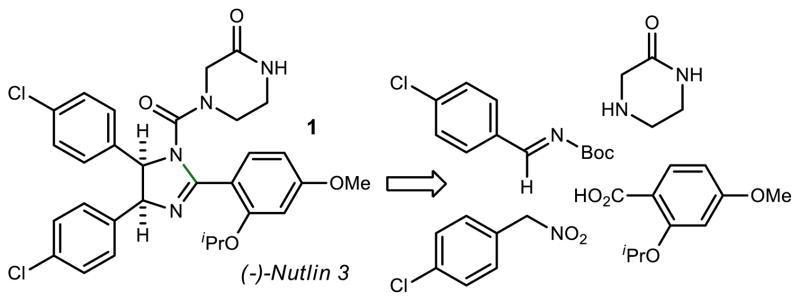
Building Blocks Required for the Enantioselective Synthesis of (−)-Nutlin 3
Several methods were available to prepare the aryl nitromethane (8, Scheme 2). The prevailing method is displacement of the corresponding bromide with nitrite.15 This preparation is often characterized by low yields due to competing O-alkylation, but it is reliably applied and a collection of diverse benzyl halides are commercially available. Alternative preparations include electrophilic nitration of the corresponding metalated toluene,16,17 electrophilic nitration and concomitant decarboxylation of phenylacetonitrile or phenyl acetic acid,18 free radical arylation of nitromethane,19 and Pd-catalyzed coupling.20 A recent advance, reported after completion of our study, is that of Kozlowski21 who described the cross-coupling of aryl halides and nitromethane to give the desired aryl nitroalkanes in high yield.
Scheme 2.
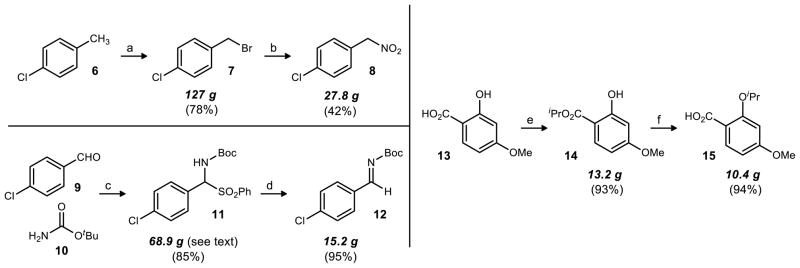
Preparation of the Synthetic Building Blocksa
a(a) NBS, AIBN, CCl4, reflux; (b)NaNO2, urea, DMF, -20°C; (c) NaSO2Ph, HCO2H, MeOH/H2O, rt, 19 d; (d) K2CO3, Na2SO4, THF, eflux; (e) K2CO3, iPrBr, DMF, reflux; (f) KOH, EtOH/H2O, reflux
In our hands, Kornblum’s procedure was superior. The arylnitroalkane is prepared in two steps from para-chloro toluene (6) with this method, although para-chloro benzyl bromide 7 is commercially available. Treatment of para-chloro toluene with N-bromosuccinimide and a substoichiometric amount of AIBN provided the desired benzyl bromide in 78% yield (127 g) after crystallization.
This benzyl bromide (7) was then treated with sodium nitrite to provide the desired nitroalkane 8. Initially, a procedure using a nitrite resin described by Gelbard22 was attempted, and although the straightforward purification by filtration was advantageous, we were unable to achieve the yield (87%) described for this substrate.
Application of the Kornblum procedure was particularly sensitive to the reaction time, as excessive time resulted in an increased ratio of benzyl alcohol to nitroalkane. Benzyl alcohol likely results from nitrite alkylation at oxygen (forming benzyl nitrite), followed by nitrosyl transfer to an incidental nucleophile (e.g. water, silica gel). This nitrosyl transfer is necessary to prevent a side reaction between benzyl nitrite and the desired nitroalkane 8 which reduces yield. Phloroglucinol, a nitrite ester trapping agent,15 was not effective in converting the nitrite ester to benzyl alcohol when used as a component of the reaction; it was not until workup that this conversion occurred. Therefore, phloroglucinol was added to the ethereal solution after an aqueous workup. Within minutes the slightly golden solution became the dark red color reportedly characteristic of nitrosophloroglucinol. This solution was washed with water, dried, and concentrated. The residue could not be purified by vacuum distillation, acid/base extraction, or recrystallization, but purification by column chromatography provided 27.8 g (42%) of the nitroalkane 8.
The N-Boc imine 12 (Scheme 2) is made in two steps that involve simple purification techniques from readily available starting materials. The N-Boc α-amido sulfone23 11 is made from the condensation of the corresponding aldehyde 9, N-Boc tert-butyl carbamate 10, and the sodium salt of phenyl sulfinic acid in the presence of formic acid.24 Synthesis of this particular sulfone has been described twice in the literature by Wulff, with yields of 23% (3 days reaction time, 1.2 equiv. aldehyde)25 and 61% (2 days reaction time, 2 equiv. aldehyde).26 Similar conditions in our hands proved reasonably general across a wide range of sulfones. In an attempt to obtain higher yields through increased conversion, we lengthened the reaction time to 9 days before the precipitated product was filtered. Trituration of the filtered solid with ether removes residual aldehyde, and provided 53.7 g of sulfone 11 (66% yield). In an attempt to further increase yield, we combined the trituration filtrate with the original filtrate, stirred the mixture in an open vessel for an additional 10 days, and collected the nascent precipitate. After trituration, an additional 15.3 g of sulfone was isolated. The total yield for this reaction on the largest scale attempted was 68.9 g after 19 days (85%). Attempts to further improve the reaction rate (e.g. temperature increase, solvent mixtures) were unsuccessful. The simplicity of the reaction conditions, ease of purification, and high yield provided an adequate platform to launch a large-scale synthesis.
Formation of the N-Boc imine (12) by treatment of the α-amido sulfone with potassium carbonate and sodium sulfate in refluxing THF was dependent on reaction time. Reactions that were stopped too long after full conversion contained aldehyde (the result of imine hydrolysis), while reactions containing unreacted α-amido sulfone could produce benzene sulfinate (or its conjugate acid) during the subsequent aza-Henry step. The latter was expected to work against the goal to lower catalyst loadings, as overprotonation of the catalyst can adversely affect reactivity and/or stereoselection.27 As a result, careful monitoring of imine formation was applied, and on the largest scale attempted, this reaction provided 15.2 grams of the N-Boc imine 12 (95%) after a 6 hour reaction period. The isolated material contained ~4% of the parent aldehyde.
The requisite carboxylic acid 15 was synthesized in two steps from commercially available 4-methoxy salicylic acid (13, Scheme 2). The dialkylated product 14 was produced in 93% yield (13.2 g) after purification. Subsequent hydrolysis of the dialkylated compound provided the desired carboxylic acid 15 in excellent yield (94%, 10.4 g).28
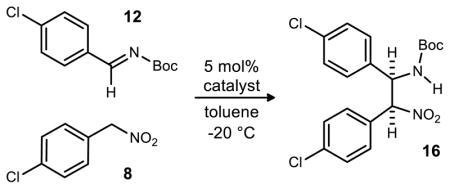
Our initial rendition of the enantioselective aza-Henry reaction employed 5 mol% 6,7(MeO)2PBAM (18, Figure 2), low temperature (−78 °C), and chromatographic purification of the desired product. In order to adapt the procedure to a large scale preparation, we sought a protocol that could be applied at higher temperature, and a purification technique that did not involve chromatography. Novel catalyst modifications were explored to achieve the high selectivity at warmer operating temperatures, while direct precipitation was used to streamline the overall protocol.
Figure 2.

Aryl Nitromethane Additions at Low Temperaturea
aAll reactions used 0.1 mmol imine and 1.1 equiv. of nitroalkane in 0.1 M toluene
The relative insolubility of the major diastereomer of the aza-Henry product under the initial conditions proved to be beneficial both in the optimization of this reaction and the scale-up of the (−)-Nutlin-3 synthesis overall. The product precipitated from solution during the reaction at more concentrated conditions (≥0.1 M, toluene). This allowed for a simple filtration of the reaction to isolate the desired diastereomer in high purity. This protocol was therefore used to facilitate an examination of reaction variables and ultimately identification of the optimal conditions. As one would anticipate, increasing the reaction temperature leads to an attenuation of stereoselection. Identification of a more selective catalyst that could operate at higher temperature became an important objective.
