

Abstract
DNA repeat expansions can result in the production of toxic RNA. RNA toxicity has been best characterised in the context of myotonic dystrophy. Nearly 20 mouse models have contributed significant and complementary insights into specific aspects of this novel disease mechanism. These models provide a unique resource to test pharmacological, anti-sense, and gene-therapy therapeutic strategies that target specific events of the pathobiological cascade. Further proof-of-principle concept studies and preclinical experiments require critical and thorough analysis of the multiple myotonic dystrophy transgenic lines available. This review provides in-depth assessment of the molecular and phenotypic features of these models and their contribution towards the dissection of disease mechanisms, and compares them with the human condition. More importantly, it provides critical assessment of their suitability and limitations for preclinical testing of emerging therapeutic strategies.
Myotonic dystrophy: a paradigm of RNA toxicity
Myotonic dystrophy (dystrophia myotonica, DM) is the most common form of adult muscular dystrophy and includes at least two genetically distinct but clinically similar disease forms. DM type 1 (DM1) accounts for the majority of DM cases (traditionally >95%), but the prevalence of DM type 2 (DM2) is probably underestimated [1]. DM1 is a truly multisystem disorder, primarily affecting skeletal muscles, the heart and the central nervous system (CNS) (Box 1). The development of DM1 transgenic mice and identification of the DM2 mutation helped to elucidate a novel disease mechanism mediated by a toxic gain-of-function RNA transcript [2]. Expanded DMPK transcripts accumulate in the nucleus of DM1 cells [3], interfering with at least two antagonistic protein families that regulate alternative splicing throughout development: the muscleblind-like (MBNL) and CUGBP/Elav-like family (CELF) proteins [4–6]. MBNL1 function is lost due to sequestration by ribonuclear aggregates or foci [4,6], and CELF1 (or CUG-binding protein 1, CUGBP1) is upregulated [5,7,8] through protein stabilisation that is mediated by hyperphosphorylation [9]. MBNL1 sequestration and CELF1 upregulation result in aberrant expression of embryonic splicing profiles of MBNL1- and/or CELF1-regulated transcripts in adult skeletal muscle and heart (Figure 1a) [10–12]. Similarly, toxic CCUG-containing CNBP/ZNF9 transcripts sequester MBNL1 and disrupt splicing in DM2 [2,6]; their effect on CELF1 is still unclear [10,13,14]. CLCN1 chloride channel missplicing in skeletal muscles results in myotonia (delayed muscle relaxation after initial contraction) [15–17], whereas abnormal splicing of the insulin receptor (INSR) might contribute to insulin resistance [7]. Other missplicing events have been described and are likely to play a role in disease manifestations (Table S1 in the supplementary material online). MBNL proteins can also participate in RNA transcription, processing and stability [18,19], whereas CELF1 regulates protein translation [20–23]. Therefore, it is conceivable that the DM1 mutation might have an impact beyond splicing deregulation.
Box 1. DM1 as a multisystem disease: clinical profile, molecular genetics and trinucleotide repeat dynamics.
The great variability of DM1 symptoms and age of onset results in three main clinical forms of the disease: late-onset, classical adultonset and congenital DM1.
Myotonia (delayed muscle relaxation after initial contraction) and progressive wasting of distal muscles are prominent features of DM1 in skeletal muscle, and are accompanied by characteristic histopathological findings [88]. The more severe congenital form of DM1 is characterised by general muscle hypotonia and respiratory distress at birth, as well as delayed motor development. A large proportion of patients suffer from cardiac conduction blocks, detected by electrocardiogram (ECG), and cardiac histological abnormalities. Progressive cardiopathy can result in complete atrioventricular (AV) block or ventricular arrhythmias and subsequent sudden death in ∼30% of DM1 patients [88]. CNS manifestations are highly debilitating and support the view that DM1 is also a brain disorder [88,89]. DM1 neuropsychological dysfunction is accompanied by histological abnormalities, as well as brain structural changes and altered metabolism, as revealed by imaging techniques [89-91]. The impact of DM1 further affects a variety of tissues and results in presenile cataracts, abnormal glucose tolerance and hyperinsulinism, gastrointestinal dysfunction and testicular atrophy (Figure I) [88].
