

Abstract
The β-catenin signaling axis is critical for normal embryonic development and tissue homeostasis in adults. We have previously shown that extracellular enzyme transglutaminase 2 (TG2) activates β-catenin signaling in vascular smooth muscle cells (VSMCs). In this study, we provide several lines of evidence that TG2 functions as an activating ligand of the LRP5/6 receptors. Specifically, we show that TG2 synergizes with LRP6 in activation of β-catenin-dependent gene expression in Cos-7 cells. Interfering with the LRP5/6 receptors attenuates TG2-induced activation of β-catenin in Cos-7 cells. Further, we show that TG2 binds directly to the extracellular domain of LRP6, which is also able to act as a substrate for TG2-mediated protein cross-linking. Furthermore, inhibitors of TG2 protein cross-linking quench the observed TG2-induced β-catenin activation, implicating protein cross-linking as a novel regulatory mechanism for this pathway. Together, our findings identify and characterize a new activating ligand of the LRP5/6 receptors and uncover a novel activity of TG2 as an agonist of β-catenin signaling, contributing to the understanding of diverse developmental events and pathological conditions in which transglutaminase and β-catenin signaling are implicated.
Keywords: Transglutaminsase 2, LRP5/6, β-catenin
1. INTRODUCTION
Wnt/β-catenin signaling is a key signal transduction pathway in normal development and disease [1]. The “canonical” pathway is activated by the binding of a secreted lipid-modified glycoprotein from the Wnt protein family [2] to a cell surface receptor complex, consisting of a serpentine receptor from the Frizzled (Fzd) family and a low-density-lipoprotein-related protein co-receptor LRP5 or LRP6 (LRP5/6). Binding of a Wnt ligand leads to LRP5/6 oligomerization and phosphorylation of its cytoplasmic tail [3–5]. This results in recruitment of Dishevelled, thereby disrupting the destruction complex containing Axin, Adenomatosis Polyposis Coli (APC) and Glycogen Synthase Kinase 3 (GSK3), which targets β-catenin for proteasomal degradation. Stabilized β-catenin can then translocate to the nucleus, where it forms a complex with transcription factors of the TCF/LEF family and activates expression of target genes [6].
Mutations in LRP5/6 signaling are associated with bone disorders, abnormal ocular vascularization, early onset cardiovascular disease and metabolic syndromes [5;7;8]. However, not all of these phenotypes may be attributable to altered activation by ligands of the Wnt family. For instance, during ocular vascularization activation of canonical β-catenin signaling is mediated by a Norrin-LRP5-Fzd interaction [9]. Activation by Norrin utilizes both the LRP5/6 and Fzd-4 co-receptors, similar to Wnt ligands; however, Fzd-independent mechanisms of β-catenin activation have also been reported. For instance, PTH activates β-catenin through the LRP6/PTHR1 complex [10], while cysteine knot proteins SOST/Sclerostin and WISE bind directly to LRP5/6 to either inhibit or activate signaling in a context-dependent manner [11–13]. Similarly, Dkk1, a protein of the Dickkopf family, binds to LRP6 and antagonizes signaling by preventing ligand binding [14]. The multiplicity of LRP5/6-regulated biological processes suggests that identification of novel interacting ligands will advance understanding of diverse and significant aspects of normal development and its pathological disruptions.
Previously, we have shown that extracellular enzyme tissue transglutaminase (TG2) activates β-catenin signaling in primary mouse vascular smooth muscle cells (VSMCs) and interacts with the LRP5 receptor on the cell surface [15]. In mammals, eight Ca2+-dependent transglutaminases (TGases) have been identified, comprising a family of enzymes that catalyze formation of intra- or intermolecular glutamyl-lysine-protein cross-links. TG2 is unique owing to its ability to catalyze more than one enzymatic reaction and interact with a number of cell-surface receptors upon externalization into the extracellular matrix (ECM) [16]. Inside the cell, TG2 is able to act as a deaminase, GTPase, protein kinase and protein disulphide isomerase [17]. While in the ECM TG2 can interact with β1 and β3 integrins [18], the atypical G-protein-coupled receptor GPR56 [19], VEGF receptor 2 [20] and LRP5 [15]. We have recently shown that the protein cross-linking catalytic activity of TG2 is critical for warfarin-induced activation of β-catenin signaling in VSMCs [21]. Taking into consideration that TG2 is often externalized and activated in response to tissue insult [22] and that this often correlates with activation of β-catenin signaling in disease, further identification and characterization of biologically relevant TG2-receptor interactions is important for a better understanding of cardiovascular disease and other conditions.