When using aryl nitromethane pronucleophiles, enantioselectivity observed when using 4-pyrrolidine bisamidine (PBAM, 17)29 is not improved by the addition of an equal amount of triflic acid, as is often the case with most other nitroalkanes.30, 31 Our hypothesis is that the acidic aryl nitroalkane serves as the Brønsted acid cocatalyst in these reactions.32, 33, 34 The assumption that the more basic 4-pyrrolidine catalysts are the best catalysts for this reaction led us to retain this element in catalyst design efforts. In one investigational line, an increase in the catalyst Brønsted basicity was sought by catalysts with electron-releasing quinoline substituents, while positioning these groups proximal to the site of presumed substrate binding. This ultimately resulted in the identification of 6,7(MeO)2PBAM (18) as the most enantioselective catalyst. Despite the high enantioselectivity produced by this catalyst (91% ee), these values could only be achieved at −78 °C.
This prompted a further investigation of catalyst modifications in order to achieve high enantioselectivity, and one aim was to explore substitution at the 8–position. Of the positions 5–8, the 8 position was expected to be the position of highest steric consequence due to its proximity to the amidine functional group. While our operational stereochemical models presume amidine protonation, followed by imine binding to the amidinium, it was not clear whether substitution at the 7- or 8-position would provide improved facial bias or hindered substrate binding. These bis(amidine) variations could be prepared using our standard approach, and illustrated for 19 (Scheme 3). An initial comparison of the free base of 8MeOPBAM (19) to PBAM (17) and 6,7(MeO)2PBAM (18) (Figure 2) under the previously optimal conditions revealed the 8-substituted catalyst to be significantly more enantioselective, providing the product in 9:1 dr and 95% ee at −78 °C without a noticeable drop in reactivity. The same trend was observed with these three catalysts when warmer conditions were used (−20 °C, Table 1). Once again, 8MeOPBAM (19) was found to be the most enantioselective catalyst of the three giving appreciably high enantioselectivity (89% ee).
Scheme 3.
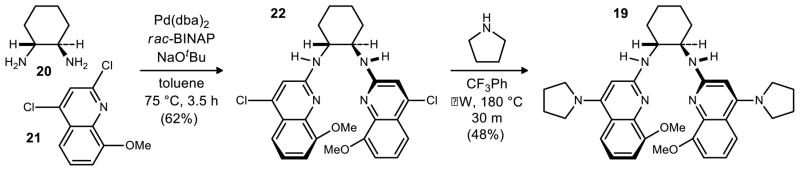
Preparation of the Pyrrolidine Bis(AMidine) Catalyst 19
Table 1.
Aryl Nitromethane Additions at −20 °Ca
| |||||
|---|---|---|---|---|---|
| entry | catalyst | drb | ee | yield | |
| 1 | PBAM | 17 | 6:1 | 83 | 99 |
| 2 | 6,7 (MeO)2PBAM | 18 | 6:1 | 80 | 91 |
| 3 | 8MeOPBAM | 19 | 7:1 | 89 | 99 |
0.1 mmol scale, 1.1 equiv. of 8, 0.1 M in toluene, 18–24 h reaction time
Diastereomer and enantiomer ratios measured by HPLC using chiral stationary phases.
Isolated yield.
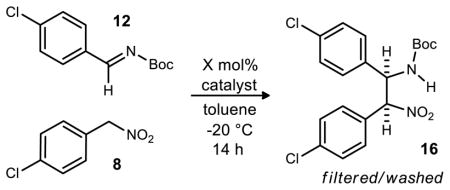
The relatively high reactivity of the aryl nitroalkanes compared to aliphatic nitroalkanes with the 4-pyrrolidine bis(amidine) class of catalysts35 offered the potential to utilize very short reaction times and/or low catalyst loadings. With this in mind, we performed a study to ascertain the lowest catalyst loading that could be tolerated without sacrificing enantioselection or reasonable reaction times.
Remarkably, a clear trend surfaced in that the enantioselectivity of each reaction increased as the catalyst loading decreased (Table 2). With PBAM (17) the enantioselectivity rose from 53% ee at 20 mol% to 74% ee at just 1 mol% loading.36 This trend was also observed using an 8-alkoxy substituted catalyst 6,8(MeO)2PBAM (23), which gives similar enantioselectivity to 8MeOPBAM (19). Enantioselectivity ranged from 62% ee at 15 mol% loading to 87% ee at both 0.38 and 0.15 mol% loadings.
Table 2.
Catalyst Loading Comparison Of Two Catalystsa
| |||||||
|---|---|---|---|---|---|---|---|
| PBAM (17) | 6,8 (MeO)2PBAM (23) | ||||||
|
| |||||||
| entry | mol% | ee (%)b | yield (%)c | entry | mol% | ee (%)b | yield (%)c |
| 1 | 20 | 53 | 39 | 1 | 15.4 | 62 | 58 |
| 2 | 10 | 65 | 59 | 2 | 7.7 | 73 | 68 |
| 3 | 5 | 70 | 71 | 3 | 3.9 | 79 | 75 |
| 4 | 2 | 73 | 76 | 4 | 1.5 | 84 | 74 |
| 5 | 1 | 74 | 76 | 5 | 0.77 | 86 | 79 |
| 6d | 1 | 80 | 78 | 6 | 0.38 | 87 | 79 |
| 7 | 0.15 | 87 | 73 | ||||
0.1 mmol scale, 1.1 equiv. of 8, 0.1 M in toluene, 18–24 h reaction time.
Determined by chiral HPLC.
Isolated yield of single diastereomer (200:1 dr by HPLC) after filtration/washing.
0.2 equiv imine added, 30 min intervals.
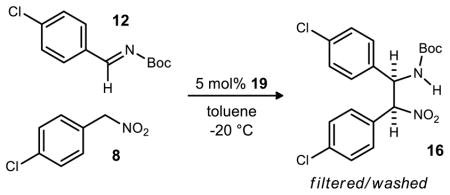
This feature resulted in a protocol that produced the aza -Henry product in sufficient yield and stereoselectivity for scale-up. 8MeOPBAM (19) was ultimately selected for further development due to its structural simplicity and ease of synthesis. On a 0.4 mmol scale, 2 mol% 8MeOPBAM (19) catalyzed the reaction leading to 91% ee and good yield. Enantioselectivity and yield remained unaffected as the catalyst loading was reduced to 0.5 mol% and the reaction was concentrated to 0.4 M (Table 3, entries 2–5). Higher concentrations could not be stirred effectively as the product precipitated from solution. Unfortunately, some initial reactions run on larger scale (2 mmol, Table 3, entry 6) were less enantioselective and variability was observed. Investigations were performed on the purities of the imine, nitroalkane, and catalyst as well as the rate of stirring but the reason for the low stereoselectivity remained elusive.
Table 3.
Aryl Nitromethane Addition Scale-up/Optimization
| |||||
|---|---|---|---|---|---|
| entry | mol% | mmol | conc. (M) | ee (%)a | yield (%)b |
| 1 | 2 | 0.4 | 0.1 | 91 | 82 |
| 2 | 1 | 0.4 | 0.1 | 92 | 78 |
| 3 | 1 | 0.4 | 0.2 | 92 | 84 |
| 4 | 1 | 0.4 | 0.4 | 91 | 84 |
| 5 | 0.5 | 0.4 | 0.4 | 91 | 81 |
| 6c | 0.5 | 2 | 0.4 | 77–94 | 81–90 |
| 7d | 0.5 | 2 | 0.4 | 86 | 86 |
| 8e | 0.5 | 20 | 0.4 | 89 | 90 |
| 9e | 0.5 | 25 | 0.4 | 89 | 89 |
Enantiomer ratios of isolated material measured by HPLC using chiral stationary phases.
Isolated yield of single diastereomer (200:1 dr by HPLC) after filtration/washing.
Results of 4 runs.
Imine added in 0.2 equiv increments.
Imine added in 0.1 equiv increments.
Ultimately, slow addition of the imine to the stirring solution containing catalyst and nitroalkane led to consistent results with high enantioselectivity. Five additions of 0.2 equivalents of imine led to 86% ee and 86% yield on 2 mmol scale (Table 3, entry 7). On 20 and 25 mmol scales, even slower additions (0.1 equiv at a time) of imine led to slightly higher levels of enantioselectivity (89% ee, Table 3, entries 8 and 9) and good yield.