DM1 is caused by expansion of a CTG trinucleotide repeat in the 3′UTR of the DM protein kinase (DMPK) gene (Figure I) [92,93]. Repeat length is typically measured in blood samples and correlates positively with disease severity and inversely with age of onset. The expanded sequence is highly unstable, with a marked tendency to further expand during intergenerational transmissions and in somatic tissues. The mean CTG repeat size is usually larger in skeletal muscle than in blood [94]. Parent-to-child instability provides the molecular basis for anticipation, whereby the age of onset decreases and the disease severity increases in successive generations [95]. Tissue-specific, age-dependent, expansion-biased somatic mosaicism may contribute to phenotypic variability and disease progression [69]. DM2 is caused by expansion of an unstable CCTG tetranucleotide repeat within the first intron of the unrelated CCHC-type zinc finger, nucleic acid binding protein (CNBP) gene, previously named zinc finger protein 9 (ZNF9) gene [2].
Figure I.

Summary of DM1 symptoms, histopathology and genetics. Symptomatology: disease manifestations have been reported in skeletal muscle, heart, CNS, smooth muscle, eye lens and in many other tissues and organ systems. Histopathology: the main histopathological findings are in the skeletal muscle, heart and CNS of DM1 patients. Genetics: the DM1 locus, showing the DMWD, DMPK and SIX5 genes. The CTG repeat sequence maps within the 3′UTR of DMPK gene, which overlaps with the promoter region of the downstream sine oculis-related homeobox 5 (SIX5) gene [100]. Immediately upstream of DMPK lies the myotonic dystrophy WD repeat-containing (DMWD) gene [101]. In the unaffected population, the size of the CTG tract varies between 5 to 37 repeats. The severity of DM1 symptoms increases with the size of the repeat expansion: expansions of ∼50–100 CTG repeats result in a mild and late-onset form of DM1; expansions of ∼100–500 CTG repeats lead to the multisystemic adult form of DM1; expansions ∼500–4000 CTG repeats often result in juvenile and congenital DM1. These boundaries are not rigid and CTG repeat sizes can overlap to some extent between clinical forms of the disease.
Figure 1.
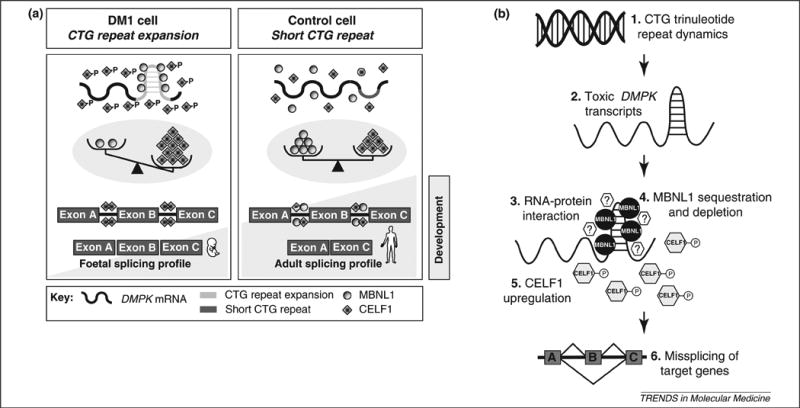
The toxic RNA gain-of-function model of DM1 molecular pathogenesis and molecular targets for rational DM1 therapies model. (a) Unaffected cells (right panel), carrying short CTG sequences, show a functional equilibrium between two antagonistic splicing regulators: muscleblind-like 1 (MBNL1) and CUGBP/Elav-like family member 1 (CELF1). The balance between MBNL1 and CELF1 plays a central role in the establishment of adult splicing profiles for a series of developmentally regulated genes. The CTG repeat expansion observed in a DM1 cell (left panel) forms an imperfect double-stranded structure that has at least two pathogenic consequences: MBNL1 sequestration by nuclear RNA foci and protein kinase C (PKC)-mediated CELF1 hyperphosphorylation and stabilisation. As a result of MBNL1 sequestration and CELF1 upregulation, the functional balance between these two splicing regulators is disturbed and the alternative splicing of a series of developmentally regulated genes reverts to a foetal profile. The abnormal expression of foetal splicing isoforms in adult skeletal muscle and heart is likely to contribute to the onset of DM1 disease symptoms. (b) Dissection of the molecular pathobiology of DM1 has identified molecular targets and potential strategies of therapeutic intervention. (1) Expansion-biased CTG trinucleotide repeat dynamics can be modified genetically or pharmacologically to stabilise the repeat or induce repeat contractions. (2) Antisense approaches can eliminate expanded myotonic dystrophy protein kinase (DMPK) transcripts. (3) Pharmacological inhibition of the interaction of transcripts with MBNL1 and other as yet unidentified proteins (question marks) or induced export to the cytoplasm can neutralise some of the pathogenic effects of the transcribed repeat. (4) MBNL1 overexpression or replacement techniques may restore levels of functional MBNL1, which is sequestered and depleted from the nucleoplasm. (5) CELF1 downregulation strategies, such as inactivation of PKC, may prove useful in reducing the levels of this splicing regulator. (6) Targeting of downstream genes and correction of alternative splicing can eliminate specific disease symptoms.
