

Abstract
Significance: Innate and adaptive immunity play fundamental roles in the development of hypertension and its complications. As effectors of the cell-mediated immune response, myeloid cells and T lymphocytes protect the host organism from infection by attacking foreign intruders with bursts of reactive oxygen species (ROS). Recent Advances: While these ROS may help to preserve the vascular tone and thereby protect against circulatory collapse in the face of overwhelming infection, aberrant elaboration of ROS triggered by immune cells in the absence of a hemodynamic insult can lead to pathologic increases in blood pressure. Conversely, misdirected oxidative stress in cardiovascular control organs, including the vasculature, the kidney, and the nervous system potentiates inflammatory responses, augmenting blood pressure elevation and inciting target organ damage. Critical Issues: Inflammation and oxidative stress thereby act as cooperative and synergistic partners in the pathogenesis of hypertension. Future Directions: Pharmacologic interventions for hypertensive patients will need to exploit this robust bidirectional relationship between ROS generation and immune activation in cardiovascular control organs to maximize therapeutic benefit, while limiting off-target side effects. Antioxid. Redox Signal. 20, 102–120.
Introduction
Agents of innate and adaptive immunity interact with reactive oxygen species (ROS) in a complex, bidirectional relationship to regulate the hypertensive response and the ensuing end-organ injury. Studies conducted more than 30 years ago hinted at a role for adaptive immunity in hypertension. However, the contributions of inflammatory cells and mediators to the pathogenesis of cardiovascular disease received more attention following the recognition that oxidized low-density lipoprotein (LDL) could act as a specific antigen to stimulate an adaptive immune response that accelerates atherogenesis (15, 165). Population studies in humans then demonstrated links between elevated markers of inflammation and the risk of developing hypertension (156), unleashing a wave of newer studies that have dissected the links between inflammation, hypertension, and target organ damage with increasing precision.
Inflammatory cells are potent sources of ROS. Recently, researchers have started to explore the pathways through which, cells participating in the innate and adaptive immune responses potentiate blood pressure elevation and end-organ injury by driving the elaboration of ROS in cardiovascular control organs, including the vasculature, the kidney, and the nervous system. Other studies have characterized a converse relationship wherein ROS generated within these cardiovascular control organs can promote activation of circulating immune cells, resulting in a dangerous positive feedback system (Fig. 1) that exaggerates hypertensive responses following an initial injury signal. In the case of hypertension, uncovering the relationships between inflammation, ROS, and blood pressure elevation has posed a challenge due to the critical role of the kidney in regulating sodium excretion and, therefore, intravascular volume. Thus, inflammatory responses or oxidative stress localized to the kidney can conceivably be the cause or the result of perturbations in blood pressure. The following review will survey some of the data implicating immune responses in the pathogenesis of hypertension and explore how inflammatory mediators and oxidative stress coordinately potentiate this epidemic disease.
FIG. 1.

Oxidative stress and inflammatory responses act synergistically in the pathogenesis of hypertension. Innate and adaptive immune responses potentiate blood pressure elevation and the ensuing end-organ injury by driving reactive oxygen species (ROS) generation in cardiovascular control organs, including the vasculature, the kidney, and the nervous system. Conversely, superoxide generated within these cardiovascular control organs can promote activation of circulating immune cells that exaggerates hypertensive responses following an initial injury signal.
Inflammatory Responses Mediate Hypertension
Epidemiologic studies
A series of epidemiologic studies have observed associations between levels of inflammatory mediators and hypertension in humans (133). Patients with modest elevations in blood pressure or prehypertension have elevated circulating levels of tumor necrosis factor-alpha (TNF-α), C reactive protein (CRP), and leukocytes compared to normotensive controls (24). Circulating levels of CRP and the inflammatory cytokine interleukin-6 (IL-6) have both correlated positively with blood pressure in cross-sectional studies (20, 195). In some populations, higher CRP levels or leukocyte counts have even predicted the onset of hypertension several years into the future (129, 156, 174). Hypertensive patients exhibit elevated levels of adhesion molecules that facilitate the exit of these mononuclear cells from blood vessels into target tissues through a rolling process known as diapedesis (20, 167). Chemokines, the mediators that recruit mononuclear cells via concentration gradients into target organs, are also upregulated in human hypertension (167). Thus, the cells that execute immune responses as well as the mediators that can organize their entry into cardiovascular control organs are present in excess in patients with hypertension, but these association studies cannot discriminate whether blood pressure elevation is caused by these mediators or whether hypertension conversely induces adaptive immune responses through hemodynamic injury.
Early animal studies pointing to immunity's role in hypertension
Before the era of transgenic models, early experiments hinted that immune responses may contribute to blood pressure elevation and its attendant complications. Although these studies did not emphasize the roles of individual immune cell populations in mediating hypertension, the experimental designs suggested that activated T lymphocytes were critical to blood pressure elevation. For example, adoptive transfer of lymph node cells from a rat made hypertensive by renal infarction recapitulated the hypertensive response in the recipient (130). Conversely, mice lacking a thymus, the organ in which T cells mature through selective processes, were protected from blood pressure elevation in a model of spontaneous hypertension (172), and athymic mice were similarly unable to sustain chronic blood pressure elevation in a mineralocorticoid-induced hypertension model (171). Moreover, proliferative responses of lymphocytes correlated with blood pressure in genetically hypertensive rats, and thymectomy in these animals reduced blood pressure (7). These studies were prescient in postulating that perivascular mononuclear cell clusters may impact vascular function, but predated the recognition that T cells and other immune cell populations could influence the course of cardiovascular disease via the generation of ROS.
Adaptive immunity in atherogenesis
Heightened interest in the contribution of inflammatory responses to cardiovascular disease emerged with the recognition that macrophages carrying pathogenic lipid are present in atherosclerotic plaques. While macrophages represent a key component of innate immunity, Hansson and colleagues further demonstrated that oxidized LDL could act as a neo-antigen inducing a specific adaptive immune response that required functional T cells for full disease progression (15, 165). As in atherosclerosis, the vasculature involved in mounting increased systemic vascular resistance during chronic hypertension undergoes remodeling, and mononuclear cell infiltrates surround large vessels in target organs damaged by blood pressure elevation, particularly in severe hypertension (58, 113). Thus, the actions of innate and adaptive immune responses in the setting of hypertension began to receive more intense scrutiny as had occurred in the study of atherogenesis.
Recent evidence implicating immune responses in the pathogenesis of hypertension
Against this historical backdrop, a wealth of experimental evidence has emerged over the past 10 years demonstrating a critical role for immunity in the pathogenesis of hypertension. First, broad pharmacologic blockade of proinflammatory signaling pathways has the capacity to limit end-organ damage in hypertension and even mitigate blood pressure elevation in some models. For example, the nuclear factor-κB (NF-κB) signaling pathway propagates gene transcription for a host of key inflammatory mediators, and inhibition of this pathway reduces blood pressure, cardiac hypertrophy, and renal disease in high-renin hypertension (124). Accordingly, suppression of the immune system through a variety of approaches limits NF-κB translocation to the nucleus in several cell lineages and thereby limits end-organ damage in diverse models of hypertension (62, 125). These studies raised questions as to which immune cell lineages and which downstream mediators were responsible for translating the inflammatory stimulus into blood pressure elevation and/or end-organ injury.
The definitive approach for exploring the functions of immune cell lineages in cardiovascular disease as in traditional immune-mediated diseases has been through adoptive transfer of these cells into immune-deficient recipients. This strategy has established that adaptive immunity plays a critical role in experimental hypertension and raises the specter of a putative neo-antigen that potentiates hypertension through a precise autoimmune mechanism just as in atherosclerosis. Mice lacking functional lymphocytes have a muted blood pressure response to hypertensive stimuli, which is restored by transfer of T, but not B lymphocytes (58). CD8 rather than CD4+ T cells appear to be the prohypertensive T cell subpopulation (175). These T cells may promote hypertension by potentiating vascular dysfunction (58) and/or sodium retention in the kidney (30), both of which involve local generation of oxidative stress as discussed below. By contrast, T regulatory cells, an immunosuppressive T cell lineage identified most specifically by Foxp3 expression, can protect from blood pressure elevation and target organ damage induced by angiotensin II (6, 85). Moreover, although angiotensin II can directly enhance lymphocyte proliferation in certain contexts (65, 127), recent studies have uncovered potentially immunosuppressive effects of type 1 angiotensin (AT1) receptors on mononuclear cells in the setting of hypertension (31, 200). Thus, whether hypertensive stimuli such as angiotensin II activate the immune system through direct stimulation of AT1 receptors on inflammatory cells remain controversial. Alternative mechanisms through which hypertensive stimuli may trigger immune activation include signals from the central nervous system (CNS) and/or the endothelium (62, 107).
The involvement of an adaptive immune response in propagating hypertension and its associated complications would suggest that innate immune defenses, including professional antigen-presenting cells (APCs) have detected and processed neo-antigens that promote the targeted clonal expansion of lymphocytes. Although, Rodriguez-Iturbe and colleagues have put forth heat shock protein-70 (HSP70) as one possible antigen (132), definitive studies cataloguing the antigens triggering immune-mediated hypertension are still forthcoming. Nevertheless, myeloid APCs may regulate blood pressure responses through alternative mechanisms unrelated to their classical APC functions. Mice depleted of monocytes, which are circulating myeloid precursors to dendritic cells and macrophages, have a blunted chronic hypertensive response to angiotensin II, and adoptive transfer of monocytes restores the blood pressure elevation in these animals (191). In turn, mice that are unable to develop splenic dendritic cells, by far the most potent APC lineage, also have blunted hypertensive responses to angiotensin II, and yet transfer of wild-type bone marrow-derived cells into these animals does not restore the hypertensive response (54). Macrophages, another critical APC population, also have the capacity to influence salt-sensitive hypertension by elaborating the vascular endothelial growth factor-C to drive lymphangiogenesis (101). These new lymphatics, in turn, regulate the nonosmotic storage of sodium. Thus, cells of the innate immune system may influence blood pressure independently of their responsibilities as APCs, and below we will discuss the possible role of ROS in this regard.
To facilitate the recruitment of cells of the innate and adaptive immune systems into cardiovascular control organs where they can influence the hypertensive response, chemokines establish gradients attracting these cells from the circulation. Adhesion models in the vasculature supplying these organs then bind to the inflammatory cells, leading to their transmigration into the tissue parenchyma. An analogous process permits entry of inflammatory cells into target organs where they can potentiate tissue damage instigated by blood pressure elevation. Thus, the accumulation of T lymphocytes in the vasculature and the kidneys of hypertensive animals (58, 125) is accompanied by local upregulation of the prototypical T cell chemokine CCL5 (58, 200). Interestingly, genetic deletion of CCL5 led to an enhanced hypertensive response at a single time point (188), but the role of CCL5 in maintaining chronic blood pressure elevation is not well established. CCL2/MCP-1 is a protypical monocyte/macrophage chemokine, and is upregulated in the kidneys during progressive damage of diverse etiologies (114). Accordingly, blockade or deletion of the receptor for CCL2 limits monocyte infiltration into the kidney and vasculature in hypertension and ameliorates progressive renal and vascular damage through a blood pressure-independent mechanism (45, 70, 93). Hypertension similarly upregulates adhesion molecules, including the intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in the renal and systemic vasculature, but a role for these molecules in regulating blood pressure is not clear (62, 113, 125). Thus, despite their paramount roles in recruiting T lymphocytes and macrophages into cardiovascular control organs, chemokines and adhesion molecules appear to regulate tissue injury responses during hypertension rather than the degree of blood pressure elevation.
Once immune cells are recruited into cardiovascular control organs and/or organs targeted for damage by hypertension, these cells can engage in direct cytotoxic activities that we will discuss below or elaborate cytokines to shape the local inflammatory response. Among these cytokines, TNF-α has been the most widely studied for its role in hypertension. Pharmacologic blockade of TNF slows the progression of target organ damage and even limits blood pressure elevation in some hypertension models (44, 46, 125, 183). Similarly, TNF-deficient mice have a muted chronic hypertensive response to angiotensin II (58, 164). However, the 2 receptors for TNF, TNFR1, and TNFR2, may have opposing effects on blood pressure regulation (22), such that, the mechanisms through which local TNF influences blood pressure are challenging to reconcile with the global TNF knockout studies (8, 157, 158).