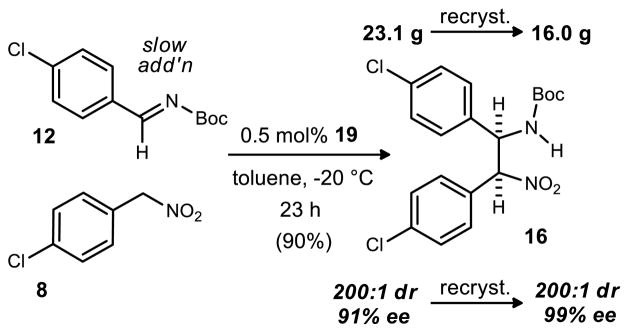
These optimized conditions produced similar results on the largest scale attempted - over 62 mmol. Features of this reaction include: 0.5 mol% catalyst loading; slow addition of imine (~0.06 equivalent/addition over 8 hours); essentially stoichiometric amounts of the two partners (1 and 1.05 equivalents of imine and nitroalkane respectively); a relatively high reaction concentration (0.4 M in toluene); exclusive precipitation of the desired diastereomer from solution and filtration to recover it; less than a day reaction time at −20 °C. A total of 23.1 g of product 16 was produced after filtration in 91% ee and >200:1 dr. This material was then recrystallized from toluene to provide 16 grams of the desired stereoisomer (>200:1 dr, 99% ee, Scheme 4).
Scheme 4.
Largest Scale Aryl Nitromethane Addition
With a reliable large scale preparation of the aza -Henry adduct developed, efforts to convert this material to (−)-Nutlin-3 commenced. The initial yield published for the NaBH4 reduction of the nitro compound on small scale was relatively low (66%, Table 4). Other common reduction methods were evaluated with mixed results: treatment with Zn/HCl provided the desired amine in modest yield (60%), whereas hydrogenations with Pd/C and Raney nickel failed to convert to product.
Table 4.
Reduction Attempts
| |
|---|---|
| conditions | yield (%) |
| CoCl2, NaBH4, MeOH, rt, 2 h | 66 |
| Zn/HCl, EtOH, 1.5 h | 60 |
| Pd/C, HCO2NH4, MeOH, rt, 24 h | no rxn |
| Raney Ni, HCO2NH4, MeOH, rt, 24 h | no rxn |
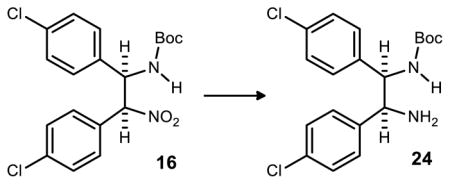
The CoCl2/NaBH4 reduction was eventually revisited for optimization. No byproducts or unreacted starting material were observed by NMR analysis of crude reaction mixtures resulting from filtration, suggesting that material could have been lost at some point in the workup. Ultimately it was found that the amine product was very insoluble and copious amounts of dichloromethane were required to fully dissolve and separate the product from the filtered salts. After this discovery, near quantitative yields were obtained across reactions of increasing scale. The largest scale reaction produced nearly 22 gramsof 24 with a 94% yield (Scheme 5).
Scheme 5.

Optimized Closing Sequence for the Synthesis of (−)-Nutlin-3
An initial attempt to couple amine 24 to carboxylic acid 15 was made with DCC, resulting in a poor yield (35%) of the desired amide. Additionally, DCC co-eluted with the product during chromatography. EDC was a better coupling reagent by measures of yield and product isolation. This reaction provided the desired product 25 (85% yield) after straightforward water and solvent washes and filtrations.
A standard TFA-promoted Boc deprotection of 25 proceeded with excellent yield after a standard quench and extractive workup. The resulting free amine 26 was converted to isocyanate by treatment with 1,1-carbonyldiimidazole. Subsequent reaction with 2-oxopiperazine (27)37 produced 28 in 99% yield. Straightforward washing with water provided 28 in sufficient purity for the final step.
The dehydrative cyclization between the pendant urea and amide followed several precedents38,39 based on Hendrickson’s work (Scheme 6).40,41 These examples demonstrated the cyclization of an amide and a sulfonamide, and an amide and a carbamate respectively. Each procedure used at least one equivalent of Tf2O and 3 equivalents of Ph3PO.42 This is not advantageous from many standpoints, but perhaps the most significant problem is the complication it presents for purification. Almqvist successfully used a polymer supported Ph3PO allowing the imidazoline products to be isolated without the use of column chromatography.39 Despite the apparent reusability of the polymer supported reagents, its cost is quite prohibitive for its use on large scale.43 Our attention therefore focused on improvement of the original Hendrickson conditions. For reasons not yet thoroughly understood, the stoichiometry could not be further optimized without sacrificing conversion. The reaction mixtures in all cases, however, were largely devoid of side products. This protocol was superior to alternatives surveyed (e.g. SOCl2, POCl3) by measures of both conversion and selectivity. Optimized conditions for the dehydration employed four equivalents of Ph3PO and two equivalents of Tf2O, and over 17 grams of the final compound 1 were produced in 89% yield after column chromatography.
Scheme 6.

Selected Applications of the Hendrickson Dehydrative Cyclization
We next tested whether the biological activity of synthetic 1 was comparable to commercially available reagent. Wild type p53-expressing human melanoma cells were used to test the ability of synthesized and commercial (−)-Nutlin-3 to block melanoma cell growth and stabilize p53. We found that addition of both compounds to the culture media caused an approximate 90% decrease in the number of viable cells over the vehicle (DMSO) control (Figure 3 top panels, compare A and B). Similarly, robust induction of p53 protein levels was detected using either compound (Figure 3A and B, bottom panels). These data demonstrate that (−)- Nutlin-3 prepared using this batch procedure performs equivalently to commercially available (−)-Nutlin-3 in downstream biological applications.
Figure 3.

Biological activity of (A) synthesized and (B) commercial (−)-Nutlin-3. Top panels: Viability test. Hs294T melanoma cells were incubated with 10μM of (−)-Nutlin-3 or vehicle control for 5 days and viable cells were counted using the trypan blue exclusion test. Bottom panels: Accumulation of p53 protein in cell lysates after 10 μM (−)-Nutlin-3 treatment, analyzed by Western Blotting. Hs294T cells were subjected to the tested compounds or vehicle control for 72 hrs. β-Actin was used as a loading control.
CONCLUSIONS
In summary, a synthesis of (−)-Nutlin-3 has been developed at the decagram scale (Scheme 7), aided by the development of the new chiral bis(amidine) catalyst 19 that allows the key step to proceed at higher temperature (−20 °C) than previously required without attenuation of selectivity. The route is 13 steps (8 steps longest linear sequence) in length from readily available starting materials. Procedures were developed in most cases using glassware that was not rigorously dried and reagents used as received. The overall yield (based on longest linear sequence) is 32% from para-chloro benzaldehyde and 13% from para-chloro toluene. The procedure is summarized in Scheme 7 with yields from the largest scale reactions attempted. aza-Henry product 16 can be produced at the 23 gram scale with low catalyst loading (0.5 mol%). A unique application of Hendrickson’s dehydrative cyclization is employed in the final step to produce the cis-imidazoline. The procedure has been used to prepare 17 g of (−)-Nutlin-3 in a single batch, with column chromatography necessary after only three of the steps (8, 2-oxo-piperazine (27), and Nutlin-3).
Scheme 7.

Overall Asymmetric Synthesis of Nutlin-3
Although the cost of (−)-Nutlin-3 has dropped 50% since our initial disclosure, the 17 grams prepared by this procedure represents a current commercial value of ~US $0.5 million. Efforts to broaden the procedure to include additional novel cis-imidazoline derivatives are underway and will be reported in due course.
EXPERIMENTAL SECTION
General Methods
All reagents and solvents were commercial grade and were used as received except when noted otherwise. Dichloromethane and tetrahydrofuran was dried by passage through a column of activated alumina as described by Grubbs.44 Organic extracts were dried using MgSO4 unless otherwise noted. Thin layer chromatography (TLC) was performed using glass-backed silica gel (250 μm) plates and flash chromatography utilized 230–400 mesh silica gel. UV light, and/or the use of potassium permanganate solutions were used to visualize products. Microwave reactions were performed in a Biotage Initiator EXP US 355302 microwave reactor in a sealed vial, at the “normal” absorption level. The temperature of microwave reactions were monitored by an external sensor.
Nuclear magnetic resonance spectra (NMR) were acquired on a 400, 500, or 600 MHz instrument. Chemical shifts are measured relative to residual solvent peaks as an internal standard set to δ7.26 and δ 77.0 (CDCl3). IR data are reported in wavenumbers (cm−1). Compounds were analyzed as neat films on a NaCl plate (transmission). Mass spectra were recorded by use of the ionization method noted and detected by TIC or TOF analysis. The data for all compounds previously reported matched data previously reported in the literature.