RNA toxicity might also underlie spinocerebellar ataxia 8 (SCA8) and fragile X-associated tremor ataxia syndrome (FXTAS), and is likely to contribute to Huntington disease-like 2 (HDL2). In these conditions, expanded transcripts accumulate in nuclear RNA foci and sequester RNA-binding proteins, such as MBNL1, and affect downstream gene expression and/or alternative splicing [24–27]. Expanded transcripts are also found in nuclear and cytoplasmic SCA10 aggregates, supporting a trans-dominant role of toxic RNA repeats in disease pathogenesis [28]. Similarly, the non-coding CAG repeat expansion in SCA12 suggests a possible mechanism of RNA toxicity [29]. Missplicing has been described in spinal bulbar muscular atrophy (SBMA) knock-in mice in the absence of RNA aggregation or MBNL1 sequestration. Despite CELF1 upregulation in SBMA, altered RNA processing probably results from an effect of androgen receptor polyglutamine expansion on hormone-regulated splicing [30]. The impact of CELF1 upregulation in oculopharyngeal muscular dystrophy (OPMD) remains elusive, but it might be indicative of molecular features shared with DM1 [31].
In this review we provide in-depth assessment of the molecular and physiological phenotypes of transgenic DM1 mouse models and their contribution towards an understanding of disease mechanisms, and we compare them with the human condition. More importantly, we review their advantages and limitations for preclinical assessment of new therapies.
Transgenic mouse models of DM: towards a methodical dissection of molecular pathogenesis
Transgenic mouse models of human neuromuscular disorders must meet genetic, molecular and physiological requirements to ensure faithful recapitulation and monitoring of the natural disease history (Box 2). DM1 transgenic lines have been developed to investigate the multisystemic pathophysiology and mechanisms of trinucleotide repeat dynamics. The mouse models available result from (i) inactivation of genes in the DM1 locus, (ii) overexpression of toxic CTG repeats, (iii) abnormal splicing regulation through Mbnl inactivation or Celf overexpression, and (iv) introduction of unstable CTG expansions (these models are described in Table S2 in the supplementary material online).
Box 2. Ideal features of transgenic mouse models of human disease.
Genetic, molecular and phenotypic characteristics are critical if a transgenic model is to provide relevant insight into the mechanisms of the disease (Table I). Molecular, cellular and organ-based features of the human disease should be examined in transgenic mice to ensure faithful recapitulation of the disease process. A thorough, systematic and multidisciplinary approach requires close interaction between researchers, clinicians, pathologists and physiologists to ascertain the relevance of the molecular abnormalities and animal model phenotypes to the understanding of disease pathogenesis.
By contrast, mouse models used in preclinical therapeutic experiments have distinctive requirements to be predictive of the efficacy of the response in humans: in vitro and in vivo data must predict the clinical outcome (Table I). Rather than recreating every element of the human disorder, it is sufficient to develop a few clinically meaningful phenotypes (either physiological or molecular) that reflect genuine elements of disease pathogenesis. The relevant phenotypes should be strong enough to be detectable and objectively quantifiable, but not so severe as to result in early and dramatic mortality. Transgene expression determines phenotype severity and progression: whereas low expression levels might not generate discernible phenotypes, high levels of expression might result in non-specific features and high mortality. In the absence of physiological readouts, the efficiency of new therapeutic schemes can be estimated in terms using disease-associated molecular deficits.