The data implicating other inflammatory cytokines in the pathogenesis of hypertension are less robust. Interferon-γ (IFN-γ), produced by activated T lymphocytes, is upregulated in the kidneys of hypertensive animals (29), but in our hands, genetic knockout studies did not confirm a role for IFN in mediating blood pressure elevation (unpublished observations, SDC). IL-1 produced by proinflammatory macrophages stimulates vasoconstriction in some vascular beds (25) and triggers generation of endothelin, another hormone that regulates both vascular function and renal sodium handling (13). Nevertheless, definitive in vivo data confirming a direct role for endogenous IL-1 in potentiating hypertension have not emerged. By contrast, activation of the IL-1 receptor leads to generation of IL-6, and deficiency of IL-6 does indeed prevent a full chronic hypertensive response to angiotensin II (87). Thus, IL-6 appears to contribute to blood pressure elevation possibly by facilitating sodium reabsorption in the renal collecting duct (91). Altogether, cells of the innate and adaptive immune systems, the chemokines that recruit them to cardiovascular control organs, and the inflammatory cytokines these immune cells produce can have profound effects on the degree of blood pressure elevation and/or the severity of target organ damage in the setting of hypertension. As discussed below, these components of the immune system can also regulate levels of oxidative stress, which may, in turn, impact blood pressure and the progression of end-organ disease.
Inflammatory Cells Generate Oxidative Stress
The notion that inflammatory cells would potentiate disease by generating excessive oxidative stress is intuitively appealing because several immune cell lineages have evolved precisely to protect the body from foreign invaders by attacking them with bursts of ROS. Within the innate immune system, neutrophils and macrophages are prototypical cell lineages, which control infection by generating local oxidative stress. Phagocytosis of microorganisms by a neutrophil leads to the phosphorylation of the cytosolic components of NADPH oxidase, p47phox, and p67phox. These components then combine with other NADPH oxidase components, including the cytochrome complex gp91phox and p22phox to form the active oxidase complex that donates an electron to oxygen to form superoxide. This superoxide can be further converted to hypochlorous acid in anticipation of the classic respiratory burst through which neutrophils destroy invading microbes by activating proteases such as cathepsin G (147, 155). The normal function of NADPH oxidase in the neutrophil and its capacity to generate ROS is thus paramount to its efficacy in eliminating pathogens. Accordingly, mutations in components of the NADPH oxidase in humans lead to chronic granulomatous disease, a syndrome in which children are overwhelmed by infections due to bacterial and fungal organisms, especially the catalase-producing bacterium Staphylcoccus aureus and Aspergillus fungal species (76, 105). In addition to their role in presenting processed antigens to adaptive immune cells, macrophages act cooperatively with neutrophils to similarly phagocytose and destroy pathogens, especially intracellular organisms, through the generation of ROS (32, 161). Accordingly, defects in NADPH oxidase function selectively within the macrophage raise the susceptibility to severe mycobacterial infections (14). Given that cells of the innate immune system protect the host from infection by elaborating ROS, inappropriate or nonspecific activation of these innate defenses in response to a hypertensive stimulus could potentiate blood pressure elevation and/or worsen target organ damage via misdirected oxidative stress.
One added complexity coloring the potential contribution of innate immune responses to oxidative stress in hypertension is that proinflammatory, or classically activated, M1 macrophages produce inducible nitric oxide synthase (iNOS) (123). In turn, iNOS increases the local availability of NO and thereby alters the level of oxidative stress at the same time as the M1 macrophage is directly mediating tissue damage or vascular dysfunction. Depending on factors at the site of production, NO can permit vasodilation when generated via endothelial nitric oxide synthase (eNOS) (9), facilitate sodium excretion when produced by kidney epithelial cells (166), or aggravate oxidative damage when the NOS isoforms, uncoupled from the cofactor tetrahydrobiopterin (BH4), increase generation of reactive nitrogen intermediates (51). Inversely, BH4 limits the production of superoxide by eNOS, thereby blunting the formation of peroxynitrite (ONOO−) from NO and superoxide (192). Whether iNOS undergoes coupling is not clear, but, overall, iNOS in macrophages appears to promote tissue damage as blockade of NO generation in autoimmune models is protective (68). Given this level of sophistication, it is not surprising that macrophages appear to have variable effects on the pathogenesis of hypertension and its complications, depending on the context and experimental approach (31, 84, 93). Indeed, pioneering researchers who confronted the field of oxidative stress predicted that nitric oxide might serve dual purposes within the inflammatory response given its cytotoxic, but also vasodilatory effects (121).
Within the adaptive immune response, T lymphocytes in particular have the capacity to enhance levels of oxidative stress via their natural function of eliminating host cells that have become infected by invading organisms (Fig. 2). Like neutrophils and macrophages, activated T cells express NADPH oxidase to generate ROS within their milieu. Conversely, these ROS impact the differentiation and function of the T cell (71). In addition, once activated, the CD8+ cytotoxic T cell binds to a target cell, inserts a pore called perforin into the target cell's membrane through which it exocytoses proteases called granzymes that kill the target cell (61). One such protease, granzyme A, damages the target cell's mitochondria by generating ROS within the cell's cytoplasm (104). Accordingly, this cytotoxic effect can be blocked by a superoxide scavenger. Although the perforin mechanism constitutes the dominant form of CD8+ T cell-mediated cytotoxicity, the Fas ligand is upregulated in the activated CD8+ T cell by superoxide and mediates apoptosis in the target cell by binding to Fas in the target cell membrane and thereby triggering activation of caspases 3 and 8 (40, 168). TNF-related apoptosis-inducing ligand (TRAIL) from the cytotoxic T cell similarly induces apoptosis in the target cell acting via caspases 8 and 10 (2) and figures prominently in the pathogenesis of vascular inflammation (152). Nevertheless, the CD8+ T cell typically exhibits exquisite control over the extent of locally generated oxidative stress as the cytotoxic T cell is capable of lysing several target cells without injuring itself in the process.
FIG. 2.
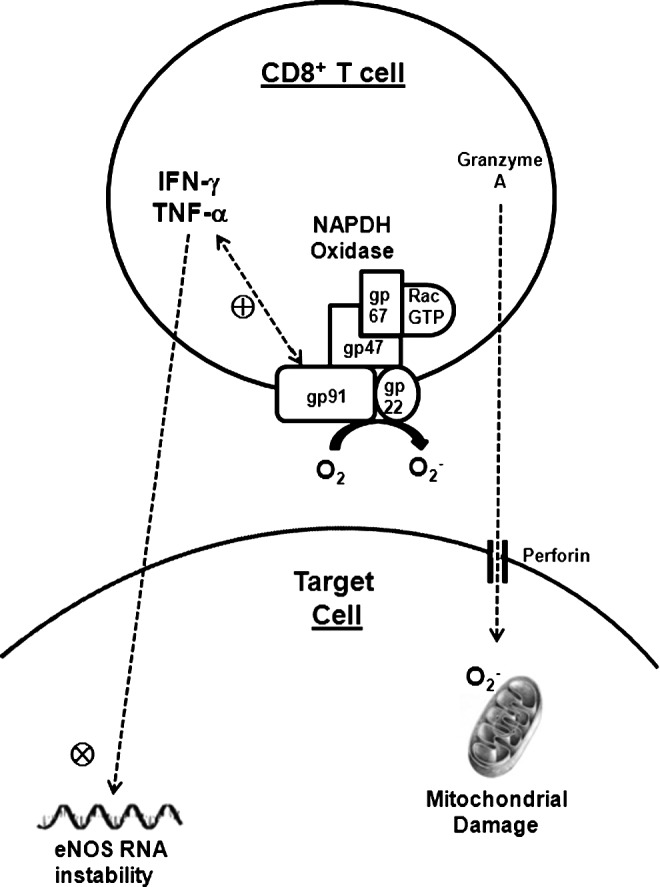
T lymphocytes mediate cytotoxic injury through the generation of ROS. CD4+ and CD8+ T cells express NADPH oxidase to generate superoxide anions in their milieu. The NADPH oxidase activity also stimulates production of the tumor necrosis factor-α (TNF-α), which can mediate proinflammatory signals in neighboring cells. CD8+ cytotoxic T cells insert perforin pores in target cell membranes. Granzyme A and other proteases from the attacking CD8+ T cells enter the target cell via the perforin pore and induce oxidative damage in the target cell mitochondria. Mitochondrial image by Gretchen Deahl of Allegheny, Health, Education & Research Foundation, 2008. Reprinted with permission.
Both CD8+ and CD4+ T lymphocytes also guide the adaptive immune response by secreting a cohort of cytokines that trigger increased levels of oxidative stress (Fig. 2). For example, TNF-α and IFN-γ, both produced by T cells and macrophages, impair the stability of mRNA for eNOS (196) and cooperatively promote oxidative stress by enhancing expression of the NADPH oxidase subunit gp91phox (116). Conversely, activation of NADPH oxidase within T cells also stimulates production of TNF-α (65). Thus, the adaptive immune system provides protection for the host organism by elaborating ROS to destroy cells infected by foreign pathogens. Accordingly, if a neo-antigen uncovered in the setting of hypertension misdirects this adaptive immune response against renal, vascular, and/or neuronal tissues, the result will be potent elevations of local oxidative stress within these cardiovascular control organs, inciting further blood pressure elevation and end-organ damage. Below, we will review some of the evidence indicating that immune responses potentiate oxidative stress, and conversely, that ROS exaggerate inflammatory responses in the setting of hypertension.
Ironically, while the elaboration of ROS by immune cells clearly serves a useful cytotoxic function to rid the host of foreign invaders, the actions of inflammatory responses to trigger ROS generation may also have evolved as a protective mechanism to raise blood pressure and thereby prevent circulatory collapse during severe infection. Septic shock is the prototypical clinical catastrophe in which the organism is overwhelmed by systemic bacterial infection. During septic shock, lipopolysaccharide, also known as endotoxin, from the bacterial cell wall induces synthesis of nitric oxide, leading to intractable hypotension (120). Accordingly, NOS inhibitors can reverse this sepsis-induced hypotension (134). Thus, endogenous effects of immune cells to potentiate oxidative stress should similarly raise blood pressure in this clinical context and thereby guard against circulatory embarrassment. Nevertheless, when inflammation triggers ROS generation in the absence of such a hemodynamic threat, the result may be a pathologic elevation in blood pressure.
Inflammatory Cells Drive ROS Generation in Cardiovascular Control Organs
The vasculature
CRP in vascular inflammation
In hypertension, inflammatory responses generate oxidative stress in the vascular wall leading to endothelial dysfunction and remodeling. These pathologies may influence the degree of blood pressure elevation, but also constitute target organ damage resulting from hypertension. For example, as the classical marker of inflammation, CRP reduces nitric oxide generation in endothelial cells. The mechanism underlying this effect is inhibition of eNOS mRNA stability, protein expression, and activity in vascular endothelial cells, leading in turn to decreased angiogenesis (184, 187). These effects of CRP on eNOS result in vasoconstriction and induction of the potent vasoactive peptide endothelin 1, adhesion molecules ICAM-1 and VCAM-1, and proinflammatory cytokine IL-6 (186). Moreover, in a vascular injury model, CRP increased ROS generation and expression of the type 1 (AT1) receptor for angiotensin II, another potent vasoconstrictor peptide (189). In human patients with hypertension and metabolic syndrome, CRP levels correlate with the level of oxidative stress in inflammatory cells (198). Thus, in addition to being a critical marker of inflammation, CRP might also participate in the pathogenesis of vascular oxidative.