The previously reported interaction of TG2 with LRP5 on the cell surface of osteogenic cells [15] supports the hypothesis that induction of the β-catenin signaling pathway by TG2 is mediated by the LRP5/6 receptors. However, recent findings implied that TG2 activates β-catenin signaling through a c-Src-dependent mechanism [23], thus putting the role of LRP5/6 into question. In this study we found that TG2 binds directly to and cross-links the LRP5/6 receptor. Moreover, we found that LRP5/6 is required for TG2-induced activation of β-catenin. These results may help us to understand the intertwined diversity of the biological functions of LRP5/6 and TG2 and suggest oligomerization of the LRP receptor by cross-linking as a mechanism of activation.
2. EXPERIMENTAL PROCEDURES
2.1 Cell Culture, Plasmid constructs, DNA Transfection and Luciferase Assay
Cos-7 cells [24] obtained from ATCC and primary mouse embryonic fibroblasts (MEFs) from LRP5 null mice and their wild type siblings were maintained in Dulbecco’s modified Eagle’s medium supplemented with 100 U/mL of penicillin G, 100 μg/mL of streptomycin, and 10% fetal bovine serum (complete DMEM) in 5% CO2 at 37°C. Cells stably carrying TCF/LEF firefly luciferase reporter and constitutively active renilla luciferase reporter were established using lentivirus-based constructs, according to the manufacturer’s instructions (SABiosciences). Puromycin-resistant colonies were expanded and maintained in complete DMEM supplemented with 1μg/ml puromycin. For assays cells were seeded at 5×103 cells/well in 96-well plates and transiently transfected with plasmids for expression of LRP5, LRP6, TG2, Dkk1 and Wnt3a, using FuGene 6 (Roche). Where indicated in text, MEFs were also transfected with dsRNA targeting LRP5 or LRP6 using HiPerfect reagent (Qiagen). Dual luciferase assay was performed two days post-transfection to measure the luciferase activity. Renilla activity was used as the internal normalization control. In separate experiments, Cos-7 cells were transiently transfected with TCF/LEF luciferase reporter construct TOP-FLASH and a β-galactosidase expression plasmid (Upstate) together with other expression constructs. In this case the β-galactosidase activity, measured using specific substrate (Promega), was used as the reference to normalize for transfection efficiency. When the medium was supplemented with TG2 protein, the medium was changed to 1% serum 24 hr post-transfection, and supplemented with purified guinea pig liver TG2 (gpTG2) (Sigma) (0.01U/ml, unless indicated otherwise) or purified recombinant human TG2 (N-zyme, 300 ng/ml) (N-Zyme), and cells cultured for 48 hours. The plasmid for transient expression of secreted human TG2 was previously described [25]. Expression constructs for GST-TG2 fusion proteins were kindly provided by Dr. A Belkin (University of Maryland). Plasmids for transient expression of human LRP5, LRP6 and Wnt3a were kindly provided by Dr. X He (Harvard Medical School). Expression construct for Dkk1 was a gift from Dr. F Long (University of St. Louis).
2.2 Western Blot Analysis and Immunoprecipitation
For Western blotting, antibodies against LRP6 (clone C-10, Santa Cruz), and phospho-LRP6 (Ser1490) (Antibody #2568, Cell Signaling) were used at a 1:1000 dilution. Secondary anti-mouse or anti-rabbit HRP-conjugated antibodies (Pierce) were used at a 1:10,000 dilution. The presence of His-tagged proteins was detected using the HRP-conjugated His probe (Pierce) at a 1:10,000 dilution. HRP-conjugated proteins were detected by the chemiluminescence method (Pierce). For immunoprecipitation, antibodies against GST (clone GST-2, Sigma) or TG2 (Abcam) were utilized and protein A/G-Sepharose (Pierce) beads were used to purify the protein complexes.
2.3 TG2 Over-Expression in Zebrafish Larvae
Expression construct encoding externalized human TG2 in RCASBP(A) vector was used [25]. The TG2 open reading frame was excised from the RCAS-TG2 vector with ClaI restriction digest and inserted into the ClaI site of the pCS2+ vector. For RNA synthetic reaction pCS2+-TG2 was linearized with NSiI and mMessage mMachine (Ambion) was used to synthesize RNA as directed by the manufacturer. RNA was cleaned with MEGAclear kit (Ambion). RNA was dissolved in water to a concentration of 60μg/mL and a final concentration of 0.1% phenol red was added to allow for visualization during injections. Approximately, 1μL of RNA was injected into zebrafish embryos at the 2–8 cell stage. Embryos were fixed in 4% PFA 24 hours after injection and analyzed by microscopy with a Nikon T-80 microscope equipped with SPOT RT slider real-time CCD camera. 83 embryos were analyzed.