Table I. Ideal features of transgenic mouse models generated to understand the molecular pathogenesis of disease or to develop preclinical assays.
| Requirements for mouse models of human neuromuscular disease | |
|---|---|
| Genetic features | Same genetic defect as the human condition |
| Homogeneous and stable background | |
| Easy colony maintenance and mouse breeding | |
| Low interindividual variability | |
| Molecular phenotype | Faithful reproduction of the pathobiological cascade |
| Physiological phenotype | Consistent, robust and quantifiable phenotypes |
| Sufficient complexity to recreate the human disease | |
| Ideal features for utility in preclinical assays | Meaningful biochemical defects |
| At least one robust, consistent and quantifiable phenotype | |
| Phenotype development within a reasonable time frame | |
| Reversible phenotype and responsiveness to treatments | |
| Prediction of the human response to treatment |
Inactivation of genes of the human DM1 locus: Dmpk and Six5 knockout mice
Reduced cytoplasmic DMPK transcripts [32], as well as decreased SIX5 mRNA levels in DM1 patients [33,34], initially suggested a role for DMPK and/or SIX5 haploinsufficiency. To test this hypothesis, Dmpk and Six5 knockout mice were generated. Dmpk−/− and Six5−/− mouse strains seemed to be overtly normal [35−38] and failed to reproduce the complex and multisystemic DM1 phenotype, suggesting that happloinsufficiency of the genes in the DM1 locus is not the primary mechanism of disease. Nevertheless, DMPK inactivation might contribute to altered ion homeostasis in muscle and heart [35,39], whereas SIX5 deficiency might increase susceptibility to cataracts [37,38], reduce male fertility [40] and affect cardiac dysfunction [41].
Transgenic models of toxic RNA gain-of-function
A second generation of transgenic lines expressing toxic RNA repeats, summarised below, has greatly contributed to dissection of this novel disease mechanism, for which DM1 became the paradigm.
Non-expanded DMPK-overexpressing mice
To investigate the consequences of DMPK overexpression, transgenic lines carrying multiple copies of the human gene with a short (CTG)11 repeat were generated (Figure 1a) [35]. The Tg26 line, with the highest copy number and DMPK expression levels, exhibits the most severe phenotype, including cardiomyopathy, reduced workload tolerance, myotonic skeletal myopathy associated with reduced CLCN1 staining and smooth muscle tone deficit [35,42]. The undistinguishable effects of RNA and protein overexpression in Tg26 mice confound the interpretation of results and limit their application in therapeutic development.
Expression of toxic CUG repeats in skeletal muscle of HSALR mice
Overexpression of untranslated CUG repeats in the skeletal muscle of HSASR (short repeat length) and HSALR (long repeat length) transgenic mice (Figure 2b) provided crucial evidence for the toxicity of expanded transcripts [43]. HSALR lines expressing high levels of RNA containing 250 CUG repeats developed the most severe phenotype, including high mortality and myotonic discharges at 4 weeks of age in the absence of histological abnormalities in skeletal muscle. Myotonic discharges are characterised by high-frequency repetitive discharges evoked by insertion or movement of an EMG needle electrode. Histological signs of myopathy were detected at a later stage [43]. MBNL1 sequestration in nuclear foci disrupted the neonatal-to-adult splicing switch of multiple target transcripts (e.g. Clcn1, Atp2a1/Serca1, Mbnl1, Ldb3/Cypher) [10]. CELF1 is not upregulated in HSALR skeletal muscle, suggesting that sequestration of MBNL protein is sufficient to trigger missplicing [10]. Muscle-specific transgene expression restricts the application of HSALR mice to the investigation of adult skeletal muscle pathology and certain aspects of the neuromuscular junction [44]. The absence of muscle weakness or wasting represents an additional limitation, as well as the lack of HSALR expression in myogenic precursor cells.
Figure 2.