Preparation of (−)-Nutlin-3 (1)
Triphenylphosphine oxide (37.14 g, 133.4 mmol) and dry CH2Cl2 (319 mL) were added to a 1 L round-bottomed flask with a stir bar and stirred for 20 min in an ice-water bath under argon. Tf2O (11.2 mL, 66.7 mmol) was added and the reaction was stirred for 20 min. The urea (20.00 g, 33.36 mmol) was added as a solid and ~5 mL of CH2Cl2 was used to rinse the solid from the sides of the flask into the reaction mixture. The mixture was stirred in the ice-water bath for 1.5 h. The mixture was slowly quenched with satd aq NaHCO3 (325 mL, added over 10 minutes). The flask was removed from the ice-water bath, stirred for 5 minutes while warming to room temperature, and transferred to a 1 L separatory funnel. The organic layer was removed and the aqueous layer was extracted twice with CH2Cl2. The combined organics were dried, filtered, and concentrated. NMR analysis of this solid indicated a 4.2:1 ratio of Ph3PO to product. The crude product was purified by column chromatography (0–4% MeOH/CH2Cl2) to provide 17.26 g (89%) of (−)-Nutlin-3.
1-(Bromomethyl)-4-chlorobenzene (7)
Note: This benzyl bromide is a potent lachrymator. This reaction should be carried out and worked up in a well-ventilated fume hood. The chlorotoluene (110.0 g, 868.8 mmol) and carbon tetrachloride (500 mL) were added to a 1 L round-bottomed flask containing a stir bar. N-Bromosuccinimide (140.6 g, 789.8 mmol) was added and the reaction was heated to reflux temperature. AIBN (25.0 mg, 150 μmol) was added. After 10 minutes, AIBN (25.0 mg, 150 μmol) was added. After another 10–15 minutes, the reaction began boiling vigorously. At this point, the reaction flask was removed from heat and it continued to boil for 15 minutes without external heat. When the reaction mixture stopped refluxing, the flask was again heated to reflux temperature and AIBN (25.0 mg, 0.15 μmol) was added. The rate of reflux did not increase after the last addition. After 15 minutes, the heat source was removed. A small aliquot was concentrated and found to be >95% conversion by 1H NMR. The reaction mixture was filtered through a glass frit and the filtered succinimide was rinsed on the frit with hexanes. The filtrate was concentrated (rotavap) and transferred to a 200 mL evaporation flask. After its concentration, the mixture was allowed to sit undisturbed overnight, resulting in crystallization of a solid out of the slightly golden solution. The flask was placed on ice for 1 h before decanting. The wet solid left in the flask was subjected to high vacuum for several hours. This solid (118.1 g) was found to be a >20:1 mixture of desired monobrominated to undesired dibrominated product, with a slight trace of toluene starting material. The decanted liquid was subjected to high vacuum briefly before placing on dry ice to promote crystallization. This material was warmed to near room temperature (10–20 °C) and filtered through a Buchner funnel. The crystalline material was dried for several minutes on the filter before subjection to high vacuum (~30 min). This produced an additional 9.1 grams of a white crystalline solid that was of essentially the same purity. Overall, the product (127.2 g, 78% yield) was produced in sufficient purity for the next step.
1-Chloro-4-(nitromethyl)benzene (8).45
Note: This benzyl bromide is a potent lachrymator. This reaction should be carried out and worked up in a well-ventilated fume hood. NaNO2 (40.3 g, 584 mmol), urea (46.8 g, 778 mmol), and DMF (589 mL) were added to a 1 L round-bottomed flask containing a stir bar and stirred until nearly dissolved. Large chunks of suspended solid were chopped up by hand with a spatula. The reaction mixture was chilled in a dry ice/acetone bath until the internal temperature was −50 °C. Bromide (80.0 g, 389 mmol, purity as described from previous reaction) was added. 5–10 mL DMF was used to rinse residual bromide into the reaction flask. The reaction was transferred to a −20 °C freezer and stirred for 4 h, 10 min. The flask was removed from the freezer and quickly poured onto ~2 L ice water in a 4 L separatory funnel. The suspension was quickly shaken before extraction (3 x diethyl ether, ~400 mL each). The combined ether extracts were dispensed into a 2 L Erlenmeyer flask containing phloroglucinol (39.2 g, 311 mmol). This suspension was stirred overnight (within minutes of stirring the suspension turned to a deep red color). The suspension was poured into a 4L separatory funnel. The Erlenmeyer flask was rinsed with ether twice to transfer the residual material into the separatory funnel. ~1.5 L of water was poured into the separatory funnel and the aqueous layer was separated without shaking to avoid emulsion. The organics were washed three times with water (~1.5 L each) and dried over MgSO4. After filtration, the dried organics were concentrated by rotavap to a red oil. This material was found to be a 53:40:7 mixture (desired nitro compound: corresponding benzyl alcohol:dibromotoluene from starting material) by 1H NMR. This residue was purified by column chromatography (0–1–2–3% ethyl acetate/hexanes) to obtain a slightly golden low-melting crystalline solid (27.9 g, 42%). This material was found to contain ~2% of the corresponding aldehyde but was otherwise pure by 1H NMR.
tert-Butyl ((4-chlorophenyl)(phenylsulfonyl)methyl)carbamate (11)
Aldehyde (45.0, 320 mmol), carbamate (25.0 g, 214 mmol), and the sodium salt (70.1 g, 427 mmol) were combined in a 1 L round-bottomed flask containing a sufficiently large stir bar. MeOH (202 mL) and water (404 mL) were added and the reaction mixture was stirred. Formic acid (16.1 mL, 427 mmol) was added to the reaction and the mixture continued to stir vigorously. After 9 days the reaction was filtered through a Buchner funnel. The contents of the reaction flask were rinsed with diethyl ether and poured onto the filter. The white solid was dried on the filter and transferred to a 500 mL Erlenmeyer flask. The solid was triturated with diethyl ether (~300 mL). The triturated suspension was filtered through a Buchner funnel and dried. This trituration, filtration, and drying sequence was repeated twice. The resulting solid was transferred to a 250 mL round-bottomed flask and subjected to high vacuum for ~2 h. This provided 53.7 g of a white, fluffy solid. 1H NMR indicated that this solid contained a trace (<1%) of aldehyde but was otherwise very pure. The combined filtrates were subjected to a stream of air to evaporate the majority of the diethyl ether and transferred back to the 1 L reaction flask (rinsed with a minimal amount of ether to fully transfer). A stream of air was blown through the reaction flask to remove ether before stirring vigorously. After 10 additional days the contents of the reaction flask were filtered through a Buchner and dried by the passage of air through the filter. The filtered solid was subjected to the trituration, filtration, and drying sequence (as described previously, but using less ether) three times. The dried solid was transferred to a 100 mL round-bottomed flask and dried under high vacuum for several hours. This produced an additional 15.2 g of white, fluffy solid that was found to be equally pure as the first sulfone batch by 1H NMR. The combined filtrates were again transferred back to the reaction flask and concentrated by a stream of air as described above. The mixture was stirred for an additional 25 days. After implementation of the same filtration and trituration sequence, 3.4 g of white, fluffy solid was obtained. This material was found to be of the same purity as previous batches by 1H NMR. Overall, 72.4 g (89%) of sulfone was isolated. This material was sufficiently pure for use in the next step.
(E)-tert-Butyl 4-chlorobenzylidenecarbamate (12)
Potassium carbonate (64.59 g, 467.3 mmol) and sodium sulfate (75.85 g, 534.0 mmol) were loaded into a 1L 3-neck round-bottomed flask with stir bar. The flask was flame-dried, backfilled with argon, and attached to a condenser. Sulfone (25.49 g, 66.75 mmol) was then added through a side neck before dry THF (556 mL) was added via cannula. The flask was place into a pre-heated oil bath and refluxed for 6 h. The flask was allowed to cool to room temperature before filtering through a glass frit. Ether was used to rinse residual solid from the reaction flask and the solid on the filter. The filtrate was concentrated by rotavap before subjection to high vacuum. 15.18 g (95%) of white solid was produced that was found to contain 4% of the corresponding but otherwise pure by 1H NMR. All characterization data matched that in the literature. This material was used in the next reaction without further purification.
Isopropyl 2-isopropoxy-4-methoxybenzoate (14).46
Potassium carbonate (32.86 g, 237.9 mmol) was dispensed into a 500 mL round-bottomed flask with a stir bar. The flask was then flame-dried under vacuum and backfilled with argon. The carboxylic acid (10.00 g, 59.47 mmol) and DMF (297 mL) were added to the flask and the mixture was stirred at rt for 15 minutes. Isopropyl bromide (22.3 mL, 238 mmol) was added to the flask. The flask was purged and backflushed with argon before placing into a sand bath. The reaction was stirred and gradually heated from room temperature to reflux temperature over 1.5 hours. The reaction was refluxed for 4 hours before removing the heat sourece and cooling to 30 °C. Potassium iodide (987.3 mg, 5.947 mmol) was added to the flask. The mixture was cooled to room temperature and stirred for 18 h. The mixture was then quenched with 3 M aq HCl and extracted three times with diethyl ether. The combined organics were washed with 2 M aq sodium carbonate and water, dried (MgSO4), filtered, and concentrated. The product was relatively pure by 1H NMR containing only a trace of monoalkylated product. The material was purified by column chromatography (0–5% ethyl acetate in hexanes). A bronze oil resulted (13.22 g, 93%) that was not noticeably purer than the crude compound by 1H NMR. All characterization data matched that in the literature.