2.4 TG Activity Assay
TG cross-linking activity was assayed as previously described [26]. 96-well microtiter plates (Maxisorp NUNC) were incubated overnight with 250μl of 1mg/ml N,N′-Dimethylcasein (Sigma-Aldrich) in 5mM Sodium Carbonate (pH 9.8), and blocked with 200μL of 0.1% bovine serum albumin (BSA) (HyClone) in 5mM Sodium Carbonate (pH 9.8) for one hour at 37°C. LRPΔC (R & D Systems), purified guinea pig liver TG2 (80ng) (Sigma-Aldrich) and Ez-link Pentylamine-Biotin (2.5mM) (Pierce) were incubated at 37°C for one hour in reaction buffer (100mM Tris-HCl pH 8.5, 6.7mM CaCl2, 13.3mM DTT) with increasing molar concentrations of LRPΔC. Incorporated Ez-link Pentylamine-Biotin was detected with 1:5000 ExtrAvidin-Peroxidase (Sigma) and Super AquaBlue ELISA Substrate (eBioscience) followed by reading the absorbance at 405nm on a Polarstar Optima plate reader.
2.5 Statistical Tests
For all figures, error bars represent Standard Errors and P values were calculated by a one-tailed student’s T test (*p<0.05, **p<0.01). At least three independent replicates were performed for each experiment.
3. RESULTS
3.1 Extracellular TG2 Activates β-Catenin Signaling in Cos-7 cells
Previously, we have shown that exogenously purified TG2 activates β-catenin signaling in mouse VSMCs [15]. In this study, we sought to identify the target position for TG2 activity in the β-catenin signaling pathway. For this, we monitored activation of this pathway using a luciferase reporter driven by TCF/LEF-binding promoter (TOP-FLASH) in Cos-7 cells, which are conventionally used for the analysis of β-catenin signaling. Similar to its effect on mouse VSMCs, extracellular TG2 purified from guinea pig liver tissue (gpTG2) induced activation of β-catenin in Cos-7 cells in a dose-dependent manner (Fig. 1A). We examined levels of gpTG2 ranging from 1.5×10−4 to 1×10−2 U/ml as this is within the range of endogenous TG2 levels in smooth muscle cells [27]. To confirm that this effect was specific to TG2 and not due to possible contaminants from the liver tissue, we showed a similar induction of the TOP-FLASH reporter with recombinant human TG2 purified from E coli (Fig. 1B, hTG2). In addition, over-expression of human TG2, fused to a secretory peptide to assure its externalization, also activated the TOP-FLASH reporter in Cos-7 cells (Fig. 1B, sTG2). In aggregate, these observations strongly indicate that extracellular TG2 can serve as an activator of β-catenin signaling.
Fig 1. Extracellular TG2 activates β-catenin signaling in Cos-7 cells.

(A) Dose-dependent activation of TOP-FLASH expression by purified gpTG2. (B) Exogenous recombinant human TG2 (hTG2, 100ng/mL) and overexpression of secreted human TG2 (sTG2) activate TOP-FLASH expression in Cos-7 cells. (C) Phosphorylation of the cytoplasmic domain of LRP6 is observed in LRP6-overexpressing Cos-7 cells treated for 4 hours in serum-free medium with 0.01U/mL purified gpTG2 or with 200 ng/mL canonical Wnt3a (Positive Control). (D) Treatment of Cos-7 cells with 100μM small molecule inhibitor FJ9 or overexpression of Dkk1 (30ng of expression plasmid/well) quenches activation of β-catenin by gpTG2.
Activation of canonical β-catenin signaling by Wnt ligands results in phosphorylation of the serine residue in the NPPPSPATE motif of the intracellular LRP6 domain [5]. This activation can be inhibited by extracellular DKK proteins [28] and by the small molecule inhibitor FJ9, which specifically disrupts intracellular interactions of the Dishevelled protein [29]. Similar to the canonical ligand Wnt3a, extracellular gpTG2 strongly induces phosphorylation of LRP6 expressed in Cos-7 cells (Fig. 1C). Moreover, over-expression of Dkk1 or addition of FJ9 to the medium blunted the response of TOP-FLASH to exogenous gpTG2 (Fig. 1D). These observations provide evidence that TG2 utilizes the canonical intracellular components to activate β-catenin signaling.
3.2 TG2-induced activation of β-Catenin is mediated by LRP6
Previous studies have shown that purified TG2 binds to the LRP5 receptor on the cell surface of VSMCs [15]. Since both LRP5 and its homologue LRP6 play a key role in canonical Wnt/β-catenin signaling, we hypothesized that these receptors also mediate activation of β-catenin by TG2. To test this we analyzed the synergistic effects of TG2 and LRP6. Singular over-expression of LRP6 in Cos-7 cells did not affect the baseline level of β-catenin activity, but it significantly augmented gpTG2-mediated induction of β-catenin activity as shown by the TOP-FLASH reporter (Fig. 2A). By transfecting increasing amounts of LRP6-coding plasmid DNA into Cos-7 cells, we show that the stimulating effect of LRP6 on gpTG2-induced activation of the TOP-FLASH reporter was dose-dependent (Fig. 2B). Thus, our experiments with over-expression of LRP6 support LRP6 as a mediator of TG2-dependent activation of the β-catenin pathway.