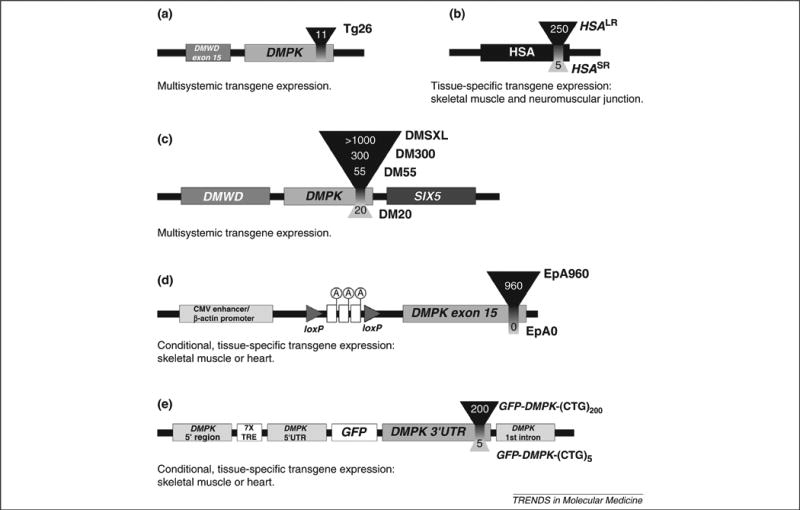
Schematic representation of CUG-expressing transgene constructs comprising multiple genetic elements. A brief description of the spatial profile of transgene expression is included for each insert. (a) Tg26 mice carry ∼25 copies of a fragment of human genomic DNA containing the DMPK gene sequence with 11 CTG repeats and the last DMWD exon. (b) HSASR and HSALR transgenic mice carry a fragment of human skeletal actin (HSA) gene, containing a short 5-CTG or an expanded 250-CTG repeat sequence inserted in its final exon, respectively. HSASR lines carry 1-20 copies of the transgene, whereas HSALR lines have 1-12. (c) DM20, DM55, DM300 and DMSXL transgenic mice carry one to three copies of a large fragment of human genomic DNA containing the three genes of the DM1 locus and a variable number of CTG repeats: 20, 55, ∼300−600 and ∼1000−1800, respectively. (d) Inducible EpA mice carry the human version of DMPK exon 15, either without repeats (EpA0 line) or with an interrupted 960-CTG expansion (EpA960 lines). Transgene expression is driven by the cytomegalovirus (CMV) enhancer and (β-actin promoter. The polyadenylation cassette located between the two loxP sites prevents DMPK exon 15 expression prior to recombination. Tamoxifen administration induces tissue-specific Cre-mediated recombination, removes the transcriptional stop and results in tissue-specific expression of control and expanded transcripts. (e) GFP-DMPK-(CTG)n mice carry a tetracycline-responsive version of the DMPK promoter to express a GFP transcript fused to the DMPK 3′′UTR containing either 5 or 200 CTG repeats. These animals also express the reverse tetracycline transactivator constitutively. In the presence of doxycycline, the transactivator binds to the tetracycline response elements (TRE) and activates expression of the GFP-DMPK-(CTG)n transgene.
Expression of expanded DMPK transcripts in the context of the human DM1 locus
Expression of expanded DMPK transcripts in multiple mouse tissues is driven by a large fragment of the human DM1 locus, which presumably contains the cis elements necessary for temporal and spatial regulation of transcription [45,46]. Mice carrying different repeat sizes have been generated (Figure 2c). Transgene expression in homozygous DM300 mice results in accumulation of ribonuclear foci in multiple tissues (e.g. skeletal muscle, heart and CNS), muscle histopathology, myotonia, progressive muscle weakness, age-dependent defects in glucose metabolism associated with Insr missplicing, growth retardation and high mortality [47–49]. The abnormal distribution of MAPT/tau protein isoforms in DM300 brains [47] resembles the tauopathy described in patients [50]. DM300 expansion-biased intergenerational instability led to DMSXL mice carrying 1000–1800 CTG repeats [51]. Homozygous DMSXL mice exhibit a more pronounced phenotype, including splicing abnormalities in skeletal muscle, heart and CNS that are more readily detected at 1 month of age [51]. The requirement for homozygosity, suggesting a dose-dependent effect of toxic RNA [47], increases the time and cost of DMSXL mouse breeding. Phenotypic evaluation will ascertain whether a single copy of a very large expanded transgene is sufficient to generate detectable and quantifiable phenotypes. DM300 and DMSXL mice recreate multisystemic DM1 features. Nonetheless, their phenotype is relatively mild, possibly as a result of lower expression levels of the CUG-containing transcripts compared to other mouse models [52].