2-Isopropoxy-4-methoxybenzoic acid (15).3
The ester (13.20 g, 52.32 mmol) was dispensed into a 500 mL round-bottomed flask with a stir bar. EtOH (145 mL), water (29 mL), and KOH (8.805 g, 157.0 mmol) were added to the flask. A condenser was attached to the flask, and the flask was placed into an oil bath (45 °C). The reaction contents were stirred as the temperature of the bath gradually increased to 95 °C over two hours. After 3.5 hours of stirring at 95 °C, the flask was removed from the heating bath and allowed to cool to ambient temperature. The reaction was concentrated by rotavap to remove most of the ethanol. The solution was diluted with water and 3M aq HCl was added until full precipitation was observed (pH was checked to be ~1). The solution was extracted three times with diethyl ether, and the combined organics were dried over MgSO4, filtered, and concentrated to an off-white solid (10.39 g, 94%). The compound was found to be pure by 1H NMR.
tert-Butyl (1R,2S)-1,2-bis(4-chlorophenyl)-2-nitroethylcarbamate (16)
Nitroalkane (11.27 g, 65.71 mmol) and catalyst (177 mg, 313 μmol) were dispensed into a 500 mL round-bottomed flask containing a stir bar (5 cm length). Toluene (300 mL) was added to the flask and the mixture was stirred at room temperature until homogeneous. In a separate vial, imine (15.00 g, 62.58 mmol) was dissolved in toluene (25 mL). The reaction flask was then placed into a dry ice/acetone bath. When the stirring reaction mixture was < −40 °C, 2 mL of the imine solution was added to the reaction. The flask was then moved into a −20 °C freezer and stirring was continued. Aliquots (2 mL) of the imine solution were added at approximately 30 min intervals while stirring in the −20 °C freezer. After the final addition, the reaction was allowed to stir an additional 15 hours (22 hour total reaction time) at −20 °C. The reaction flask was quickly removed from the freezer and filtered through a Buchner funnel (12.5 cm diameter). The flask was rinsed with ~100 mL of hexanes to transfer the remaining solid to the filter. The solid was rinsed with 150 mL of cold toluene (4 °C) and 150 mL of hexanes (room temperature) before drying for 15 min by the passage of air through the filter. The solid was transferred to a 250 mL round-bottomed flask and dried under vacuum for 2h. The crude solid (23.07 g, 90 %) was found to be 200:1 dr, 91% ee by chiral HPLC. The material was transferred to a 2 L Erlenmeyer flask and toluene (1.13 L, 49 mL/gram of crude product) was added to the flask. The suspension was swirled while being heated in a 70 °C water bath until fully dissolved. The flask was then placed on the benchtop to cool. After 130 min, the crystallized suspension was filtered through a Buchner funnel into a 2 L filter flask. The crystallized material was dried on the filter for at least 15 minutes before transfer to a 250 mL round-bottomed flask and subjection to vacuum for 2 h. This produced 12.49 g of crystallized product that was found to be 200:1 dr, 99% ee by chiral HPLC. The filtrate from this recrystallization was redissolved with heat in a 70 °C water bath and left to cool to room temperature and age 19 h. The mother liquor was separated from the newly formed globular crystals by filtration through fluted filter paper into a 2 L Erlenmeyer flask. This filtrate was placed in an ice water bath for 1 h. The contents of the flask were filtered through a Buchner funnel (8.5 cm diameter). After the crystalline material was dried on the filter by the passage of air, it was transferred to a 250 mL round-bottomed flask and placed under high vacuum for 3 h. This produced 3.46 additional grams of product that was found to be 200:1 dr and 99% ee. The combined yield from the recrystallizations was 15.98 g (62%) of diastereo- and enantiomerically pure compound.
8MeOPBAM (19)
A 0.5–2.0 mL microwave vial was charged with the corresponding 4ClBAM (200.0 mg, 402.1 μmol), pyrrolidine (132 μL, 1.61 mmol), and trifluoromethyl benzene (1.2 mL). This suspension was heated at 180 °C and stirred in the microwave for 30 min. The reaction mixture was purified by column chromatography (2–5% methanol in dichloromethane with 0.5% AcOH) to provide a light brown solid. This material was dissolved in dichloromethane and then washed with 3 M aq NaOH. The combined organic layers were dried over MgSO4 and concentrated. The resulting solid was dissolved in EtOAc and washed with water, dried over MgSO4, filtered, and concentrated to a light brown solid (108.4 mg, 48%). +320 (c 0.14, CHCl3); Rf = 0.24 (10% MeOH/CH2Cl2); IR (film) 3244, 2933, 2857, 1637, 1593 cm-1; 1H NMR (400 MHz, CDCl3) δ7.49 (d, J = 8.4 Hz, 2H), 6.93 (dd, J = 7.6, 7.6 Hz, 2H), 6.88 (d, J = 7.2 Hz, 2H), 5.78 (br s, 2H), 5.52 (br s, 2H), 4.10–4.00 (m, 2H), 4.00 (s, 6H), 3.41–3.29 (m, 4H), 3.28–3.15 (m, 4H), 2.40–2.27 (m, 2H), 1.90–1.73 (m, 8H), 1.73–1.65 (m, 2H), 1.60–1.40 (m, 4H); 13C NMR (150 MHz, CDCl3, 325 K) ppm 157.5, 153.8, 153.7, 141.6, 119.5, 118.5, 117.3, 109.0, 93.3, 56.5, 56.3, 51.7, 33.1, 25.4, 24.9; HRMS (ESI): Exact mass calcd for C34H43N6O2[M+H] +567.3448, found 567.3431.
2,4-Dichloro-8-methoxyquinoline (21)
Phosphorus(V) oxychloride (50 mL) was added through a running condenser into a 3-neck round bottom flask equipped with a stir bar containing malonic acid (13.0 g, 125 mmol) at room temperature. While stirring, o-anisidine (14.1 mL, 125 mmol) was added in small portions over a period of 15 minutes through an open neck of the round bottom flask. The reaction mixture was heated and stirred at reflux for 7 h. The reaction mixture was allowed to cool to room temperature before it was poured over crushed ice. Soxhlet extraction of the crude solid with hexanes for ~24 h followed by cooling of the obtained extract yielded a yellow solid precipitate (2.6135 g, 9%) that was pure by 1H NMR. Mp 127.5–128.5 °C; Rf = 0.08 (10% EtOAc/hexanes); IR (film) 3097, 3008, 1576, 1561 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 1H), 7.57 (dd, J = 8.0, 8.0 Hz, 1H), 7.55 (s, 1H), 7.15 (d, J = 7.5 Hz, 1H), 4.08 (s, 3H); 13C NMR (100 MHz, CDCl3) ppm 154.8, 148.9, 144.3, 139.9, 128.1, 126.4, 122.7, 115.6, 109.8, 56.3; HRMS (CI): Exact mass calcd for C10H7Cl2NO [M]+226.9899, found 226.9890.
4Cl8MeOQuinBAM (22)
A 100 mL round bottom flask was charged with Pd(dba)2 (25.2 mg, 43.8 μmol), rac- BINAP (27.3 mg, 43.8 μmol), sodium tert-butoxide (632.0 mg, 6.576 mmol), (R,R)-diaminocyclohexane (250.3 mg, 2.192 mmol), and the quinoline (1.0000 g, 4.385 mmol).47 Toluene (22 mL) was added, and the reaction mixture was stirred at 70 °C for 3.5 h. The reaction was cooled to room temperature, diluted with CH2Cl2, and filtered through Celite. The filtrate was concentrated and purified by column chromatography (25–50% ethyl acetate in hexanes) to provide a yellow solid (642.6 mg, 62%). +530 (c0.16, CHCl3); Rf = 0.31 (50% EtOAc/hexanes); IR (film) 3240, 2933, 1607, 1545 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.54 (dd, J = 8.4, 0.8 Hz, 2H), 7.17 (dd, J = 7.6, 0.8 Hz, 2H), 7.01 (dd, J = 7.6, 0.8 Hz, 2H), 6.59 (s, 2H), 6.38 (br s, 2H), 4.15–3.95 (m, 2H), 4.04 (s, 6H), 2.45–2.30 (m, 2H), 1.85–1.70 (m, 2H), 1.50–1.30 (m, 4H); 13C NMR (150 MHz, CDCl3) ppm 155.9, 153.2, 142.1, 140.0, 122.2, 121.9, 116.2, 112.7, 109.8, 56.6, 56.2, 32.5, 24.7; HRMS (ESI): Exact mass calcd for C26H27Cl2N4O2[M+H] +497.15 11, found 497.1521.