Fig 2. TG2-induced activation of β-catenin is mediated by LRP6.
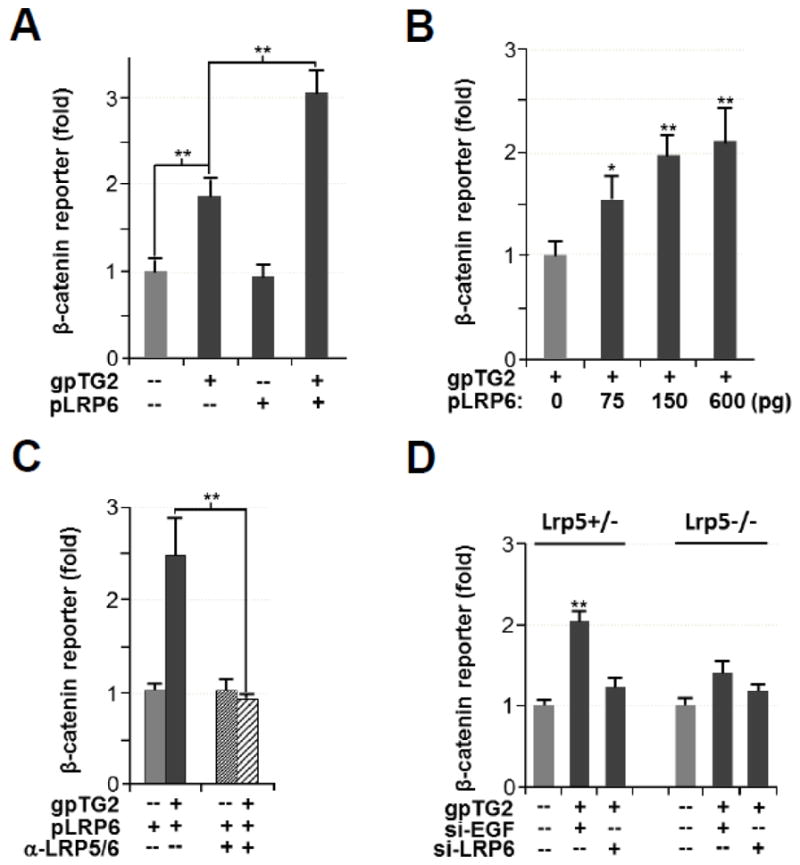
(A) Addition of exogenous purified gpTG2 activates β-catenin as shown by TOP-FLASH expression, which is augmented by overexpression of LRP6 in Cos-7 cells. (B) Dose-dependent induction of gpTG2-mediated β-catenin activation by LRP6. (C) LRP6 augmentation of gpTG2-induced β-catenin activation is inhibited by a monoclonal anti-LRP5/6 antibody 1A12, which blocks LRP6 receptor binding. (D) gpTG2-mediated activation of β-catenin signaling by LRP5+/− and LRP5−/− fibroblasts. Treatment with LRP6 shRNA shows that both LRP5 and LRP6 are required for this activation.
To complement this gain-of-function approach we performed loss-of-function studies, in which we employed a monoclonal anti-LRP5/6 antibody 1A12 to block LRP6 receptor overexpressed in Cos-7 cells [13]. As expected, blocking the LRP5/6 receptor with this antibody quenched the responsiveness of β-catenin signaling to exogenous gpTG2 (Fig. 2C). Next we employed LRP5+/− and LRP5−/− mouse fibroblasts [30] to determine the contribution of individual LRP5 and LRP6 receptors in mediating the effects of TG2. Exogenous gpTG2 significantly induced β-catenin reporter expression in LRP5+/− fibroblasts and when they were treated with LRP6 siRNA this induction was abrogated (fig. 2D). This observation suggests that LRP6 is a mediator of TG2-induced β-catenin signaling. Comparison of gpTG2-mediated induction between the LRP5+/− and LRP5−/− shows a reduced but still present induction in β-catenin reporter expression, suggesting that both LRP5 and LRP6 participate in TG2-mediated β-catenin activation.
In conclusion, these experiments demonstrate that LRP5/6 mediate extracellular TG2 induced activation of β-catenin signaling.