Inducible tissue-specific expression of expanded CUG repeats
The Cre-lox system allows tissue-specific expression of large interrupted CTG repeats within DMPK exon 15 (Figure 2d). Expression in heart tissue resulted in severe histopathological, functional and electrophysiological features of DM1, leading to premature death within 2 weeks [53]. Transgene expression in skeletal muscle triggered myotonia, histopathology and progressive muscle impairment [54]. Acute myofibre degeneration was detected 4 weeks after tamoxifen-induced transgene expression [54]. The physiological changes detected in the heart and skeletal muscle of EpA960 mice were accompanied by sequential RNA foci accumulation, MBNL1 sequestration, CELF1 hyperphosphorylation and upregulation, and reversion to embryonic splicing profiles (Figure S1 in the supplementary material online) [9,53,54]. Whereas HSALR mice do not show CELF1 upregulation or muscle weakness, the expression of CUG repeats within the DMPK 3′UTR results in increased levels of CELF1 and development of a dystrophic scenario in EpA960 mice. Hence, there is a hypothesis that the genomic context of CTG expansion and CELF1 upregulation contributes, at least in part, to muscle wasting [54].
Leaky EpA960 transgene expression introduces a confounding limitation in the dissection of tissue- and/or cellspecific mechanisms of DM1. In addition, the interrupted nature of the EpA960 repeat might trigger specific molecular events and unusual DM1 symptomatology [55,56].
Inducible and reversible expression of the DMPK 3′UTR
The reversible nature of RNA toxicity was demonstrated using an inducible model overexpressing the DMPK 3′UTR (Figure 2e). Transgene expression in hemizygous GFP-DMPK-(CTG)200 mice was high enough to result in RNA foci accumulation and MBNL1 sequestration, but insufficient to generate a discernible phenotype [52]. Unlike the expanded version, the GFP-DMPK-(CTG)5 short repeat line did not show RNA foci or MBNL1 sequestration, but exhibited profound myotonia and severe heart block on transgene expression, as well as histopathology in skeletal muscle [52]. GFP-DMPK-(CTG)5 mice failed to model skeletal muscle atrophy, possibly because of the small repeat size or premature and sudden death from severe cardiac pathology. Modest missplicing was observed in skeletal muscle but was absent in cardiac tissue. Unlike HSALR mice, splicing defects in GFP-DMPK-(CTG)5 mice were associated with CELF1 upregulation [52]. An increase of the transcriptional factor NKX2-5 in both cardiac and skeletal muscle suggests ongoing transcriptional abnormalities in mice overexpressing the DMPK 3′UTR [57].
The CUG dose effect on myogenesis was reported in a transgenic model overexpressing the DMPK 3′UTR containing either 11 or 91 CTG repeats in multiple tissues under the control of the DMPK regulatory elements [58]. Both DMPK-GFP-(CTG)11 and DMPK-GFP-(CTG)91 strains displayed defective muscle differentiation, but the phenotype was exacerbated by the CTG repeat expansion in DMPK-GFP-(CTG)91 mice, suggesting a cooperative role between DMPK 3′UTR sequences and toxic expanded repeats in the full expression of DM1-associated pathology [58]. The more severe phenotype of GFP-DMPK-(CTG)5 mice probably reflects the very high expression levels of CUG-containing transcripts compared to other mouse models [52]. Importantly, transgene silencing in these mice resulted in phenotype reversion, providing crucial support to therapeutic neutralisation of expanded DMPK transcripts.
In addition to the expression of toxic CUG-expanded RNA repeats, mouse models of abnormal splicing regulation have recreated relevant features of DM1.
Models of abnormal splicing regulators
The characterisation of the molecular and physiological phenotypes resulting from MBNL inactivation and CELF upregulation in mice, independently of repeat expansion, has illustrated the central role of these two families of splicing regulators in DM1 pathogenesis.