Mononuclear cells generate ROS in the vascular wall
Once recruited to the vascular wall, myeloid cells and T lymphocytes can influence vascular damage and function by directly generating ROS or, perhaps, more importantly, by altering the local cytokine milieu. As detailed above, myeloid cells and T cells express activated NADPH oxidase (71) through which they can exert direct oxidative injury. The hypertensive hormone angiotensin II further stimulates NADPH expression in monocytes (59). Accordingly, the protection from experimental hypertension afforded by selective depletion of circulating monocytes results in attenuated generation of superoxide in the vascular wall (191). In the setting of angiotensin II-induced hypertension, T cells also express higher levels of the NADPH oxidase subunits p47phox, p22phox, and Nox2 (58). Conversely, adoptive transfer of T cells lacking NADPH oxidase leads to blunted superoxide generation in the aorta and a muted hypertensive response during chronic angiotensin II infusion (58). Indeed, the generation of maximal oxidative stress in the vascular wall during hypertension requires vascular infiltration of both macrophages and T lymphocytes (58, 81). In addition, ROS produced by macrophages infiltrating the vessel wall permeate the extracellular matrix and activate matrix metalloproteinases (MMPs) to direct vascular remodeling (141), such that, the effects of macrophages on vascular responses versus disease may depend on the layer of the vascular wall in which these inflammatory cells localize (173). In sum, myeloid cells and T lymphocytes invade the vascular wall, accentuate local oxidative stress, and potentiate the blood pressure response to hypertensive stimuli.
Although mononuclear cells produce superoxide, their secretion of inflammatory cytokines likely has an even more profound impact to augment vascular oxidative stress. The role of cytokines in regulating vascular ROS has therefore received considerable attention. In vitro, TNF-α stimulates hypertrophy of cardiac myocytes that is blocked by antioxidants (126). In vascular endothelial cells, TNF destabilizes mRNA for eNOS leading to decreased eNOS expression (196) and reducing NO bioavailability (80). TNF also potentiates assembly of NADPH oxidase in these cells (197). In vivo, the protection from hypertension afforded by TNF-α blockade is associated with reduced generation of superoxide in the vascular wall (58) and reduced vascular stiffness (3), suggesting that TNF drives endothelial dysfunction and, in turn, blood pressure elevation through its effects on vascular oxidative stress. Accordingly, TNF blockade restores endothelial-dependent vasodilation in humans (102). TNF also induces expression of ICAM-1 in endothelial cells through a ROS-dependent mechanism (4). TNF activates NADPH oxidase directly in vascular smooth muscle cells (VSMCs) (35). TNF and IL-1 drive superoxide generation from human fibroblasts (112) and vascular endothelial cells, leading in the latter to upregulation of VCAM-1 (106). Although not considered inflammatory cells per se, platelets contain stores of IL-1 poised to act on the vascular endothelium to which the platelets adhere following vascular injury (95), yielding another potential source of IL-1-induced oxidative stress. The transforming growth factor-β1 (TGF-β1) stimulates ROS generation in VSMCs and release of hydrogen peroxide from human fibroblasts (159, 176). Thus, cytokines produced by infiltrating mononuclear cells or cells intrinsic to several layers of the vessel wall augment local levels of oxidative stress, in turn promoting recruitment of additional inflammatory cells via expression of adhesion molecules in the vascular endothelium.
Vascular inflammation induced by aldosterone promotes oxidative stress
Aldosterone has nongenomic effects to promote vascular inflammation that could explain some of the benefits of mineralocorticoid antagonists (MRAs) in slowing the progression of target organ damage in human clinical trials (100, 135, 143). While MRAs may provide some protection due to lowering of blood pressure, recent studies suggest that aldosterone provokes vascular dysfunction by accentuating oxidative stress in the vasculature (89, 100). Aldosterone enhances expression of NADPH oxidase subunit p22phox in mononuclear cells, and T cells mediate aldosterone-induced superoxide generation in the vessel wall (17, 58). Among the cytokines secreted by T cells is IL-6, which raises blood pressure (87) and may impart some of aldosterone's proinflammatory effects on the vasculature (100). By contrast, adoptive transfer of T regulatory cells, which suppresses inflammation, limits superoxide generation and mononuclear cell accumulation in the vasculature and kidney during aldosterone infusion, such that, the effects of aldosterone to modulate oxidative stress in the vasculature through T cell activation depend on the subpopulation of T cells (78).
Aldosterone also regulates ROS generation via non-T cell-dependent mechanisms. Aldosterone upregulates c-Src in VSMCs leading to generation of oxidative stress and induction of inflammation via the mitogen-activated protein (MAP) kinase pathway (16). In addition, mineralocorticoids can inhibit the glucose-6-phosphate dehydrogenase (G6PD) activity (28, 94). As G6PD is a primary source of NADPH, G6PD inhibition causes ROS accumulation, but also uncoupling of eNOS resulting in further ROS generation in endothelial cells (88). Accordingly, infusion of the proinflammatory hormone aldosterone in vivo impairs the G6PD activity and reduces NO bioavailability leading to increased oxidative stress in the vessel wall and endothelial cell dysfunction (89). Inversely, MRA administration or G6PD overexpression during aldosterone infusion restores vascular reactivity. As will be discussed below, the oxidative stress induced by proinflammatory signals due to NF-κB translocation, T cell activation, TNF, aldosterone, and other inflammatory mediators can then exaggerate the immunologic response in the vessel well, resulting in a vicious cycle in which inflammation begets oxidative stress and vice versa with progressive deterioration in vascular function (Fig. 1).
Autoantibodies elevate placental ROS levels in pre-eclampsia
While the literature supports a more transparent contribution of cell-mediated immunity than humoral immunity to the pathogenesis of hypertension, B cells may play a prominent role in blood pressure elevation related to pre-eclampsia (Fig. 3). Thus, although the adoptive transfer of B lymphocytes does not restore the chronic hypertensive response in Rag1-deficient animals (58), B cell production of autoantibodies to the AT1 receptor predicts the onset of pre-eclampsia in humans (38, 39). Regarding a possible mechanism, these autoantibodies stimulate intracellular calcium signaling that leads to cardiomyocyte contraction (177) and can reduce uterine blood flow by facilitating the production of NAPDH oxidase in trophoblast cells (185). Accordingly, pre-eclamptic animals manifest elevated levels of placental ROS, and scavenging these ROS with tempol reduces both maternal hypertension and ischemic damage to the fetus (66). The importance of B cells to the pathogenesis of pre-eclampsia does not preclude an important contribution of T lymphocytes in this setting. B cells can function, although weakly, as APCs to facilitate T cell activation. This interaction also leads to T cell-dependent B cell activation (131). Moreover, T lymphocytes from human patients with pre-eclampsia show heightened levels of activation (115). Finally, in human patients with pre-eclampsia, circulating mononuclear cells have compromised uptake of L-arginine that could result in a lack of NO bioavailability (109). Elevated levels of myeloperoxidase in the placentas of pre-eclamptic women may consume local NO, and thereby, further contribute to pathologic NO deficiency (49). Available data therefore indicate that preventing maladaptive oxidative stress induced by autoantibodies in pre-eclamptic individuals may limit blood pressure elevation and target organ damage through effects on the vasculature.
FIG. 3.

Fundamental role of B lymphocytes in the pathogenesis of pre-eclampsia. In pre-eclamptic patients, activated B cells differentiate into plasma cells that secrete autoantibodies specific for the type 1 angiotensin receptor (AT1 AutoAb). These antibodies stimulate production of NADPH oxidase in the trophoblast cells of the placenta. In addition, B lymphocytes acting as antigen-presenting cells (APCs) present processed antigen in the context of a class II major histocompatibility complex (MHC II) to the T cell receptor (TCR), activating the T cell. Activated T cells display heightened NADPH oxidase activity. Conversely, CD4+ T cells also potentiate B lymphocyte activation through the same MHC II↔TCR interaction in a process termed T cell-dependent B cell activation. Superoxide (O2−) accumulating in the placenta causes vascular endothelial dysfunction resulting in decreased uterine blood flow and placental ischemia.
The kidney
Inflammatory signals promote generation of oxidative stress within the kidney leading to distortions in renal sodium handling that result in blood pressure elevation. The impact of ROS on renal blood flow and sodium reabsorption provides a key link between oxidative stress in the kidney and the development of hypertension (27, 193), and there are several inflammatory signals that drive renal generation of ROS. Dahl salt-sensitive (SS) rats on a high salt diet have robust infiltration of T cells into the kidney. These T cells have enhanced expression of the p67phox, gp91phox, and p47phox NADPH subunits, and suppression of renal T cell accumulation by a calcineurin inhibitor blunts ROS excretion and lowers blood pressure (36). Furthermore, tubulointerstitial damage associated with mononuclear cell infiltration in the kidney causes local downregulation of eNOS in genetic and pharmacologic models of hypertension with subsequent increases in salt sensitivity (74, 75). In the T cells and macrophages infiltrating the kidney, NF-κB activation triggers transcription of TNF-α. Intrinsic kidney epithelial cells similarly have the capacity to elaborate this cytokine (1, 103). Accordingly, one mechanism linking mononuclear cell infiltration, NF-κB activation, and eNOS suppression in the kidney may be enhanced local TNF production. Indeed, recent evidence has emerged that TNF suppresses eNOS expression and NO levels in the thick ascending limb via a Rho kinase-dependent pathway (142). Inversely, blocking activation of this proinflammatory NF-κB signaling pathway in spontaneously hypertensive rats (SHR) attenuates mitochondrial oxidative stress in the kidney (43). Thus, mononuclear cells invade the kidney in hypertension, activate proinflammatory signaling pathways, and augment ROS generation both directly and via eNOS suppression.
Studies using mycophenolate mofetil (MMF) to suppress lymphocyte proliferation similarly illustrate a role for immune cells to promote renal oxidative stress. For example, suppression of immune cell infiltration into the kidney with MMF during hypertension prevents the salt sensitivity that results following nitric oxide suppression, consistent with a role for inflammatory cells to drive sodium retention by promoting oxidative stress in the kidney (139). Similarly, MMF reduces the number of superoxide-secreting cells in the kidney and urinary excretion of the oxidative stress marker malondialdehyde (MDA) during angiotensin II infusion, also resulting in subsequent protection from salt senstivity (144). In human patients with hypertension associated with immune activation, blood pressures correlate with urinary levels of MDA (63). Finally, treatment of genetically hypertensive rats with MMF suppresses blood pressure elevation, activation of NF-κB in the kidney, and renal parameters of oxidative stress (145, 178). Off-target effects of MMF to block ROS generation in the endothelium rather than via effects on inflammatory cells cannot be completely excluded (82). Barring that criticism, these models indicate that lymphocytes in the kidney promote ROS generation and thereby potentiate blood pressure elevation.
Mouse models of autoimmunity present a novel approach to illustrate how activation of immune responses drives renal oxidative stress in the pathogenesis of hypertension. NZBWF1 mice develop a syndrome mimicking human systemic lupus erythematosus with robust hypertension and marked ROS generation in the vasculature and kidney (149, 183). Antioxidant therapy with tempol and apocynin abrogates renal oxidative stress in this model, lowers blood pressure, and ameliorates renal damage as evidenced by reduced albuminuria (108). Blockade of TNF-α affords similar reductions in blood pressure, renal NADPH oxidase activity, renal macrophage accumulation, and proteinuria (183). Thus, aberrant activation of immune responses promotes ROS generation in the kidney culminating in renal damage and hypertension. These experiments support the possibility that essential hypertension accrues, in part, from misdirected immune responses against neo-antigens uncovered during an initial hypertensive insult (132).
Studies with immunodeficient mice also support the notion that T lymphocytes drive blood pressure elevation through their effects on kidney function. First, T cell deficiency in the Rag1−/− mice mentioned above protects them from mineralocorticoid-induced hypertension, a model in which renal sodium retention drives blood pressure elevation (58). Second, scid mice lacking functional T and B lymphocytes have an exaggerated pressure natriuresis resulting in a blunted increase in blood pressure during chronic angiotensin II infusion (30). We have also found that anesthetized scid mice have preserved blood pressure responses to acute angiotensin II infusion (unpublished observations, SDC), suggesting that T cells do not potentiate vascular dysfunction in these experiments. Consistent with a role for T cells to incite renal generation of ROS, scid mice have enhanced eNOS expression levels in the kidney leading to augmented urinary excretion of NO and suppressed renal generation of the oxidative stress marker, 8-isoprostane (30). In addition to direct effects of NO to promote sodium excretion, upregulation of cyclooxygenase 2 (COX-2) in the scid kidneys culminating in enhanced synthesis of the prostaglandins PGE2 and PGI2 may have also contributed to the enhanced scid natriuresis in this model (56, 64, 150, 151). In sum, T cells augment oxidative stress in the kidney resulting in pathologic sodium retention and salt-sensitive hypertension.