3.3 TG2 Over-Expression Mirrors β-Catenin Over-Activation Phenotype in Zebrafish
Previous studies have observed that over-activation of β-catenin signaling in zebrafish larvae results in a ventral phenotype of small or no eyes [31]. Here we tested whether the demonstrated TG2-mediated activation of β-catenin signaling has similar biological effects. Zebrafish embryos were injected with GFP (negative control) or secreted human TG2 (hTG2) RNA before they reached the 8-cell stage and the resulting phenotype was analyzed at 1day post fertilization (dpf), paying particular attention to the eyes. Upon injection of hTG2 we observed two phenotypes: the Wnt-like phenotype of small or no eyes (Figure 3A, middle-right panels) and an additional phenotype of curled tails (Figure 3A, top-right panel). Interestingly, a small population of hTG2 injected larvae showed both phenotypes (Figure 3A, bottom-right panel). Using a chi squared statistical analysis we were able to determine that these two phenotypes were statistically independent events, indicating that TG2 is affecting other developmental pathways in addition to β-catenin signaling and that the curled tail phenotype is independent of the Wnt-like phenotype of small/no eyes. Quantification of these injected larvae showed that approximately 50% of TG2 injected embryos showed a mutated phenotype, and of these embryos approximately 15% demonstrated a non-Wnt-like phenotype whereas approximately 85% showed a no/small eyes phenotype (Wnt-like phenotype) (Figure 3B). Importantly, these results demonstrate the in vivo activation of β-catenin signaling through TG2.
Fig 3. TG2 Over-Expression Mirrors β-Catenin Over-Activation Phenotype in Zebrafish.
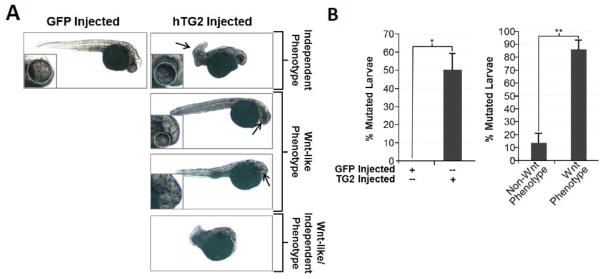
(A) Representative images of GFP (negative control) and hTG2 RNA injected larvae 1day post fertilization with arrows highlighting resulting phenotypes. (B) Quantitation of mutant phenotype occurrence in GFP and TG2 injected embryos. Left panel: 50% of TG2 injected embryos show a mutated phenotype compared to the 0% in GFP injected embryos. Right panel: Of the 50% mutated TG2 injected embryos, ~85% showed a no/small eye phenotype (Wnt Phenotype), while ~15% showed non-Wnt-like phenotype.
3.4 TG2 binds to the extracellular domain of LRP6 in vitro
Our data suggest that extracellular TG2 acts as an agonist of β-catenin signaling through an interaction with the LRP6 receptors. To determine whether TG2 binds directly to LRP6 or regulates signaling through other LRP5/6-interacting protein(s), we employed a co-immunoprecipitation analysis using purified gpTG2 and purified recombinant extracellular domain of human LRP6 (LRP6 C) carrying a His-tag. We found that incubation of LRP6 C and gpTG2 resulted in LRP6 C specifically co-precipitating with an anti-TG2 antibody while no precipitation occurred in the absence of gpTG2 (Fig. 4A), thus demonstrating that TG2 directly binds to the extracellular domain of LRP6. To further investigate which domain of TG2 is involved in these interactions, we analyzed binding of the LRP6 C protein to two truncated TG2 mutants along with full-sized TG2 (Fig 4B, upper panel, [32]). TG2 variants were expressed as GST fusions in E. coli and purified by affinity chromatography. These proteins were incubated with purified LRP6 C and co-immunoprecipitated with an anti-GST antibody. Co-immunoprecipitation of LRP6 C and TG2 was observed for full-size TG2-GST and for the truncated mutant lacking the two C-terminal β-barrel domains (Fig. 4B, bottom panel). However, the TG2N-GST mutant protein lacking both the β-barrel domains and the catalytic core domain failed to co-precipitate LRP6 C. Thus, the central catalytic core domain of TG2 is essential for binding of this protein to the extracellular domain of LRP6 raising the question of whether LRP6 is able to act as a substrate for TG2 cross-linking.
Fig 4. TG2 binds to the extracellular domain of LRP6 in vitro.

(A) Purified, recombinant His-tagged extracellular LRP6 domain (LRP C, 1 μg) was incubated with purified gpTG2 (1 μg). Precipitation with anti-TG2 antibody and detection by western blot with anti-His antibody showed an interaction between TG2 and LRP C. (B) Two truncated forms of TG2, TG2 C-GST and TGN-GST, or full length TG2 were incubated with LRP C. Precipitation with anti-GST antibody and detection by western blot with anti-His antibody revealed that TG2 core domain is necessary for this interaction.