6,8(MeO)2PBAM (23)
A 0.5–2.0 mL microwave vial was charged with the corresponding 4ClBAM (125.0 mg, 224.2 μmol), pyrrolidine (200 μL, 2.44 mmol), and trifluoromethylbenzene (600 μL). This suspension was heated at 180 °C and stirred in the microwave for 10 min. The reaction was then concentrated and purified by column chromatography (0–10% methanol in dichloromethane) to provide a light brown solid. This material was dissolved in dichloromethane and then washed with 3 M aq NaOH. The combined organic layers were dried over MgSO4 and concentrated to afford a brown/red powder (100.9 mg, 72%). mp 112.0–114.0 °C; +274 (c 1.03, CHCl3); Rf = 0.14 (10% MeOH/0.5% AcOH/CH2Cl2); IR (film) 3257, 2930, 2858, 1607, 1538 cm− 1; 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 2.4 Hz, 2H), 6.58 (d, J = 2.4 Hz, 2H), 5.71 (br s, 2H), 5.60 (s, 2H), 4.05–3.92 (m, 2H), 3.97 (s, 2H), 3.82 (s, 6H), 3.34–3.23 (m, 2H), 3.23–3.12 (m, 4H), 2.35–2.24 (m, 2H), 1.93–1.82 (m, 8H), 1.82–1.74 (m, 2H), 1.55–1.35 (m, 4H); 13C NMR (100 MHz, CDCl3) ppm 156.5, 154.4, 153.6, 152.6, 136.7, 119.0, 100.2, 96.8, 94.4, 56.3, 56.2, 55.3, 51.6, 33.1, 25.4, 24.9; HRMS (CI): Exact mass calcd for C36H46N6O4[M] +626.3575, found 626.3550.
tert-Butyl (1R,2S)-2-amino-1,2-bis(4-chlorophenyl)ethylcarbamate (24)
The nitroalkane (25.00 g, 60.78 mmol) was dispensed into a 2 L 3-neck round-bottomed flask with stir bar, and MeOH was added (500 mL) before stirring at room temperature. CoCl2 (7.891 g, 60.78 mmol) was added and the reaction mixture was chilled (0 °C). A septum was attached to each of the three necks and a needle attached to an empty balloon was inserted into each septum. NaBH4 (11.50 mg, 303.9 mmol) was slowly added over a one hour period. The reaction mixture was stirred at 0 °C for an additional hour (starting material consumed by TLC) before the mixture was quenched with 325 mL water, 550 mL 1 M aq HCl, and 375 mL satd aq NH4OH. The pH of the reaction mixture was found to be ~10. The mixture was filtered through a Buchner funnel (17 cm diameter). Water (600 mL) was used to rinse the flask and wash the filtered solid. The solid was dried on the filter for 15 minutes. The filtered flask was changed before rinsing the filtered solid with CH2Cl2 (950 mL in several portions). This CH2Cl2 filtrate was transferred to a 1 L separatory funnel and the water was separated. The organic layer was dried, filtered, concentrated by rotavap, and subjected to high vacuum (5 h). This produced a white solid (21.74 g, 94%) which was judged to be of high purity by 1H NMR.
tert-Butyl (1R,2S)-1,2-bis(4-chlorophenyl)-2-(2-isopropoxy-4-methoxybenzamido)ethylcarbamate (25)
The amine (21.5 g, 56.4 mmol) and carboxylic acid (11.85 g, 56.39 mmol) were dissolved in CH2Cl2 (500 mL) at room temperature in a 1 L round-bottomed flask. The solution was chilled to 0 °C in an ice/water bath and EDC (14.05 g, 73.30 mmol) and DMAP (688 mg, 5.64 mmol) were added. The reaction mixture was stirred vigorously (reaction thickens over time) and allowed to gradually warm to room temperature. After 16 h, the reaction mixture was diluted with CH2Cl2 (125 mL) and the precipitated solid was divided and stirred with a spatula by hand. The reaction was filtered through a Buchner funnel. The reaction flask was rinsed and poured onto the filter with CH2Cl2 (300 mL). An additional 300 mL of CH2Cl2 was used to wash the solid on the filter. This solid was transferred to a 500 mL round-bottomed flask and triturated with 300 mL CH2Cl2 by manual stirring with a spatula. The solid was filtered through a Buchner funnel and washed out of the flask with an additional 200 mL CH2Cl2. The solid was transferred to a 200 mL round-bottomed flask and dried under vacuum overnight. This produced 24.4 g of white solid with some light blue color and was determined to be an ~6:1 mixture of product to EDC (~4% EDC urea by mass) by 1H NMR. This 24.4 g of material (solid A) was set aside for later purification. The combined filtrates were poured into a 1 L separatory funnel and washed 3 times with water, dried, and concentrated to a solid. This solid was transferred to a 250 mL Erlenmeyer flask and triturated with 150 mL of CH2Cl2. The solid was filtered through a Buchner funnel and washed two times with CH2Cl2 (50 mL each) and once with hexanes (50 mL). After transfer to an evaporation flask, the solid was subjected to high vacuum overnight. This produced 6.87 g of white-yellow solid which contained a trace of EDC and some other impurities. This material was then transferred to a 125 mL Erlenmeyer flask and triturated with 50/50 CH2Cl2/hexanes (70 mL). The solid was filtered through a Buchner funnel and washed with 50 mL (50/50 CH2Cl2/hexanes). After transfer to a 100 mL round-bottomed flask, the solid (solid B) was subjected to high vacuum and set aside for later purification. The 24.4 g of material isolated from the first filtration (solid A) was added to a 500 mL Erlenmeyer flask and triturated with 400 mL of 50/50 CH2Cl2/hexanes with the aid of manual stirring by hand. This material was filtered by Buchner funnel and washed with hexanes. This solid was combined with solid B in a 1 L Erlenmeyer flask and triturated by stir bar and manual spatula stirring (dividing large, undispersed chunks) by hand with 600 mL 50/50 CH2Cl2/hexanes. This material was filtered (Buchner funnel, large chunks were divided on the filter) and washed with 300 mL 50/50 CH2Cl2/hexanes. After drying on the filter for 1 hour, the solid was transferred to a 250 mL evaporation flask and subjected to high vacuum for 3 hours. This produced 28.6 g of white solid with only a very faint blue tint. By 1H NMR, this material was a 6:1 mixture of desired product to EDC urea. This solid was transferred to a 1 L Erlenmeyer flask and triturated with a suspension of 500 mL 4:1 water:hexanes. After filtration through a Buchner funnel, the solid was washed successively on the filter with 200 mL hexanes, 200 mL water, and 200 mL hexanes. The solid was dried on the filter for 20 minutes before transferring to a flask and subjecting to high vacuum. This produced 26.7 g (83%) of a white solid that was found to be a >20:1 mixture of desired product to EDC urea.
N-((1S,2R)-2-Amino-1,2-bis(4-chlorophenyl)ethyl)-2-isopropoxy-4-methoxybenzamide (26)
The amide (26.4 g, 46.0 mmol) was dispensed into a 1 L round-bottomed flask containing a stir bar. Dichloromethane (455 mL) was added and the reaction mixture was stirred at room temperature. TFA (~38 mL) was added over a period of 10 minutes via an addition funnel to the stirred suspension (after the addition of the first ~5 mL the mixture became homogenous). After stirring for 2.5 h, the reaction mixture was checked by TLC and found to still contain starting material. Approximately 15 mL TFA (53 mL total, 710 mmol) was added over 2 minutes. After stirring for 1.5 h, the reaction mixture was found to no longer contain starting material by TLC. The reaction flask was placed into an ice water bath and 300 mL satd aq NaHCO3 was slowly added to the stirring mixture. After transfer to a 2 L Erlenmeyer flask with 50 mL satd aq NaHCO3 and 50 mL CH2Cl2, the following amounts of solutions and solvents were carefully and successively added to the vigorously stirring mixture: 50 mL 3 M aq NaOH, 100 mL CH2Cl2, 50 mL 3 M aq NaOH, 100 mL CH2Cl2, 200 mL water, 100 mL 3 M aq NaOH, 100 mL CH2Cl2, 200 mL water. The suspension was vigorously stirred for 1.5 hours. After transfer to a 4 L separatory funnel, the organic layer was removed. The aqueous layer was extracted twice with CH2Cl2 (400 mL each). The combined organic layers were dried, filtered, and concentrated by rotavap and high vacuum. This produced 20.0 g (92%) of white solid that contained a trace of EDC urea and CH2Cl2 but was otherwise pure by 1H NMR. This material was sufficiently pure for use in the next reaction.