Oxidative stress induced by macrophages in the kidney contributes to hypertensive renal damage. For example, deficiency of CCR2, the receptor for the macrophage chemokine CCL2, attenuates macrophage accumulation in the kidney, blunts renal expressions of nitrotyrosine and gp91phox, and reduces proteinuria following chronic angiotensin II (93). Nevertheless, these effects of CCR2 on ROS generation in the kidney do not regulate the chronic hypertensive response (70, 93). Thus, oxidative stress accruing from the presence of macrophages in the kidney accentuates target organ damage through a blood pressure-independent mechanism.
The CNS
Nuclei within the CNS that regulate cardiovascular responses contribute to hypertension via the elaboration of ROS. Lying outside the blood–brain barrier, the circumventricular organs (CVO) in the brain transmit neuroendocrine signals between the systemic circulation and cardiovascular control centers in the brain stem and hypothalamus, including the paraventricular nucleus (PVN) (33, 37). Neurons in these CNS nuclei express NADPH oxidase, and reducing superoxide levels in these nuclei with a targeted approach attenuates the blood pressure response to hypertensive stimuli (202). Recent evidence suggests that activation of inflammatory responses in the PVN contributes to local ROS generation that drives centrally mediated blood pressure elevation. Specifically, targeted blockade of the NF-κB signaling pathway via infusion of pyrrolidine dithiocarbamate into the intracerebral ventricle, infusion of an NF-κB decoy oligodeoxynucleotide directly into the PVN, or PVN injection with adenovirus carrying a mutated IκB blunts generation of superoxide and peroxynitrite within the PVN. Moreover, each of these forms of NF-κB blockade in the PVN prevents blood pressure elevation in response to systemic angiotensin II infusion (18, 77). Similarly, targeted activation of NF-κB in the mediobasal hypothalamus raises blood pressure significantly in obesity-related hypertension, whereas suppression of the NF-κB activity in the mediobasal hypothalamus via a dominant negative strategy lowers blood pressure (137). In vitro experiments localize key NF-κB activation sites within the hypothalamus to the proopiomelanocortin (POMC) neurons (137). POMC neurons, which project to the PVN, appear to regulate blood pressure through effects on sympathetic outflow (55, 83). These studies point to a role for cooperative actions of oxidative stress and inflammation in sustaining the hypertension seen in the metabolic syndrome.
By contrast, mediators that temper inflammation within the PVN may conversely protect against neurogenic hypertension by relieving oxidative stress within PVN neurons. The macrophage migration inhibitory factor (MIF) limits ROS generation in PVN neurons via thiol protein oxidoreductase and blunts the chronotropic effects of these neurons (169). Normotensive, but not SHR increase MIF expression in the PVN in response to angiotensin II, and injection of MIF into the PVN of SHRs lowers blood pressure (90). MIF in the PVN thus protects from hypertension by alleviating oxidative stress and thereby slowing the heart rate.
Oxidative Stress Provokes Inflammation in Hypertension
The literature summarized above highlights a clear role for inflammatory cells and mediators to augment blood pressure elevation and target organ damage by promoting ROS generation in the vasculature, the kidney, and the nervous system. However, robust evidence has also emerged placing oxidative stress upstream of inflammatory responses in the pathogenesis of hypertension. Below, we review the experiments illustrating how elevated ROS levels in cardiovascular control organs elicit targeted immune responses that potentiate chronic systemic hypertension and its complications.
The vasculature
ROS activate inflammatory signaling pathways
ROS influence inflammatory responses within the vasculature at the level of gene transcription by regulating several intracellular signaling pathways that are central to immunity, including Nrf2 and NF-κB. Within the vascular wall, Nox4 limits the level of oxidative stress by promoting eNOS expression and NO generation (154). Accordingly, Nox4 deficiency permits ROS accumulation in the vessel wall leading to blunted activation of the antioxidant Nrf2 pathway and heightened vascular inflammation during angiotensin II-induced hypertension (154). Mitochondrial ROS have also been found to modulate activation of Nrf2 in other cell lineages (69). Similarly, oxidative stress in the cytoplasm of vascular endothelial cells facilitates the translocation of NF-κB to the nucleus, where it drives transcription of a broad array of inflammatory mediators (190). The activation of NF-κB by ROS occurs, in part, through the redox factor-1 (Ref-1), a redox modulator whose N-terminal contains a redox regulatory domain and a nuclear localization sequence domain. Through these 2 domains, Ref-1 senses rising levels of oxidative stress and activates key inflammatory signaling cascades, including NF-κB and AP-1 (48, 96). In the vascular wall, this NF-κB activation leads to mononuclear cell accumulation via upregulation of adhesion molecules and chemokines culminating in severe target organ damage in hypertension (41, 62, 122, 190).
A third transcription factor linking oxidative stress to vascular inflammation in hypertension is Ets-1. Ets-1 deficiency or knockdown limits induction of the NADPH oxidase subunit p47phox and vascular superoxide generation in angiotensin II-dependent hypertension (128). Accordingly, Ets-1−/− animals have blunted vascular ROS levels associated with reduced expression of the macrophage chemokine CCL2 and attenuation of macrophage and T cell accumulation in the vessel wall (199). Ets-1 deficiency prevents angiotensin II-induced cardiac hypertrophy, but blood pressure elevation is preserved in these animals (199). Angiotensin II therefore promotes ROS generation in the vasculature through the Ets-1 signaling pathway, causing augmented vascular inflammation and target organ damage through blood pressure-independent mechanisms.
Finally, the p38 MAP kinase pathway provides a further link between ROS generation and vascular inflammation. Disruption of this pathway via deletion of its downstream target MAP kinase-activated protein kinase 2 (Mk2) blocks superoxide generation in the vasculature and delays the blood pressure increase induced by in vivo angiotensin II infusion (42). Analogous in vitro studies with VSMCs illustrate Mk2 is required for delivery of p47phox to the cell membrane and, in turn, full NADPH oxidase activity. Further, the local induction of oxidative stress through MAP kinase and Mk2 potentiates vascular inflammation, evidenced by upregulation of ICAM-1 and CCL2. In sum, ROS trigger activation in the vasculature of several key proinflammatory signaling pathways, including Nrf2, NF-kB, Ets-1, and Mk2. Activation of these pathways culminates in the expression of chemokines and adhesion molecules that recruit inflammatory cells into the vascular wall, resulting in augmented blood pressure elevation and/or target organ damage.
Oxidative stress in the vascular endothelium initiates an inflammatory cascade
The vascular endothelium first senses changes in blood pressure as shear stress and responds by triggering adaptive remodeling of the vascular wall. Following changes in shear stress, endothelial cells generate ROS that serve as mechanotransducers, activating proinflammatory pathways to an extent commensurate with the mechanical stress that confronts the cell (110, 117, 180). The ROS derive from NADPH oxidase, NOS, xanthine oxidase, and the mitrochondrial electron transport chain in intrinsic vascular cells (52, 119, 138, 140), infiltrating mononuclear cells (141), and platelets adherent to an activated vascular endothelium (26, 53). The dominant source of ROS in circulating mononuclear cells is NADPH oxidase. These ROS directly regulate vascular remodeling and dysfunction (19, 140). In addition, however, ROS activate the NF-κB pathway in endothelial cells (118) and augment expression of the chemokine CCL2 in vascular smooth muscle (23), leading to further recruitment of inflammatory cells into the vessel wall and local cytokine production (67, 190). Conversely, NO inhibits recruitment of inflammatory cells into the vascular wall (10). To facilitate infiltration of immune cells into sites of vascular injury, ROS drive activation of phosphoinositide 3-kinase-γ resulting in upregulation of VCAM-1 (182).
Oxidative stress further provokes local inflammation by increasing endothelial permeability and thus allowing infiltration of proinflammatory neo-antigens that can incite circulating elements of the innate and adaptive immune responses (12, 162). Among the infiltrating mononuclear cells, macrophages play a paramount role in guiding vascular remodeling (5, 34). Blocking NADPH oxidase limits infiltration of macrophages into the vascular wall and prevents angiotensin II-induced vascular remodeling (97). Similarly, in the same model, NADPH oxidase-deficient mice have blunted accumulation of T lymphocytes in the perivascular space and are protected from blood pressure elevation (58). Indeed, NAPDH in T cells is required for full T lymphocyte activation marked by CD69 expression as well as T cell production of the proinflammatory cytokine TNF-α (58), which in turn can directly mediate blood pressure elevation and/or target organ damage as discussed above. In addition, oxidative stress enhances T cell proliferation and amplifies production of IFN-γ and IL-2 in activated T cells (58, 60, 181). Thus, ROS produced in the vasculature recruit macrophages and T cells to invade the vessel wall and activate these mononuclear cells to potentiate injury signals in hypertension.
Another inflammatory cytokine produced by immune cells or the injured vasculature is IL-1. IL-1 further stimulates NF-κB and production of other inflammatory mediators via a myeloid differentiation protein (Myd88)-signaling pathway (111). Elegant transplant studies have established that Myd88 from intrinsic vascular cells is critical for local cytokine and chemokine production, whereas signals via Myd88 in vascular cells and in the infiltrating macrophages facilitate adaptive vascular remodeling (173). Moreover, Myd88 signals are required for maximal generation of ROS in the vascular wall (173). The altered vascular flow model therefore represents another context in which ROS are both a promoter and a product of local inflammatory responses. In sum, changes in flow through the vessel provoke oxidative stress in the endothelium, which in turn produces inflammatory mediators and recruits mononuclear cells to drive vascular remodeling.
Cyclophilin A links oxidative stress to vascular inflammation
A novel mechanism through which oxidative stress promotes inflammation in the vascular wall involves the chaperone protein cyclophilin A (CypA). ROS stimulate secretion of CypA from VSMCs (73, 92). In turn, CypA drives vascular remodeling, recruitment of mononuclear cells, and upregulation of adhesion molecules in the vascular endothelium to facilitate transmigration of inflammatory cells through the vascular wall (73, 79, 170). Accordingly, Apoe-deficient mice lacking CypA were protected from abdominal aortic aneurysm (AAA) induced by chronic infusion of angiotensin II, despite levels of blood pressure elevation similar to controls (153). Bone marrow transplant studies further illustrated that vascular CypA recruits CD45+ inflammatory cells into the vessel wall. Through its chaperone function, CypA also drives activation of MMPs, also key mediators of vascular injury in the AAA model. Thus, oxidative stress induced by angiotensin II in the vasculature increases local CypA expression, which drives invasion of inflammatory cells into the vessel wall and activation of MMPs where they exaggerate vascular injury (153).
The kidney
In his seminal descriptions of vascular damage induced by malignant hypertension, Harry Goldblatt hypothesized that arteriolar necrosis in the kidney occurred due to a “hypothetical toxic substance or substances in the blood which result from the renal insufficiency” (50). In retrospect, some of these toxic substances were certainly ROS. Moreover, several of the injured renal vessels shown in Goldblatt's panels were surrounded by inflammatory cell infiltrates. Within the kidney, generation of oxidative stress by hypertensive stimuli provokes a local inflammatory response that can worsen renal damage and blood pressure elevation (Table 1). For example, salt loading augments the NADPH oxidase activity and suppresses eNOS in the kidney leading to increased renal expression of CCL5 and enhanced NF-κB activation within the proximal tubule of the nephron, all of which is reversible with tempol therapy (47, 86, 148). Inhibition of NO generation causes hypertensive scarring in the kidney glomerulus known as glomerulosclerosis, detected functionally by the loss of albumin in the urine (9). Elegant servo-control experiments indicate that the oxidative stress and renal macrophage infiltration seen with NO inhibition does not depend on blood pressure elevation (136). Moreover, cytokines induced by ROS such as TNF-α are directly toxic to the kidney glomerulus (11). Thus, some of the glomerular damage resulting from oxidant stress-induced hypertension occurs secondarily through upregulated local inflammatory responses.