3.5 Extracellular domain of LRP6 is cross-linked by TG2
TG2 was originally characterized as a protein cross-linking enzyme, in which it catalyzes the formation of an isopeptide bond between lysine and glutamine residues [33]. To examine whether LRP6 is a substrate for TG2-mediated cross-linking we performed a competition assay with LRP6. For this we tested whether purified LRP6ΔC acts as a competitive inhibitor of TG2 enzymatic activity using an ELISA-based assay of pentylamine cross-linking to casein by purified gpTG2. This assay showed that irreversible cross-linking of biotin-labeled EZ-link pentylamine to casein-coated plates was reduced in a dose-dependent manner by the addition of purified recombinant LRP6ΔC (Fig. 5A). The addition of LRP6ΔC to the EZ-link substrate mixture at 2:1 ratio caused an approximately 50% reduction in incorporation of EZ-link amide and increasing this ratio to 4:1 almost completely inhibited incorporation of EZ-link pentylamine. These data suggest that LRP6ΔC acts as a competitive inhibitor to the pentylamine substrate and is likely cross-linked by TG2. This was confirmed by Western blot analysis, in which high molecular weight-complexes of LRP6ΔC formed in the presence of purified gpTG2 (Fig. 4B, arrows). Further, we showed that the cross-linking enzymatic activity of TG2 is required for LRP6-mediated activation of β-catenin signaling by TG2. Treatment of LRP6-over-expressing Cos-7 cells with the TG2-specific irreversible small molecule inhibitor KCC-009 [34] significantly attenuated the activating effect of TG2 on TOP-FLASH expression (Fig. 4C), restoring it to baseline. Altogether these results suggest that LRP6 is a substrate for TG2-mediated cross-linking and that this cross-linking activity is pertinent to activation of β-catenin signaling.
Fig 5. Extracellular domain of LRP6 is cross-linked by TG2.

(A) TG2 activity assay performed using stable levels of TG2 and Ez-link pentylamine with increasing doses of LRP6 shows that LRP6 inhibits TG2 cross-linking activity. (B) Western blot analysis of LRP6ΔC alone (left panel) or an LRP6ΔC/TG2 mixture (right panel) reveals formation of LRP6 complexes in the presence of gpTG2. (C) Induction of β-catenin activity by TG2 can be blocked by treatment with a TG2-specific inhibitor, KCC009.
4. DISCUSSION
Our previous finding that extracellular TG2 activates canonical β-catenin signaling in VSMCs [15] suggested that TG2 is able to bind to a receptor pertinent to β-catenin signaling. The receptors and corresponding ligands that regulate the molecular pathways of β-catenin activation and signaling are diverse [35]; however, our studies strongly indicate that TG2 acts through the canonical LRP5/6 receptors. TG2 shows synergy with LRP6 in activation of β-catenin-dependent gene expression in the Cos-7 model system. Blocking LRP5/6 receptors by antibody incubation or Dkk1over-expression - a known inhibitor of LRP5/6-mediated signaling - quenches the effect of TG2 on β-catenin activity. Similarly, in VSMCs blocking LRP5/6 receptors with an antibody or inhibition of LRP5/6-mediated signaling with the soluble extracellular domain of LRP6 attenuates TG2-induced mineralization, which is associated with β-catenin activation in these cells [15]. These observations are consistent with LRP5/6 being the major receptors for TG2-regulated β-catenin activation. Indeed, our results demonstrate direct binding of TG2 to the extracellular domain of LRP6 in vitro and indicate that this binding requires the TG2 catalytic core domain. Intriguingly, the catalytic domain of TG2 has also been implicated in binding to LRP1, another member of the LRP family [36]. While LRP1 has rather weak sequence similarity with LRP5/6, 35% identity in the most conserved regions, the organization of the extracellular domains of these receptors shows similar structural motifs including β-propeller/EGF repeat modules and LDLR repeats [37]. Even though the LRP1 domain capable of TG2 binding has not been defined [36], our finding that TG2 directly interacts with the extracellular domain of LRP6 in combination with previous observations of a LRP1-TG2 interaction suggests that TG2 interacts with the structurally similar extracellular domains of the LRP family and thus has the potential to regulate the diverse biological activities of these receptors.
We observed that binding of TG2 to the extracellular domain of LRP6 results in phosphorylation of its intracellular domain suggesting that the mechanism of LRP5/6-mediated signaling by TG2 is similar to that of the canonical ligands (e.g. canonical Wnts). Inhibition of TG2-induced β-catenin signaling by the small molecule inhibitor FJ9, which disrupts Dishevelled-dependent nuclear translocation of β-catenin without affecting the non-canonical JNK-mediated β-catenin pathway [29] further supports that TG2 acts in a manner similar to that of canonical ligands. Binding of the TG2 catalytic domain to other proteins can lead to TG2-mediated cross-linking of the binding partners, as indicated by studies on human papillomavirus protein E7 [38]. We found that LRP6 is also a substrate for TG2-mediated cross-linking. Incubation of the extracellular domain of LRP6 with TG2 resulted in two bands of higher molecular weight and one band at the correct size for LRP6. It is probable that the band around 200 kDa is TG2 cross-linked to LRP6, based on the molecular weight, while the highest band is likely to be a multimer of LRP6. The biological significance of this modification is suggested by the quenching effect of cross-linking inhibitors on TG2-induced activation of β-catenin. Moreover, in an artificial model system oligomerization of LRP6 can activate β-catenin signaling and bypass the requirement for the Fzd co-receptors [3;4]. While further experiments will determine whether this mechanism is indeed in place for TG2-induced β-catenin activation, our studies identify extracellular TG2 as an activating ligand of the LRP5/6 receptors. This finding has broad significance for the understanding of diverse developmental processes and pathological conditions where both transglutaminase and β-catenin signaling have been implicated, including vascular calcification [15], osteogenesis [39;40], neurodegeneration [41;42] and cancers such as melanoma [43;44], prostate cancer [45;46] and breast cancer [47;48]. Specifically, the identification of a novel activator for β-catenin signaling presents a new target for pharmacological treatment of such ailments. This is particularly advantageous as identification of specific biological inhibitors for Wnt-mediated β-catenin signaling have yet to be identified [49].