2-Oxo-piperazine (27).37
This compound was prepared according to the patent procedure on a scale using 19.6 g of ethyl chloroacetate (160 mmol). After a 45 h reaction time, the reaction mixture was concentrated by rotary evaporator with water bath heating (60–70 °C). The resulting orange oil was purified by column chromatography (3–10% MeOH in CH2Cl2 w/1% NH4OH). A yellow solid (9.9 g) was obtained. This material was recrystallized from acetone (40 mL) to obtain a yellow-brown solid (3.75 g, 23%) that was sufficiently pure by 1H NMR.
N-((1R,2S)-1,2-Bis(4-chlorophenyl)-2-(2-isopropoxy-4-methoxybenzamido)ethyl)-3-oxopiperazine-1-carbox-amide (28)
The amine (20.30 g, 42.87 mmol) was dispensed into a 500 mL round-bottomed flask equipped with a stir bar. Dry dichloromethane (214 mL) was added and the reaction mixture was stirred at room temperature. 1,1-Carbonyldiimidazole (8.342 g, 51.45 mmol) was added and stirring continued for 1.5 h. TLC indicated complete consumption of starting material (Rf= 0.36 in 5% MeOH/CH2Cl2) and a new spot had appeared (Rf= 0.43 in 5% MeOH/CH2Cl2). The oxopiperazine (6.800 g, 67.93 mmol) was added to the reaction and stirring continued for 18 h. TLC indicated the complete consumption of isocyanate. The reaction was diluted with CH2Cl2 (750 mL) and washed three times with water (500 mL each) in a 4 L separatory funnel. After shaking the last wash, 200 mL of brine was added to alleviate an emulsion in the aqueous layer. The combined organic layers were dried over MgSO4 in a 2 L Erlenmeyer flask and filtered through a Buchner funnel. Some solid was present in the filtrate before the filtrate was concentrated by rotavap. The material was redissolved in 600 mL CH2Cl2 and filtered through Celite on a glass frit. This filtrate was concentrated to a white solid (25.5 g, 99%) that was found to be pure by 1H NMR.
2,4-Dichloro-6,8-dimethoxyquinoline (S1)
Compound was prepared using 6,8-dimethoxy aniline (10.0 g, 65.3 mmol), malonic acid (6.79 g, 65.3 mmol), and POCl3 (26 mL) according to the procedure for S5. The reaction time was 2 hours. The crude solid was extracted (soxhlet) with hexanes for 3 d to provide the title compound as a green-brown solid (1.376 g, 8%). Mp 183.5–184.5 °C; Rf = 0.09 (10% EtOAc/hexanes); IR (film) 3080, 2964, 1615, 1562 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 6.98 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 2.4 Hz, 1H), 4.03 (s, 3H), 3.95 (s, 3H); 13C NMR (100 MHz, CDCl3) ppm 159.4, 155.8, 146.1, 142.7, 136.6, 127.1, 122.9, 103.0, 93.7, 56.3, 55.7; HRMS (ESI): Exact mass calcd for C11H10Cl2NO2[M+H] +258.0089, found 258.0094.
4Cl6,8(MeO)2QuinBAM (S2)
A 25 mL round bottom flask was charged with Pd(dba)2 (22.3 mg, 38.7 μmol), rac-BINAP (24.1 mg, 38.7 μmol), sodium tert-butoxide (558.4 mg, 5.811 mmol), (R,R)-diaminocyclohexane (221.2 mg, 1.937 mmol), and the quinoline (1.000 g, 3.874 mmol). Toluene (13 mL) was added, and the reaction mixture was stirred at 85 °C. The reaction mixture was monitored by TLC, and complete conversion was observed after 65 min. The reaction was cooled to room temperature, diluted with CH2Cl2, and filtered through Celite. The filtrate was concentrated and purified by column chromatography (25–30% ethyl acetate in hexanes) to provide a yellow/orange solid (775.7 mg, 72%). mp 120.0–122.0 °C; +434 (c1.28, CHCl3); Rf = 0.09 (25% EtOAc/hexanes); IR (film) 3245, 2934, 2856, 1606, 1545 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.86 (d, J = 2.4 Hz, 2H), 6.68 (d, J = 2.4 Hz, 2H), 6.57 (s, 2H), 6.09 (s, 2H), 4.01 (s, 6H), 3.98–3.90 (m, 2H), 3.88 (m, 6H), 2.42–2.28 (m, 2H), 1.86–1.70 (m, 2H), 1.52–1.35 (m, 4H); 13C NMR (100 MHz, CDCl3) ppm 155.0, 154.7, 154.2, 141.2, 136.0, 122.1, 112.6, 102.2, 94.7, 56.6, 56.2, 55.4, 32.5, 24.7; HRMS (ESI): Exact mass calcd for C28H31Cl2N4O4 [M+H]+ 557.1722, found 557.1738.
Biological activity test
A stock solution of 30 mM (−)-Nutlin-3 was prepared in DMSO. Hs294T cells were obtained from ATCC and maintained in DMEM/F12 media supplemented with 10% Fetal Bovine Serum, 100U/mL penicillin and 100ug/mL streptomycin. For viability, test cells were incubated with 10 μM (−)-Nutlin-3 for 5 days, trypsinized and mixed 9:1 with 0.4% buffered trypan blue. Viable trypan blue negative cells were counted in a haemocytometer. For the analysis of p53 protein levels Hs294T cells were incubated with 10 μM (−)-Nutlin-3 or an equivalent volume of vehicle DMSO for 3 days. Cells lysates were analyzed by Western Blotting with p53-specific (DO-1, Calbiochem) and β-actin-specific (Cell Signaling) antibodies.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) GM084333 (JNJ) and CA116021-S1 (AR). We are grateful to Brandon Vara for assistance with purification of the final compound.
Footnotes
The authors declare no competing financial interest.
Includes 1H and 13C NMR spectra of all new compounds not previously reported, as well as HPLC traces for 16. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Vassilev LT. Trends in Molecular Medicine. 2007;13:23. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]; (b) Shangary S, Wang S. Annu Rev Pharmacol Toxicol. 2009;49:223. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vassilev Lyubomir T. J Med Chem. 2005;48:4491. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]; (d) Zhong H, Carlson Heather A. Proteins. 2005;58:222. doi: 10.1002/prot.20275. [DOI] [PubMed] [Google Scholar]; (e) Tanaka A, Tsuganesawa K, Utada R. 2008 [Google Scholar]; (f) McCubrey JA, Abrams SL, Ligresti G, Misaghian N, Wong EWT, Steelman LS, Baesecke J, Troppmair J, Libra M, Nicoletti F, Molton S, McMahon M, Evangelisti C, Martelli AM. Leukemia. 2008;22:2080. doi: 10.1038/leu.2008.207. [DOI] [PubMed] [Google Scholar]; (g) Corallini F, Celeghini CJ. Leukocyte Biol. 2008;84:651. doi: 10.1189/jlb.0408222. [DOI] [PubMed] [Google Scholar]; (h) Nazabal AT, Wenzel RJ. 2008-EP53085. 2008
- 2.Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, Cicero SA, Schulman BA, Jochemsen AG, Guy RK, Dyer MA. J Biol Chem. 285:10786. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis TA, Johnston JN. Chem Sci. 2011;2:1076. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kong N, Liu EA, Vu BT. 2003051360. 2003; (b) Bartkovitz DJ, Cai J, Chu X-J, Li H, Lovey AJ, Vu BT, Zhao C. 2008-EP63053. 2009
- 5.(a) Best OG, Gardiner AC, Majid A, Walewska R, Austen B, Skowronska A, Ibbotson R, Stankovic T, Dyer MJS, Oscier DG. Leukemia. 2008;22:1456. doi: 10.1038/sj.leu.2405092. [DOI] [PubMed] [Google Scholar]; (b) De Winter HLJ, Langenaeker WGR. 2007-EP7681. 2008; (c) Dyer MA, Marine J-C, Jochemsen AG. 2007-US74149. 2008; (d) Chen J, Hu B. 2008-US55116. 2008
- 6.(a) Seyfried I, Hofbauer S, Stoecher M, Greil R, Tinhofer I. Blood. 2008;112:2168. doi: 10.1182/blood-2008-05-158634. [DOI] [PubMed] [Google Scholar]; (b) Kranz D, Dohmesen C, Dobbelstein M. J Cell Biol. 2008;182:197. doi: 10.1083/jcb.200712014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, Liu J-J, Zhao C, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, Graves B. ACS Medicinal Chemistry Letters. 4:466. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Estimate based on 2013 price listed by CaymanChemical (25mg/US$750. 2013 Mar 21; www.caymanchem.com.