Table 1.
Oxidative Stress Potentiates Inflammatory Responses in the Kidney
| Model | Oxidant/antioxidant effect or intervention | Inflammatory effects in kidney | Reference |
|---|---|---|---|
| Salt-loaded rats |
Renal NADPH oxidase induction/eNOS suppression |
Proteinuria; TGF-β induction; NF-κB activation; macrophage infiltration; CCL5 induction in proximal tubule |
45, 84, 146 |
| L-NAME-infused rats |
eNOS inhibition |
Glomerulosclerosis; albuminuria; macrophage infiltration |
8, 134 |
| Dahl salt-sensitive rats |
Vitamins C and E |
Attenuation of macrophages in glomeruli and interstitium; reduced TNF-α and MCP-1 expression; blunted damage to glomeruli/interstitium; reduced BP and proteinuria |
175 |
| Spontaneously hypertensive rats |
Antioxidant-rich diet |
Reduction in infiltration of T cells, macrophages, and Ang II-producing cells; blunted BP elevation |
144 |
| |
Bradykinin Infusion (reduces NADPH oxidase activity) |
Reduction in macrophage infiltration, TGF-β expression, and proteinuria |
20 |
| DOCA-salt | HO-1 induction (scavenges ROS) | Reduced glomerulosclerosis, albuminuria, mononuclear cell infiltration; lowered levels of NF-κB, AP-1, TGF-β, and fibronectin | 70 |
Although immune responses clearly induce renal oxidative stress, the converse effects of superoxide to stimulate inflammation in the kidney are evident in the experiments summarized here.
eNOS, endothelial nitric oxide synthase; NF-κB, nuclear factor-κB; ROS, reactive oxygen species; TNF, tumor necrosis factor; TGF-β transforming growth factor-β; DOCA, deoxycorticosterone acetate.
By contrast, interventions that limit renal oxidative stress ameliorate blood pressure elevation and/or inflammatory kidney damage in models of hypertension. For example, antioxidant therapy in Dahl salt sensitive and SHRs limits renal expression of chemokines and accumulation of macrophages in the kidney and in turn lowers blood pressure (146, 179). Infusion of bradykinin into these hypertensive rats suppresses the renal NADPH oxidase activity and superoxide generation. This control of oxidative stress in turn reduces proteinuria, renal macrophage infiltration, and structural damage to the kidney glomeruli and tubules (21). Induction of heme oxygenase during deoxycorticosterone acetate (DOCA)-salt hypertension promotes ROS scavenging in the kidney and accordingly reduces 8-isoprostane excretion, blunts accumulation of mononuclear cells in the kidney, and attenuates renal structural damage (72). Angiotensin converting enzyme 2 (ACE2) counteracts the oxidative damage in the kidney caused by angiotensin II, normalizing renal levels of oxidative stress, blunting T cell accumulation in the kidney, and ameliorating renal fibrosis (201). Control of ROS generation in the kidney therefore provides renoprotection in part by preventing downstream escalation of immune responses.
The CNS
Generation of oxidative stress in key regions of the CNS can influence systemic inflammation to alter blood pressure responses and aggravate target organ damage. As discussed above, the CVO transfer systemic afferent signals to cardiovascular control nuclei in the brain stem and hypothalamus. Preventing degradation of ROS in the CVO via conditional deletion of extracellular superoxide dismutase (ecSOD) yields a robust blood pressure increase and augments ROS generation even in the systemic vasculature following administration of angiotensin II at a dose that does not alter blood pressure in normal animals (98). This enhanced oxidative stress in the CNS and in the vasculature is associated with increased expression of activation markers on circulating T lymphocytes. However, whether the exaggerated T cell activation accrues from the blood pressure elevation in these CNS ecSOD knockout mice or directly from the effects of ROS accumulation in the CVO or the periphery is unclear. Blocking the hypertensive response of wild-type animals to a higher dose of angiotensin II with hydralazine prevents circulating T cell activation, pointing to an important effect of blood pressure to stimulate cell-mediated immune responses (107).
ROS in the CNS also elevate blood pressure by stimulating the proinflammatory NF-κB signaling pathway within key CNS nuclei. For example, systemic infusion of angiotensin II raises gp91phox levels in the PVN of the hypothalamus leading to enhanced phosphorylation of NF-κB regulator IKKβ and subunit p50, respectively (77) (Fig. 4). As in other cell lineages, translocation of NF-κB to the nucleus in cells of the PVN triggers increased generation of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 (77). Production of TNF within the PVN is associated with enhanced sympathetic nerve activation, providing a link between central inflammation and blood pressure elevation (57). Moreover, injection of IL-1β directly into the PVN raises blood pressure (99, 160), and blockade of cytokine production in the PVN with minocycline attenuates angiotensin II-induced hypertension (160). Antioxidant therapy with tempol introduced directly into the cerebrospinal fluid reverses NF-κB activation and inflammatory cytokine production within the PVN, in turn, blunting sympathetic outflow and abrogating the hypertensive response to angiotensin II (77). These studies illustrate that the induction of the proinflammatory NF-κB signaling pathway in the PVN and its actions to raise blood pressure depends on local ROS generation.
FIG. 4.
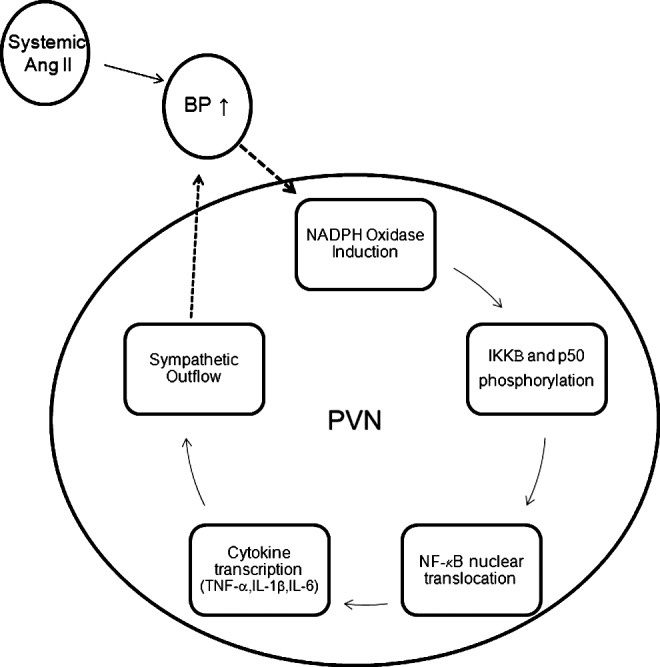
Feed-forward induction of oxidative stress and nuclear factor-κB (NF-κB) activation in the paraventricular nucleus (PVN) during angiotensin II-dependent hypertension. Systemic angiotensin II infusion modeling activation of the renin-angiotensin system raises blood pressure and induces subunits of the NADPH oxidase complex in cells of the PVN within the hypothalamus. Superoxide generation within the PVN triggers phosphorylation of regulators and subunits of the NF-κB signaling complex. Free to translocate from the cytoplasm to the nucleus of the cell, NF-κB induces transcription of inflammatory cytokines, including TNF-α and interleukin-1β (IL-1β) that drive sympathetic outflow from the central nervous system and further elevate blood pressure, sustaining a prohypertensive, proinflammatory cycle.
ACE2 in the CNS has protective effects to limit local oxidative stress just as in other cardiovascular control organs. Accordingly, following angiotensin II infusion, ACE2-deficiency augments ROS generation in the PVN and in the rostral ventrolateral medulla, whereas ACE2 gene therapy into the PVN ameliorates this oxidative stress (194). Moreover, these actions of ACE2 to suppress ROS in the CNS protect against local inflammation as overexpression of ACE2 in the PVN reduces levels of TNF, IL-1β, and IL-6 and in turn attenuates angiotensin II-induced hypertension (163).
Conclusion
Inflammatory responses heighten oxidative stress, and, conversely, ROS generation potentiates immune activation cooperatively in the pathogenesis of hypertension. Separating these 2 phenomena as we have done is instructive for clarifying inflammatory cascades and signaling pathways. However, in the intact organism, the interactions between oxidative stress and inflammation are in all likelihood constitutively bidirectional (Fig. 1). Pharmacologic interventions to lower blood pressure commonly improve markers of both inflammation and oxidative stress in the organ undergoing analysis such that study designs typically do not reveal whether ROS lay upstream of immune responses or vice versa. Experiments that carefully discriminate the effects of oxidative stress versus those of immunity point to a feed-forward mechanism in which one begets the other (i.e., Fig. 4). As we have discussed, oxidative stress induces CypA in the vasculature leading to vascular inflammation, but CypA deficiency limits ROS generation in the vessel wall. ROS activate the Myd88 inflammatory signaling pathway in immune cells and the vasculature, which in turn propagates additional oxidative stress. ROS generation in the PVN activates NF-κB leading to inflammatory cytokine production; blocking NF-κB translocation in the PVN reduces oxidative stress and lowers blood pressure. The robust interactions between ROS generation and immune activation would suggest that interventions targeting one may also limit the other. This paradigm holds promise for potent therapeutics that may nevertheless have unintended off-target consequences. Therefore, future research will need to define with greater precision the interrelationships during hypertension between oxidative stress and inflammation in specific cell lineages within cardiovascular control organs and within specialized subtypes of myeloid and lymphoid inflammatory cells. For example, although T lymphocytes in general enhance ROS generation in the vasculature and the kidney, T regulatory cells, which tend to suppress inflammation, can limit oxidative stress and blunt the chronic hypertensive response to angiotensin II (6). The human organism utilizes this rare, but potent population of T regulatory cells to exercise exquisite control over immune responses. Our analogous interventions to contain pathologic oxidative stress and inflammation in hypertensive patients, while preserving important protective functions of ROS in immune cells, will similarly require daunting precision.
Abbreviations Used
- AAA
abdominal aortic aneurysm
- ACE2
angiotensin converting enzyme 2
- APC
antigen-presenting cell
- AT1
type 1 angiotensin
- BH4
tetrahydrobiopterin
- CNS
central nervous system
- COX-2
cyclooxygenase 2
- CRP
C reactive protein
- CVO
circumventricular organs
- CypA
cyclophilin A
- ecSOD
extracellular superoxide dismutase
- eNOS
endothelial nitric oxide synthase
- G6PD
glucose-6-phosphate dehydrogenase
- HSP70
heat shock protein-70
- ICAM-1
intercellular adhesion molecule-1
- IFN-γ
interferon-γ
- IL-1
interleukin-1
- iNOS
inducible nitric oxide synthase
- LDL
low-density lipoprotein
- MAP
mitogen-activated protein
- MDA
malondialdehyde
- MIF
macrophage migration inhibitory factor
- MMF
mycophenolate mofetil
- MMPs
matrix metalloproteinases
- MRAs
mineralocorticoid antagonists
- NF-κB
nuclear factor-κB
- PDTC
pyrrolidine dithiocarbamate
- POMC
proopiomelanocortin
- PVN
paraventricular nucleus
- Ref-1
redox factor-1
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rats
- TCR
T cell receptor
- TGF
transforming growth factor
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- VCAM-1
vascular cell adhesion molecule-1
- VSMCs
vascular smooth muscle cell
Acknowledgments
This work was supported by a National Institutes of Health Grant DK087783, the Medical Research Service of the Department of Veterans Affairs, and the Edna and Fred L. Mandel Center for Hypertension and Atherosclerosis Research.