5. CONCLUSIONS
In conclusion we have shown that extracellular TG2 is able to activate the canonical β-catenin signaling pathway and that this activation is mediated by the LRP5/6 receptors. Further, we have shown that activation likely occurs through TG2-mediated cross-linking of the LRP5/6 receptors. These findings may provide an interesting new target for pharmacological treatment of pathologies linked to TG2 and β-catenin.
Highlights.
Extracellular transglutaminase 2 is able to activate β-catenin signaling
Extracellular transglutaminase 2 activates β-catenin signaling through the LRP5/6 receptors
LRP5/6 serves as a substrate for transglutaminase 2-mediated protein cross-linking
Acknowledgments
This study was supported by National Institute of Health, grant HL093305 (to M.N.). We are very grateful to Dr. B. Williams for sharing the LRP5 null embryonic fibroblasts used in these studies, to Dr. X He for providing the LRP6-expression vector and to Dr. S Du for providing zebrafish and facilities to perform injections.
Abbreviation List
- Fzd
Frizzled
- LRP5/6
LRP5 or LRP6
- APC
Adenomatosis Polyposis Coli
- GSK3
Glycogen Synthase Kinase 3
- TG2
Tissue Transglutaminase
- VSMCs
Vascular Smooth Muscle Cells
- TGases
Transglutaminases
- ECM
Extracellular Matrix
- MEFs
Mouse Embryonic Fibroblasts
- gpTG2
Guinea Pig Liver TG2
- hTG2
Hu man TG2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Nusse R. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 2.Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. Nature. 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- 3.Cong F, Varmus H. Proc Natl Acad Sci USA. 2004;101:2882–2887. doi: 10.1073/pnas.0307344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- 5.Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, He X. Mol Cell. 2004;13:149–156. doi: 10.1016/s1097-2765(03)00484-2. [DOI] [PubMed] [Google Scholar]
- 6.Kikuchi A, Yamamoto H, Kishida S. Cell Signal. 2007;19:659–671. doi: 10.1016/j.cellsig.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D, Lifton RP. Science. 2007;315:1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujino T, Asaba H, Kang MJ, Ikeda Y, Sone H, Takada S, Kim DH, Ioka RX, Ono M, Tomoyori H, Okubo M, Murase T, Kamataki A, Yamamoto J, Magoori K, Takahashi S, Miyamoto Y, Oishi H, Nose M, Okazaki M, Usui S, Imaizumi K, Yanagisawa M, Sakai J, Yamamoto TT. Proc Natl Acad Sci USA. 2003;100:229–234. doi: 10.1073/pnas.0133792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Cell. 2004;%19;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 10.Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Genes Dev. 2008;22:2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenov MV, He X. J Biol Chem. 2006;281:38276–38284. doi: 10.1074/jbc.M609509200. [DOI] [PubMed] [Google Scholar]
- 12.Itasaki N, Jones CM, Mercurio S, Rowe A, Domingos PM, Smith JC, Krumlauf R. Development. 2003;130:4295–4305. doi: 10.1242/dev.00674. [DOI] [PubMed] [Google Scholar]
- 13.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R. J Bone Miner Res. 2006;21:1738–1749. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 14.Semenov MV, Zhang X, He X. J Biol Chem. 2008;283:21427–21432. doi: 10.1074/jbc.M800014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faverman L, Mikhaylova L, Malmquist J, Nurminskaya M. FEBS Lett. 2008;582:1552–1557. doi: 10.1016/j.febslet.2008.03.053. [DOI] [PubMed] [Google Scholar]
- 16.Nurminskaya MV, Belkin AM. Int Rev Cell Mol Biol. 2012;294:1–97. doi: 10.1016/B978-0-12-394305-7.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorand L, Graham RM. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 18.Akimov SS, Krylov D, Fleischman LF, Belkin AM. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Begum S, Hearn JD, Hynes RO. Proc Natl Acad Sci USA. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dardik R, Inbal A. Exp Cell Res. 2006;312:2973–2982. doi: 10.1016/j.yexcr.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Beazley KE, Deasey S, Lima F, Nurminskaya MV. Arterioscler Thromb Vasc Biol. 2012;32:123–130. doi: 10.1161/ATVBAHA.111.237834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. PLoS One. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condello S, Cao L, Matei D. FASEB J. 2013 doi: 10.1096/fj.12-222620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gluzman Y. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 25.Nurminsky D, Shanmugasundaram S, Deasey S, Michaud C, Allen S, Hendig D, Dastjerdi A, Francis-West P, Nurminskaya M. Mech Dev. 2011;128:234–245. doi: 10.1016/j.mod.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trigwell SM, Lynch PT, Griffin M, Hargreaves AJ, Bonner PL. Anal Biochem. 2004;330:164–166. doi: 10.1016/j.ab.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 27.Ou H, Haendeler J, Aebly MR, Kelly LA, Cholewa BC, Koike G, Kwitek-Black A, Jacob HJ, Berk BC, Miano JM. Circ Res. 2000;87:881–887. doi: 10.1161/01.res.87.10.881. [DOI] [PubMed] [Google Scholar]
- 28.Niehrs C. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 29.Fujii N, You L, Xu Z, Uematsu K, Shan J, He B, Mikami I, Edmondson LR, Neale G, Zheng J, Guy RK, Jablons DM. Cancer Res. 2007;67:573–579. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]
- 30.Riddle RC, Diegel CR, Leslie JM, Van Koevering KK, Faugere MC, Clemens TL, Williams BO. PLoS One. 2013;8:e63323. doi: 10.1371/journal.pone.0063323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van de Water S, van de Wetering M, Joore J, Esseling J, Bink R, Clevers H, Zivkovic D. Development. 2001;128:3877–3888. doi: 10.1242/dev.128.20.3877. [DOI] [PubMed] [Google Scholar]
- 32.Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Blood. 2005;105:3561–3568. doi: 10.1182/blood-2004-10-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iismaa SE, Mearns BM, Lorand L, Graham RM. Physiol Rev. 2009;89:991–1023. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- 34.Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 35.MacDonald BT, Semenov MV, He X. Cell. 2007;131:1204. doi: 10.1016/j.cell.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 36.Zemskov EA, Mikhailenko I, Strickland DK, Belkin AM. J Cell Sci. 2007;120:3188–3199. doi: 10.1242/jcs.010397. [DOI] [PubMed] [Google Scholar]
- 37.Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman AR, Metzker ML, Caskey CT, Todd JA, Hess JF. Biochem Biophys Res Commun. 1998;248:879–888. doi: 10.1006/bbrc.1998.9061. [DOI] [PubMed] [Google Scholar]
- 38.Jeon JH, Choi KH, Cho SY, Kim CW, Shin DM, Kwon JC, Song KY, Park SC, Kim IG. EMBO J. 2003;22:5273–5282. doi: 10.1093/emboj/cdg495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurminskaya M, Magee C, Faverman L, Linsenmayer TF. Dev Biol. 2003;263:139–152. doi: 10.1016/s0012-1606(03)00445-7. [DOI] [PubMed] [Google Scholar]
- 40.Baron R, Rawadi G, Roman-Roman S. Curr Top Dev Biol. 2006;76:103–127. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- 41.Muma NA. J Neuropathol Exp Neurol. 2007;66:258–263. doi: 10.1097/nen.0b013e31803d3b02. [DOI] [PubMed] [Google Scholar]
- 42.Inestrosa N, De Ferrari GV, Garrido JL, Alvarez A, Olivares GH, Barria MI, Bronfman M, Chacon MA. Neurochem Int. 2002;41:341–344. doi: 10.1016/s0197-0186(02)00056-6. [DOI] [PubMed] [Google Scholar]
- 43.Fok JY, Ekmekcioglu S, Mehta K. Mol Cancer Ther. 2006;5:1493–1503. doi: 10.1158/1535-7163.MCT-06-0083. [DOI] [PubMed] [Google Scholar]
- 44.Larue L, Delmas V. Front Biosci. 2006;11:733–742. doi: 10.2741/1831. [DOI] [PubMed] [Google Scholar]
- 45.Davies G, Ablin RJ, Mason MD, Jiang WG. J Exp Ther Oncol. 2007;6:257–264. [PubMed] [Google Scholar]
- 46.Verras M, Sun Z. Cancer Lett. 2006;237:22–32. doi: 10.1016/j.canlet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 47.Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Oncogene. 2007;26:2459–2470. doi: 10.1038/sj.onc.1210035. [DOI] [PubMed] [Google Scholar]
- 48.Lindvall C, Bu W, Williams BO, Li Y. Stem Cell Rev. 2007;3:157–168. doi: 10.1007/s12015-007-0025-3. [DOI] [PubMed] [Google Scholar]
- 49.Voronkov A, Krauss S. Curr Pharm Des. 2013;19:634–664. doi: 10.2174/138161213804581837. [DOI] [PMC free article] [PubMed] [Google Scholar]