- 9.(a) Glover-Cutter K, Kim S, Espinosa J, Bentley DL. Nat Struct Mol Biol. 2008;15:71. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Glover-Cutter K, Kim S, Espinosa J, Bentley DL. Nat Struct Mol Biol. 2008;15:71. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Jonca M, Lambros T, Ferguson S, Goodnow R. J Pharm Biomed Anal. 2007;45:720. doi: 10.1016/j.jpba.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 11.De CK, Seidel D. J Am Chem Soc. 2011;133:14538. doi: 10.1021/ja2060462.Application to trans-stilbene diamines: Min C, Mittal N, De CK, Seidel D. Chem Commun (Cambridge, U K) 2012;48:10853. doi: 10.1039/c2cc36361e.
- 12.For recent examples of the application of bis(amidine) catalysis to the synthesis of potential therapeutics, see: Davis TA, Danneman MW, Johnston JN. Chem Commun (Cambridge, U K) 2012;48:5578. doi: 10.1039/c2cc32225k.Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Org Lett. 2012;14:6322. doi: 10.1021/ol303092v.Villalta F, Dobish MC, Nde PN, Kleshchenko YY, Hargrove TY, Johnson CA, Waterman MR, Johnston JN, Lepesheva GI. J Infect Dis. 2013;208:504. doi: 10.1093/infdis/jit042.
- 13.Wallace Valerie A. Nature. 2006;444:45. doi: 10.1038/444045a. [DOI] [PubMed] [Google Scholar]
- 14.Fotouhi N, Haley GJ, Simonsen KB, Vu BT, Webber SE. 2005-EP5046. 2005
- 15.Kornblum N, Larson HO, Blackwood RK, Mooberry DD, Oliveto EP, Graham GE. J Am Chem Soc. 1956;78:1497. [Google Scholar]
- 16.Feuer H, Lawrence JP. J Org Chem. 1972;37:3662. [Google Scholar]
- 17.Feuer H, Friedman H. J Org Chem. 1975;40:187. [Google Scholar]
- 18.(a) Black AP, Babers FH. Org Synth. 1939;19:73. [Google Scholar]; (b) Hauser FM, Baghdanov VM. J Org Chem. 1988;53:2872. [Google Scholar]
- 19.Kurz ME, Ngoviwatchai P, Tantrarant T. J Org Chem. 1981;46:4668. [Google Scholar]
- 20.Vogl EM, Buchwald SL. J Org Chem. 2002;67:106. doi: 10.1021/jo010953v. [DOI] [PubMed] [Google Scholar]
- 21.Walvoord RR, Berritt S, Kozlowski MC. Org Lett. 2012;14:4086. doi: 10.1021/ol301713j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gelbard G. Synthesis. 1977:113. [Google Scholar]
- 23.(a) Yin B, Zhang Y, Xu L-W. Synthesis. 2010:3583. [Google Scholar]; (b) Petrini M. Chem Rev (Washington, DC, U S) 2005;105:3949. doi: 10.1021/cr050528s. [DOI] [PubMed] [Google Scholar]
- 24.Yu Q. 2007-SG58. 2007
- 25.Rampalakos C, Wulff WD. Adv Synth Catal. 2008;350:1785. doi: 10.1002/adsc.200800214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang L, Wulff WD. J Am Chem Soc. 2011;133:8892. doi: 10.1021/ja203754p. [DOI] [PubMed] [Google Scholar]
- 27.Davis TA, Wilt JC, Johnston JN. J Am Chem Soc. 2010;132:2880. doi: 10.1021/ja908814h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hattori T, Shimazumi Y, Goto H, Yamabe O, Morohashi N, Kawai W, Miyano S. J Org Chem. 2003;68:2099. doi: 10.1021/jo026747k. [DOI] [PubMed] [Google Scholar]
- 29.Davis TA, Dobish MC, Schwieter KE, Chun AC, Johnston JN. Org Synth. 2012;89:380. doi: 10.15227/orgsyn.089.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027. doi: 10.1038/nature09125.Dobish MC, Johnston JN. J Am Chem Soc. 2012;134:6068. doi: 10.1021/ja301858r.Shen B, Johnston JN. Org Lett. 2008;10:4397. doi: 10.1021/ol801797h.Wilt JC, Pink M, Johnston JN. Chem Commun (Cambridge, U K) 2008:4177. doi: 10.1039/b808393b.Singh A, Johnston JN. J Am Chem Soc. 2008;130:5866. doi: 10.1021/ja8011808.Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418. doi: 10.1021/ja031906i.(g) ref. 12a. (h) ref. 19a.
- 31.Exceptions: Dobish MC, Johnston JN. Org Lett. 2010;12:5744. doi: 10.1021/ol1025712.(b) ref. 12b.
- 32.For the experimental determination of pKa for organocatalysts, see: Hess AS, Yoder RA, Johnston JN. Synlett. 2006:147.Christ P, Lindsay AG, Vormittag SS, Neudörfl JM, Berkessel A, O’Donoghue AC. Chem Eur J. 2011;17:8524. doi: 10.1002/chem.201101157.
- 33.For a discussion of hydrogen bonding vs. ion pairing with organocatalysts, see: Fleischmann M, Drettwan D, Sugiono E, Rueping M, Gschwind RM. Angew Chem Int Ed. 2011;50:6364. doi: 10.1002/anie.201101385.
- 34.(a) Akiyama T. Chem Rev. 2007;107:5744. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]; (b) Terada M. Synthesis-Stuttgart. 2010:1929. [Google Scholar]
- 35.Generally, additions of simple aliphatic nitroalkanes (such as nitroethane) were complete in one day at −20 °C. Under similar conditions, additions of aryl nitroalkanes were complete in one day at −78 °C. See references 3 and 27.
- 36.It is presumed that purity differences in the batches of imine used could have caused the lower enantioselectivity observed in this series of reactions relative to those listed in Table 1.
- 37.2-Oxopiperazine was synthesized according to the following patent procedure, using chloroethyl acetate and ethylene diamine in the presence of sodium ethoxide. Purification by column chromatography provided 10.1g (63%) of material that was recrystallized from acetone to give 3.8 g (23%) of pure 2-oxopiperazine; Elmaleh, D. R., Choi, S.-W. Fishman, A. J. 2005-US0222166–2005.
- 38.You SL, Kelly JW. Org Lett. 2004;6:1681. doi: 10.1021/ol049439c. [DOI] [PubMed] [Google Scholar]
- 39.Stuhmer T, Bargou Ralf C. Cell Cycle. 2006;5:39. doi: 10.4161/cc.5.1.2281. [DOI] [PubMed] [Google Scholar]
- 40.Fairfull-Smith KE, Jenkins ID, Loughlin WA. Org Biomol Chem. 2004;2:1979. doi: 10.1039/b406770c. [DOI] [PubMed] [Google Scholar]
- 41.Petersson MJ, Jenkins ID, Loughlin WA. J Org Chem. 2008;73:4691. doi: 10.1021/jo800447v. [DOI] [PubMed] [Google Scholar]
- 42.(a) Hendrickson JB, Schwartzman SM. Tetrahedron Lett. 1975;16:277. [Google Scholar]; (b) Hendrickson JB, Hussoin MS. J Org Chem. 1989;54:1144. [Google Scholar]; (c) Hendrickson JB, Hussoin MS. J Org Chem. 1987;52:4137. [Google Scholar]
- 43.$83/gram, Sigmaaldrich.com, Dec 2012. Attempts to precipitate or recrystallize 1 from toluene were not sufficiently successful in separating the product from Ph3PO.
- 44.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 45.A modification of the procedure of Kornblum: Kornblum N, Larson HO, Blackwood RK, Mooberry DD, Oliveto EP, Graham GE. J Am Chem Soc. 1956;78:1497.
- 46.Adapted from: Hattori T, Shimazumi Y, Goto H, Yamabe O, Morohashi N, Kawai W, Miyano S. J Org Chem. 2003;68:2099. doi: 10.1021/jo026747k.
- 47.Adapted from Wagaw S, Rennels R, Buchwald S. J Am Chem Soc. 1997;119:8451–8458.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.