References
- 1.Abdullah HI, Pedraza PL, McGiff JC, and Ferreri NR. CaR activation increases TNF production by mTAL cells via a Gi-dependent mechanism. Am J Physiol Renal Physiol 294: F345–F354, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Almasan A. and Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14: 337–348, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Angel K, Provan SA, Gulseth HL, Mowinckel P, Kvien TK, and Atar D. Tumor necrosis factor-{alpha} antagonists improve aortic stiffness in patients with inflammatory arthropathies: a controlled study. Hypertension 55: 333–338, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Arai T, Kelly SA, Brengman ML, Takano M, Smith EH, Goldschmidt-Clermont PJ, and Bulkley GB. Ambient but not incremental oxidant generation effects intercellular adhesion molecule 1 induction by tumour necrosis factor alpha in endothelium. Biochem J 331 (Pt 3): 853–861, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakker EN, Matlung HL, Bonta P, de Vries CJ, van Rooijen N, and Vanbavel E. Blood flow-dependent arterial remodelling is facilitated by inflammation but directed by vascular tone. Cardiovasc Res 78: 341–348, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, and Schiffrin EL. T Regulatory lymphocytes prevent angiotensin ii–induced hypertension and vascular injury. Hypertension 57: 469–476, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Bataillard A, Freiche JC, Vincent M, Sassard J, and Touraine JL. Antihypertensive effect of neonatal thymectomy in the genetically hypertensive LH rat. Thymus 8: 321–330, 1986 [PubMed] [Google Scholar]
- 8.Battula S, Hao S, Pedraza PL, Stier CT, and Ferreri NR. Tumor necrosis factor-α is an endogenous inhibitor of Na+-K+-2Cl− cotransporter (NKCC2) isoform A in the thick ascending limb. Am J Physiol Renal Physiol 301: F94–F100, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baylis C, Mitruka B, and Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest 90: 278–281, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behrendt D. and Ganz P. Endothelial function. From vascular biology to clinical applications. Am J Cardiol 90: 40L–48L, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, and Remuzzi G. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol 134: 419–430, 1989 [PMC free article] [PubMed] [Google Scholar]
- 12.Bjorkbacka H. Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr Opin Lipidol 17: 527–533, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Boesen EI, Sasser JM, Saleh MA, Potter WA, Woods M, Warner TD, Pollock JS, and Pollock DM. Interleukin-1beta, but not interleukin-6, enhances renal and systemic endothelin production in vivo. Am J Physiol Renal Physiol 295: F446–F453, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, and Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 12: 213–221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caligiuri G, Paulsson G, Nicoletti A, Maseri A, and Hansson GK. Evidence for antigen-driven T-cell response in unstable angina. Circulation 102: 1114–1119, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Callera GE, Montezano AC, Yogi A, Tostes RC, He Y, Schiffrin EL, and Touyz RM. c-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats. Hypertension 46: 1032–1038, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Calo LA, Zaghetto F, Pagnin E, Davis PA, De Mozzi P, Sartorato P, Martire G, Fiore C, and Armanini D. Effect of aldosterone and glycyrrhetinic acid on the protein expression of PAI-1 and p22(phox) in human mononuclear leukocytes. J Clin Endocrinol Metab 89: 1973–1976, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Cardinale JP, Sriramula S, Mariappan N, Agarwal D, and Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-kappaBin the paraventricular nucleus. Hypertension 59: 113–121, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castier Y, Brandes RP, Leseche G, Tedgui A, and Lehoux S. p47phox-dependent NADPH oxidase regulates flow-induced vascular remodeling. Circ Res 97: 533–540, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Chae CU, Lee RT, Rifai N, and Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension 38: 399–403, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Chao J, Li HJ, Yao YY, Shen B, Gao L, Bledsoe G, and Chao L. Kinin infusion prevents renal inflammation, apoptosis, and fibrosis via inhibition of oxidative stress and mitogen-activated protein kinase activity. Hypertension 49: 490–497, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Chen CC, Pedraza PL, Hao S, Stier CT, and Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol 299: F1141–F1150, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen XL, Tummala PE, Olbrych MT, Alexander RW, and Medford RM. Angiotensin II induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circ Res 83: 952–959, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Chrysohoou C, Pitsavos C, Panagiotakos DB, Skoumas J, and Stefanadis C. Association between prehypertension status and inflammatory markers related to atherosclerotic disease: The ATTICA Study. Am J Hypertens 17: 568–573, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Cooper AL. and Beasley D. Hypoxia stimulates proliferation and interleukin-1alpha production in human vascular smooth muscle cells. Am J Physiol 277: H1326–H1337, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Cooper D, Stokes KY, Tailor A, and Granger DN. Oxidative stress promotes blood cell-endothelial cell interactions in the microcirculation. Cardiovasc Toxicol 2: 165–180, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Cowley AW, Jr., Mori T, Mattson D, and Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol 284: R1355–R1369, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Criss WE. and McKerns KW. Inhibitors of the catalytic activity of bovine adrenal glucose-6-phosphate dehydrogenase. Biochim Biophys Acta 184: 486–494, 1969 [DOI] [PubMed] [Google Scholar]
- 29.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, and Coffman TM. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol 295: F515–F524, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, and Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux PL, and Ruiz P. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II-dependent hypertension. Hypertension 55: 99–108, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dale DC, Boxer L, and Liles WC. The phagocytes: neutrophils and monocytes. Blood 112: 935–945, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Davisson RL. Physiological genomic analysis of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 285: R498–R511, 2003 [DOI] [PubMed] [Google Scholar]
- 34.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, and Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113, 2005 [DOI] [PubMed] [Google Scholar]
- 35.De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, and Griendling KK. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J 329 (Pt 3): 653–657, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Miguel C, Guo C, Lund H, Feng D, and Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Wardener HE. The hypothalamus and hypertension. Physiol Rev 81: 1599–1658, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Dechend R, Muller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J, and Luft FC. AT1 receptor agonistic antibodies, hypertension, and preeclampsia. Semin Nephrol 24: 571–579, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Dechend R, Viedt C, Muller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, Fiebeler A, Homuth V, Dietz R, Haller H, Kreuzer J, and Luft FC. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 107: 1632–1639, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Devadas S, Zaritskaya L, Rhee SG, Oberley L, and Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation. J Exp Med 195: 59–70, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhawan S, Singh S, and Aggarwal BB. Induction of endothelial cell surface adhesion molecules by tumor necrosis factor is blocked by protein tyrosine phosphatase inhibitors: role of the nuclear transcription factor NF-kappa B. Eur J Immunol 27: 2172–2179, 1997 [DOI] [PubMed] [Google Scholar]
- 42.Ebrahimian T, Li MW, Lemarie CA, Simeone SM, Pagano PJ, Gaestel M, Paradis P, Wassmann S, and Schiffrin EL. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: regulation of oxidative stress. Hypertension 57: 245–254, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elks CM, Mariappan N, Haque M, Guggilam A, Majid DS, and Francis J. Chronic NF-{kappa}B blockade reduces cytosolic and mitochondrial oxidative stress and attenuates renal injury and hypertension in SHR. Am J Physiol Renal Physiol 296: F298–F305, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, and Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elmarakby AA, Quigley JE, Olearczyk JJ, Sridhar A, Cook AK, Inscho EW, Pollock DM, and Imig JD. Chemokine receptor 2b inhibition provides renal protection in angiotensin II salt hypertension. Hypertension 50: 1069–1076, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elmarakby AA, Quigley JE, Pollock DM, and Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension 47: 557–562, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Fiore MC, Jimenez PM, Cremonezzi D, Juncos LI, and Garcia NH. Statins reverse renal inflammation and endothelial dysfunction induced by chronic high salt intake. Am J Physiol Renal Physiol 301: F263–F270, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Fritz G, Grosch S, Tomicic M, and Kaina B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology 193: 67–78, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Gandley RE, Rohland J, Zhou Y, Shibata E, Harger GF, Rajakumar A, Kagan VE, Markovic N, and Hubel CA. Increased myeloperoxidase in the placenta and circulation of women with preeclampsia. Hypertension 52: 387–393, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldblatt H. Studies on Experimental Hypertension: Vii. The production of the malignant phase of hypertension. J Exp Med 67: 809–826, 1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goligorsky MS. and Noiri E. Duality of nitric oxide in acute renal injury. Semin Nephrol 19: 263–271, 1999 [PubMed] [Google Scholar]
- 52.Granger DN, Benoit JN, Suzuki M, and Grisham MB. Leukocyte adherence to venular endothelium during ischemia-reperfusion. Am J Physiol 257: G683–G688, 1989 [DOI] [PubMed] [Google Scholar]
- 53.Granger DN, Vowinkel T, and Petnehazy T. Modulation of the inflammatory response in cardiovascular disease. Hypertension 43: 924–931, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Gratze P, Dechend R, Stocker C, Park JK, Feldt S, Shagdarsuren E, Wellner M, Gueler F, Rong S, Gross V, Obst M, Plehm R, Alenina N, Zenclussen A, Titze J, Small K, Yokota Y, Zenke M, Luft FC, and Muller DN. Novel role for inhibitor of differentiation 2 in the genesis of angiotensin II-induced hypertension. Circulation 117: 2645–2656, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, and Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med 360: 44–52, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hebert RL, and Breyer MD. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest 102: 194–201, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guggilam A, Haque M, Kerut EK, McIlwain E, Lucchesi P, Seghal I, and Francis J. TNF-alpha blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol 293: H599–H609, 2007 [DOI] [PubMed] [Google Scholar]
- 58.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, and Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hazan-Halevy I, Levy T, Wolak T, Lubarsky I, Levy R, and Paran E. Stimulation of NADPH oxidase by angiotensin II in human neutrophils is mediated by ERK, p38 MAP-kinase and cytosolic phospholipase A2. J Hypertens 23: 1183–1190, 2005 [DOI] [PubMed] [Google Scholar]
- 60.Hehner SP, Breitkreutz R, Shubinsky G, Unsoeld H, Schulze-Osthoff K, Schmitz ML, and Droge W. Enhancement of T cell receptor signaling by a mild oxidative shift in the intracellular thiol pool. J Immunol 165: 4319–4328, 2000 [DOI] [PubMed] [Google Scholar]
- 61.Henkart PA. and Catalfamo M. CD8+ effector cells. Adv Immunol 83: 233–252, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Henke N, Schmidt-Ullrich R, Dechend R, Park J-K, Qadri F, Wellner M, Obst M, Gross V, Dietz R, Luft FC, Scheidereit C, and Muller DN. Vascular endothelial cell specific NF-{kappa}B suppression attenuates hypertension-induced renal damage. Circ Res 101: 268–276, 2007 [DOI] [PubMed] [Google Scholar]
- 63.Herrera J, Ferrebuz A, Macgregor EG, and Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17: S218–S225, 2006 [DOI] [PubMed] [Google Scholar]
- 64.Hill TW. and Moncada S. The renal haemodynamic and excretory actions of prostacyclin and 6-oxo-PGF1 alpha in anaesthetized dogs. Prostaglandins 17: 87–98, 1979 [DOI] [PubMed] [Google Scholar]
- 65.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, and Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoffmann DS, Weydert CJ, Lazartigues E, Kutschke WJ, Kienzle MF, Leach JE, Sharma JA, Sharma RV, and Davisson RL. Chronic tempol prevents hypertension, proteinuria, and poor feto-placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 51: 1058–1065, 2008 [DOI] [PubMed] [Google Scholar]
- 67.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, and Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem 278: 47291–47298, 2003 [DOI] [PubMed] [Google Scholar]
- 68.Ialenti A, Moncada S, and Di Rosa M. Modulation of adjuvant arthritis by endogenous nitric oxide. Br J Pharmacol 110: 701–706, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Imhoff BR. and Hansen JM. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem J 424: 491–500, 2009 [DOI] [PubMed] [Google Scholar]
- 70.Ishibashi M, Hiasa K-i, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, and Egashira K. Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res 94: 1203–1210, 2004 [DOI] [PubMed] [Google Scholar]
- 71.Jackson SH, Devadas S, Kwon J, Pinto LA, and Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol 5: 818–827, 2004 [DOI] [PubMed] [Google Scholar]
- 72.Jadhav A, Torlakovic E, and Ndisang JF. Hemin therapy attenuates kidney injury in deoxycorticosterone acetate-salt hypertensive rats. Am J Physiol Renal Physiol 296: F521–F534, 2009 [DOI] [PubMed] [Google Scholar]
- 73.Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, and Berk BC. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ Res 87: 789–796, 2000 [DOI] [PubMed] [Google Scholar]
- 74.Johnson RJ, Gordon KL, Giachelli C, Kurth T, Skelton MM, and Cowley AW., Jr.Tubulointerstitial injury and loss of nitric oxide synthases parallel the development of hypertension in the Dahl-SS rat. J Hypertens 18: 1497–1505, 2000 [DOI] [PubMed] [Google Scholar]
- 75.Johnson RJ, Gordon KL, Suga S, Duijvestijn AM, Griffin K, and Bidani A. Renal injury and salt-sensitive hypertension after exposure to catecholamines. Hypertension 34: 151–159, 1999 [DOI] [PubMed] [Google Scholar]
- 76.Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, Goldblatt D, Parker L, and Cant AJ. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol 152: 211–218, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, and Francis J. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res 82: 503–512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, and Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension 59: 324–330, 2012 [DOI] [PubMed] [Google Scholar]
- 79.Khromykh LM, Kulikova NL, Anfalova TV, Muranova TA, Abramov VM, Vasiliev AM, Khlebnikov VS, and Kazansky DB. Cyclophilin A produced by thymocytes regulates the migration of murine bone marrow cells. Cell Immunol 249: 46–53, 2007 [DOI] [PubMed] [Google Scholar]
- 80.Kim F, Gallis B, and Corson MA. TNF-alpha inhibits flow and insulin signaling leading to NO production in aortic endothelial cells. Am J Physiol Cell Physiol 280: C1057–C1065, 2001 [DOI] [PubMed] [Google Scholar]
- 81.Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, and Schiffrin EL. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol 292: H1789–H1795, 2007 [DOI] [PubMed] [Google Scholar]
- 82.Krotz F, Keller M, Derflinger S, Schmid H, Gloe T, Bassermann F, Duyster J, Cohen CD, Schuhmann C, Klauss V, Pohl U, Stempfle H-U, and Sohn H-Y. Mycophenolate acid inhibits endothelial NAD(P)H oxidase activity and superoxide formation by a rac1-dependent mechanism. Hypertension 49: 201–208, 2007 [DOI] [PubMed] [Google Scholar]
- 83.Kuo JJ, da Silva AA, Tallam LS, and Hall JE. Role of adrenergic activity in pressor responses to chronic melanocortin receptor activation. Hypertension 43: 370–375, 2004 [DOI] [PubMed] [Google Scholar]
- 84.Kvakan H, Dahlman A, Machnik A, van Rooijen N, Qadri F, Dechend R, Titze J, Luft FC, and Muller DN. Macrophage depletion accelerates angiotensin II-induced target organ damage. Hypertension 52: e45, 2008 [Google Scholar]
- 85.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, and Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119: 2904–2912, 2009 [DOI] [PubMed] [Google Scholar]
- 86.Lara LS, McCormack M, Semprum-Prieto LC, Shenouda S, Majid DS, Kobori H, Navar LG, and Prieto MC. AT1 receptor-mediated augmentation of angiotensinogen, oxidative stress, and inflammation in ANG II-salt hypertension. Am J Physiol Renal Physiol 302: F85–F94, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr., Fleming C, Pollock JS, Manhiani M, Imig JD, and Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006 [DOI] [PubMed] [Google Scholar]
- 88.Leopold JA, Cap A, Scribner AW, Stanton RC, and Loscalzo J. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J 15: 1771–1773, 2001 [DOI] [PubMed] [Google Scholar]
- 89.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, and Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 13: 189–197, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H, Gao Y, Qi Y, Katovich MJ, Jiang N, Braseth LN, Scheuer DA, Shi P, and Sumners C. Macrophage migration inhibitory factor in hypothalamic paraventricular nucleus neurons decreases blood pressure in spontaneously hypertensive rats. FASEB J 22: 3175–3185, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, and Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol 299: R590–R595, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liao DF, Jin ZG, Baas AS, Daum G, Gygi SP, Aebersold R, and Berk BC. Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J Biol Chem 275: 189–196, 2000 [DOI] [PubMed] [Google Scholar]
- 93.Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, and Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 52: 256–263, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liew CC. and Gornall AG. Effects of aldosterone on blood pressure and glucose-6-phosphate dehydrogenase activity of heart muscle. Can J Physiol Pharmacol 47: 193–197, 1969 [DOI] [PubMed] [Google Scholar]
- 95.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, and Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol 154: 485–490, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu H, Colavitti R, Rovira II, and Finkel T. Redox-dependent transcriptional regulation. Circ Res 97: 967–974, 2005 [DOI] [PubMed] [Google Scholar]
- 97.Liu J, Yang F, Yang XP, Jankowski M, and Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol 23: 776–782, 2003 [DOI] [PubMed] [Google Scholar]
- 98.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, and Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 55: 277–283, 276p following 283, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu Y, Chen J, Yin X, and Zhao H. Angiotensin II receptor 1 involved in the central pressor response induced by interleukin-1 beta in the paraventricular nucleus. Neurol Res 31: 420–424, 2009 [DOI] [PubMed] [Google Scholar]
- 100.Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, and Brown NJ. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor–dependent mechanism. Hypertension 48: 1050–1057, 2006 [DOI] [PubMed] [Google Scholar]
- 101.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park J-K, Beck F-X, Muller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt K-U, Luft FC, Kerjaschki D, and Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med 15: 545–552, 2009 [DOI] [PubMed] [Google Scholar]
- 102.Maki-Petaja KM, Hall FC, Booth AD, Wallace SM, Yasmin , Bearcroft PW, Harish S, Furlong A, McEniery CM, Brown J, and Wilkinson IB. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy. Circulation 114: 1185–1192, 2006 [DOI] [PubMed] [Google Scholar]
- 103.Markewitz BA, Michael JR, and Kohan DE. Cytokine-induced expression of a nitric oxide synthase in rat renal tubule cells. J Clin Invest 91: 2138–2143, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinvalet D, Zhu P, and Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22: 355–370, 2005 [DOI] [PubMed] [Google Scholar]
- 105.Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, Rossi P, Gattorno M, Rabusin M, Azzari C, Dellepiane RM, Pietrogrande MC, Trizzino A, Di Bartolomeo P, Martino S, Carpino L, Cossu F, Locatelli F, Maccario R, Pierani P, Putti MC, Stabile A, Notarangelo LD, Ugazio AG, Plebani A, and De Mattia D. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: an Italian multicenter study. Clin Immunol 126: 155–164, 2008 [DOI] [PubMed] [Google Scholar]
- 106.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, and Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest 92: 1866–1874, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marvar PJ, Lob H, Vinh A, Zarreen F, and Harrison DG. The central nervous system and inflammation in hypertension. Curr Opin Pharmacol 11: 156–161, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, and Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension 59: 673–679, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McCord N, Ayuk P, McMahon M, Boyd RC, Sargent I, and Redman C. System y+ arginine transport and NO production in peripheral blood mononuclear cells in pregnancy and preeclampsia. Hypertension 47: 109–115, 2006 [DOI] [PubMed] [Google Scholar]
- 110.McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, and Harrison DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol 285: H2290–H2297, 2003 [DOI] [PubMed] [Google Scholar]
- 111.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, and Janeway CA., Jr.MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2: 253–258, 1998 [DOI] [PubMed] [Google Scholar]
- 112.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, and Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J 263: 539–545, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mervaala EM, Muller DN, Park JK, Schmidt F, Lohn M, Breu V, Dragun D, Ganten D, Haller H, and Luft FC. Monocyte infiltration and adhesion molecules in a rat model of high human renin hypertension. Hypertension 33: 389–395, 1999 [DOI] [PubMed] [Google Scholar]
- 114.Mezzano S, Droguett A, Burgos ME, Ardiles LG, Flores CA, Aros CA, Caorsi I, Vio CP, Ruiz-Ortega M, and Egido J. Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int Suppl S64–S70, 2003 [DOI] [PubMed] [Google Scholar]
- 115.Minagawa M, Narita J, Tada T, Maruyama S, Shimizu T, Bannai M, Oya H, Hatakeyama K, and Abo T. Mechanisms underlying immunologic states during pregnancy: possible association of the sympathetic nervous system. Cell Immunol 196: 1–13, 1999 [DOI] [PubMed] [Google Scholar]
- 116.Mir M, Asensio VJ, Tolosa L, Gou-Fabregas M, Soler RM, Llado J, and Olmos G. Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants. Neuroscience 162: 959–971, 2009 [DOI] [PubMed] [Google Scholar]
- 117.Mohan S, Koyoma K, Thangasamy A, Nakano H, Glickman RD, and Mohan N. Low shear stress preferentially enhances IKK activity through selective sources of ROS for persistent activation of NF-kappaB in endothelial cells. Am J Physiol Cell Physiol 292: C362–C371, 2007 [DOI] [PubMed] [Google Scholar]
- 118.Mohan S, Mohan N, and Sprague EA. Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am J Physiol 273: C572–C578, 1997 [DOI] [PubMed] [Google Scholar]
- 119.Mohazzab KM, Kaminski PM, and Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol 266: H2568–H2572, 1994 [DOI] [PubMed] [Google Scholar]
- 120.Moncada S. The 1991 Ulf von Euler Lecture. The L-arginine: nitric oxide pathway. Acta Physiol Scand 145: 201–227, 1992 [DOI] [PubMed] [Google Scholar]
- 121.Moncada S. and Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002–2012, 1993 [DOI] [PubMed] [Google Scholar]
- 122.Moriuchi H, Moriuchi M, and Fauci AS. Nuclear factor-kappa B potently up-regulates the promoter activity of RANTES, a chemokine that blocks HIV infection. J Immunol 158: 3483–3491, 1997 [PubMed] [Google Scholar]
- 123.Mosser DM. and Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muller DN, Dechend R, Mervaala EMA, Park J-K, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, and Luft FC. NF-{kappa}B inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35: 193–201, 2000 [DOI] [PubMed] [Google Scholar]
- 125.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, and Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161: 1679–1693, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, and Namba M. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 98: 794–799, 1998 [DOI] [PubMed] [Google Scholar]
- 127.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, and Coffman TM. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 104: 1693–1701, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ni W, Zhan Y, He H, Maynard E, Balschi JA, and Oettgen P. Ets-1 is a critical transcriptional regulator of reactive oxygen species and p47phox gene expression in response to angiotensin II. Circ Res 101: 985–994, 2007 [DOI] [PubMed] [Google Scholar]
- 129.Niskanen L, Laaksonen DE, Nyyssonen K, Punnonen K, Valkonen VP, Fuentes R, Tuomainen TP, Salonen R, and Salonen JT. Inflammation, abdominal obesity, and smoking as predictors of hypertension. Hypertension 44: 859–865, 2004 [DOI] [PubMed] [Google Scholar]
- 130.Okuda T. and Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med 25: 257–264, 1967 [PubMed] [Google Scholar]
- 131.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol 11: 331–360, 1993 [DOI] [PubMed] [Google Scholar]
- 132.Parra G, Quiroz Y, Salazar J, Bravo Y, Pons H, Chavez M, Johnson RJ, and Rodriguez-Iturbe B. Experimental induction of salt-sensitive hypertension is associated with lymphocyte proliferative response to HSP70. Kidney Int Suppl S55–S59, 2008 [DOI] [PubMed] [Google Scholar]
- 133.Pauletto P. and Rattazzi M. Inflammation and hypertension: the search for a link. Nephrol Dial Transplant 21: 850–853, 2006 [DOI] [PubMed] [Google Scholar]
- 134.Petros A, Bennett D, and Vallance P. Effect of nitric oxide synthase inhibitors on hypotension in patients with septic shock. Lancet 338: 1557–1558, 1991 [DOI] [PubMed] [Google Scholar]
- 135.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, and Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341: 709–717, 1999 [DOI] [PubMed] [Google Scholar]
- 136.Polichnowski AJ, Lu L, and Cowley AW. Renal injury in angiotensin II+l-NAME-induced hypertensive rats is independent of elevated blood pressure. Am J Physiol Renal Physiol 300: F1008–F1016, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Purkayastha S, Zhang G, and Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med 17: 883–887, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Quintero M, Colombo SL, Godfrey A, and Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A 103: 5379–5384, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Quiroz Y, Pons H, Gordon KL, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ, and Rodriguez-Iturbe B. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 281: F38–F47, 2001 [DOI] [PubMed] [Google Scholar]
- 140.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, and Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, and Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 98: 2572–2579, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ramseyer VD, Hong NJ, and Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension 59: 1145–1150, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rocha R. and Stier CT., Jr.Pathophysiological effects of aldosterone in cardiovascular tissues. Trends Endocrinol Metab 12: 308–314, 2001 [DOI] [PubMed] [Google Scholar]
- 144.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, and Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001 [DOI] [PubMed] [Google Scholar]
- 145.Rodríguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chávez M, Herrera-Acosta J, Johnson RJ, and Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 282: F191–F201, 2002 [DOI] [PubMed] [Google Scholar]
- 146.Rodriguez-Iturbe B, Zhan CD, Quiroz Y, Sindhu RK, and Vaziri ND. Antioxidant-rich diet relieves hypertension and reduces renal immune infiltration in spontaneously hypertensive rats. Hypertension 41: 341–346, 2003 [DOI] [PubMed] [Google Scholar]
- 147.Roos D, de Boer M, Kuribayashi F, Meischl C, Weening RS, Segal AW, Ahlin A, Nemet K, Hossle JP, Bernatowska-Matuszkiewicz E, and Middleton-Price H. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood 87: 1663–1681, 1996 [PubMed] [Google Scholar]
- 148.Roson MI, Della Penna SL, Cao G, Gorzalczany S, Pandolfo M, Toblli JE, and Fernandez BE. Different protective actions of losartan and tempol on the renal inflammatory response to acute sodium overload. J Cell Physiol 224: 41–48, 2010 [DOI] [PubMed] [Google Scholar]
- 149.Ryan MJ. and McLemore GR., Jr.Hypertension and impaired vascular function in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 292: R736–R742, 2007 [DOI] [PubMed] [Google Scholar]
- 150.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, and Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A 90: 7240–7244, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Salvemini D, Seibert K, Masferrer JL, Misko TP, Currie MG, and Needleman P. Endogenous nitric oxide enhances prostaglandin production in a model of renal inflammation. J Clin Invest 93: 1940–1947, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sato K, Niessner A, Kopecky SL, Frye RL, Goronzy JJ, and Weyand CM. TRAIL-expressing T cells induce apoptosis of vascular smooth muscle cells in the atherosclerotic plaque. J Exp Med 203: 239–250, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Satoh K, Nigro P, Matoba T, O'Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J-i, Illig KA, and Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med 15: 649–656, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, and Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217–1225, 2012 [DOI] [PubMed] [Google Scholar]
- 155.Segal BH, Leto TL, Gallin JI, Malech HL, and Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 79: 170–200, 2000 [DOI] [PubMed] [Google Scholar]
- 156.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, and Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA 290: 2945–2951, 2003 [DOI] [PubMed] [Google Scholar]
- 157.Shahid M, Francis J, and Majid DSA. Tumor necrosis factor-{alpha} induces renal vasoconstriction as well as natriuresis in mice. Am J Physiol Renal Physiol 295: F1836–F1844, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shahid M, Francis J, Matrougui K, and Majid DSA. Involvement of tumor necrosis factor-{alpha} in natriuretic response to systemic infusion of nitric oxide synthase inhibitor in anesthetized mice. Am J Physiol Renal Physiol 299: F217–F224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sharma K, Cook A, Smith M, Valancius C, and Inscho EW. TGF-β impairs renal autoregulation via generation of ROS. Am J Physiol Renal Physiol 288: F1069–F1077, 2005 [DOI] [PubMed] [Google Scholar]
- 160.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, and Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension 56: 297–303, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Silva MT. When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol 87: 93–106, 2010 [DOI] [PubMed] [Google Scholar]
- 162.Sima AV, Stancu CS, and Simionescu M. Vascular endothelium in atherosclerosis. Cell Tissue Res 335: 191–203, 2009 [DOI] [PubMed] [Google Scholar]
- 163.Sriramula S, Cardinale JP, Lazartigues E, and Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res 92: 401–408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sriramula S, Haque M, Majid DSA, and Francis J. Involvement of tumor necrosis factor-{alpha} in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, and Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A 92: 3893–3897, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stoos BA, Garcia NH, and Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol 6: 89–94, 1995 [DOI] [PubMed] [Google Scholar]
- 167.Stumpf C, John S, Jukic J, Yilmaz A, Raaz D, Schmieder RE, Daniel WG, and Garlichs CD. Enhanced levels of platelet P-selectin and circulating cytokines in young patients with mild arterial hypertension. J Hypertens 23: 995–1000, 2005 [DOI] [PubMed] [Google Scholar]
- 168.Suda T, Okazaki T, Naito Y, Yokota T, Arai N, Ozaki S, Nakao K, and Nagata S. Expression of the Fas ligand in cells of T cell lineage. J Immunol 154: 3806–3813, 1995 [PubMed] [Google Scholar]
- 169.Sun C, Li H, Gao Y, Matsuura T, Upchurch PA, Raizada MK, and Sumners C. Lack of macrophage migration inhibitory factor regulation is linked to the increased chronotropic action of angiotensin II in SHR neurons. Hypertension 49: 528–534, 2007 [DOI] [PubMed] [Google Scholar]
- 170.Suzuki J, Jin ZG, Meoli DF, Matoba T, and Berk BC. Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ Res 98: 811–817, 2006 [DOI] [PubMed] [Google Scholar]
- 171.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A 84: 523–528, 1976 [DOI] [PubMed] [Google Scholar]
- 172.Svendsen UG. Spontaneous hypertension and hypertensive vascular disease in the NZB strain of mice. Acta Pathol Microbiol Scand A 85: 548–554, 1977 [DOI] [PubMed] [Google Scholar]
- 173.Tang PCY, Qin L, Zielonka J, Zhou J, Matte-Martone C, Bergaya S, van Rooijen N, Shlomchik WD, Min W, Sessa WC, Pober JS, and Tellides G. MyD88-dependent, superoxide-initiated inflammation is necessary for flow-mediated inward remodeling of conduit arteries. J Exp Med 205: 3159–3171, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tatsukawa Y, Hsu WL, Yamada M, Cologne JB, Suzuki G, Yamamoto H, Yamane K, Akahoshi M, Fujiwara S, and Kohno N. White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens Res 31: 1391–1397, 2008 [DOI] [PubMed] [Google Scholar]
- 175.Thabet SR, Wu J, Chen W, Marvar PJ, Gongora MC, Madhur MS, Blinder Y, Weyand C, and Harrison DG. The role of CD8+ T cells, IP-10 and MMP12 in hypertension. Hypertension 58: e49 Abstract, 2011 [Google Scholar]
- 176.Thannickal VJ. and Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem 270: 30334–30338, 1995 [DOI] [PubMed] [Google Scholar]
- 177.Thway TM, Shlykov SG, Day MC, Sanborn BM, Gilstrap LC, 3rd, Xia Y, and Kellems RE. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation 110: 1612–1619, 2004 [DOI] [PubMed] [Google Scholar]
- 178.Tian N, Gu JW, Jordan S, Rose RA, Hughson MD, and Manning RD., Jr.Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 292: H1018–H1025, 2007 [DOI] [PubMed] [Google Scholar]
- 179.Tian N, Moore RS, Braddy S, Rose RA, Gu JW, Hughson MD, and Manning RD., Jr.Interactions between oxidative stress and inflammation in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 293: H3388–H3395, 2007 [DOI] [PubMed] [Google Scholar]
- 180.Ungvari Z, Wolin MS, and Csiszar A. Mechanosensitive production of reactive oxygen species in endothelial and smooth muscle cells: role in microvascular remodeling? Antioxid Redox Signal 8: 1121–1129, 2006 [DOI] [PubMed] [Google Scholar]
- 181.van der Veen RC, Dietlin TA, Karapetian A, Holland SM, and Hofman FM. Extra-cellular superoxide promotes T cell expansion through inactivation of nitric oxide. J Neuroimmunol 153: 183–189, 2004 [DOI] [PubMed] [Google Scholar]
- 182.Vecchione C, Patrucco E, Marino G, Barberis L, Poulet R, Aretini A, Maffei A, Gentile MT, Storto M, Azzolino O, Brancaccio M, Colussi GL, Bettarini U, Altruda F, Silengo L, Tarone G, Wymann MP, Hirsch E, and Lembo G. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kgamma. J Exp Med 201: 1217–1228, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, and Ryan MJ. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Venugopal SK, Devaraj S, Yuhanna I, Shaul P, and Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation 106: 1439–1441, 2002 [DOI] [PubMed] [Google Scholar]
- 185.Verlohren S, Muller DN, Luft FC, and Dechend R. Immunology in hypertension, preeclampsia, and target-organ damage. Hypertension 54: 439–443, 2009 [DOI] [PubMed] [Google Scholar]
- 186.Verma S, Li SH, Badiwala MV, Weisel RD, Fedak PW, Li RK, Dhillon B, and Mickle DA. Endothelin antagonism and interleukin-6 inhibition attenuate the proatherogenic effects of C-reactive protein. Circulation 105: 1890–1896, 2002 [DOI] [PubMed] [Google Scholar]
- 187.Verma S, Wang CH, Li SH, Dumont AS, Fedak PW, Badiwala MV, Dhillon B, Weisel RD, Li RK, Mickle DA, and Stewart DJ. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation 106: 913–919, 2002 [DOI] [PubMed] [Google Scholar]
- 188.Vital SA, Terao S, Nagai M, and Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation 17: 641–649, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, Szmitko P, Li RK, Mickle DA, and Verma S. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation 107: 1783–1790, 2003 [DOI] [PubMed] [Google Scholar]
- 190.Weber C, Erl W, Pietsch A, Strobel M, Ziegler-Heitbrock HW, and Weber PC. Antioxidants inhibit monocyte adhesion by suppressing nuclear factor-kappa B mobilization and induction of vascular cell adhesion molecule-1 in endothelial cells stimulated to generate radicals. Arterioscler Thromb 14: 1665–1673, 1994 [DOI] [PubMed] [Google Scholar]
- 191.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, and Münzel T. Lysozyme M–positive monocytes mediate angiotensin II–induced arterial hypertension and vascular dysfunction/clinical perspective. Circulation 124: 1370–1381, 2011 [DOI] [PubMed] [Google Scholar]
- 192.Wever RM, van Dam T, van Rijn HJ, de Groot F, and Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun 237: 340–344, 1997 [DOI] [PubMed] [Google Scholar]
- 193.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol 289: R913–R935, 2005 [DOI] [PubMed] [Google Scholar]
- 194.Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, Navar LG, and Lazartigues E. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One 6: e22682, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yamada S, Gotoh T, Nakashima Y, Kayaba K, Ishikawa S, Nago N, Nakamura Y, Itoh Y, and Kajii E. Distribution of serum C-reactive protein and its association with atherosclerotic risk factors in a Japanese population: Jichi Medical School Cohort Study. Am J Epidemiol 153: 1183–1190, 2001 [DOI] [PubMed] [Google Scholar]
- 196.Yan G, You B, Chen SP, Liao JK, and Sun J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ Res 103: 591–597, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yang B. and Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 292: H954–H962, 2007 [DOI] [PubMed] [Google Scholar]
- 198.Yasunari K, Maeda K, Nakamura M, and Yoshikawa J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and C-reacting protein. Hypertension 39: 777–780, 2002 [DOI] [PubMed] [Google Scholar]
- 199.Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, and Oettgen P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest 115: 2508–2516, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, and Crowley SD. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res 110: 1604–1617, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhong J, Guo D, Chen CB, Wang W, Schuster M, Loibner H, Penninger JM, Scholey JW, Kassiri Z, and Oudit GY. Prevention of angiotensin II–mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 57: 314–322, 2011 [DOI] [PubMed] [Google Scholar]
- 202.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, and Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91: 1038–1045, 2002 [DOI] [PubMed] [Google Scholar]