Mbnl1 knockout mice
Disruption of Mbnl1 exon 3 eliminates CUG-binding isoforms and mimics the DM1 situation, in which MBNL1 is extensively sequestered and functionally unavailable [59]. Mbnl1Δ3/Δ3 mice exhibit overt myotonia associated with abnormal CLCN1 splicing, reduced protein staining and chloride currents in skeletal muscle [10,59,60]. Mbnl1Δ3/Δ3 mice develop myopathy but no signs of muscle degeneration [59].
Loss of MBNL1 affects other tissues, resulting in typical DM1 cataracts [59], motivation deficits and apathy [61], as well as conduction defects and missplicing in heart (M. Swanson, unpublished data). Muscle weakness was not detected and might result from different mechanisms, such as CELF1 upregulation. MBNL1 has been implicated in the regulation of RNA transcription, processing and stability of downstream targets, possibly involved in DM pathogenesis [18]. Mbnl1Δ3/Δ3 mice provide a unique tool for identifying such targets.
Mbnl2 knockout mice
In addition to MBNL1, MBNL2 and MBNL3 also colocalise with DMPK and ZFN9 ribonuclear inclusions [6], suggesting a combinatorial loss-of-function of muscleblind-like proteins. To elucidate the role of MBNL2, two knockout lines were generated. Myopathy and myotonia associated with mild Clcn1 missplicing were detected in one line [62], but another independent Mbnl2-deficient mouse was overtly normal [10]. These contradictory results raised questions about the role of Mbnl2 in muscle physiology. Mbnl2 knockout lines may help elucidate the impact of Mbnl2 inactivation in other tissues and organ systems.
CELF1 overexpression
Sequestration and functional inactivation of both MBNL1 and MBNL2 proteins in HSALR muscle is not sufficient to recreate all muscle abnormalities, suggesting a role for additional disease intermediates. The contribution of CELF1 upregulation to DM1 pathogenesis has been confirmed by the generation of CELF1-overexpressing lines. The CUGBP1-TR line showed growth retardation, delayed myogenesis, and histological and molecular abnormalities, which correlated with the degree of CELF1 upregulation [20]. MCKCUG-BP1 mice have normally sized stillborn pups showing histological abnormalities and missplicing in skeletal muscle [63]. Both models suggest that CELF1 overexpression is sufficient to induce a developmental phenotype resembling congenital DM1. Their application is limited by high mortality and breeding difficulties. Conditional CELF1 overexpression in TRECUGBP1 mice has circumvented this limitation. Induced CELF1 expression in adult skeletal muscle recreates DM1 histopathology and functional impairment [12]. Upregulation in adult heart results in DM1-like cardiac conduction defects and histological abnormalities, leading to animal death within 2 weeks [11]. Both tissue-specific phenotypes were associated with embryonic splicing profiles.
In addition to RNA toxicity and missplicing, CTG repeat instability is another hallmark of DM1. Mouse models carrying unstable repeat expansions have been generated to investigate repeat dynamics.
Mouse models of unstable CTG repeats
Intergenerational and somatic instability of trinucleotide repeats was recreated in Dmt-D transgenic mice carrying a (CTG)162 repeat tract within the DMPK 3′UTR [64,65] and in DM55 and DM300 mice carrying a moderate (CTG)55 or large (CTG)300 expansion, respectively, in the context of the human DM1 locus [46,51,66]. Important features of intergenerational instability were reproduced in these mice, such as the effects of sex and age of the transmitting parent [46,64]. As in humans, expansion-biased somatic mosaicism was tissue-specific and age-dependent [46,65,66]. The “humanised” Dmpk(CTG)84 knock-in mice exhibited very low intergenerational instability but high somatic mosaicism [67]. No overt CTG repeat instability has been reported in other DM1 transgenic mice.
Mouse models of unstable DM1 repeats have revealed cis- and trans-acting modifiers of trinucleotide dynamics [68], indicating a role for functional components of the mismatch repair system in CTG repeat expansion by mechanisms of DNA misrepair [69,70]. These lines provide suitable model systems for investigating repeat size mutation and the contribution of somatic mosaicism to phenotypic variability and disease progression.
Transgenic mouse models of congenital DM1
It is unknown to what extent congenital DM1 shares molecular mechanisms with the adult disease form. Specific animal models of congenital DM1 are therefore required. Developmental abnormalities have been reported in DMSXL lines [47,51] and CELF1-overexpressing mice [20,63]. Further studies are required to determine whether the phenotypes detected recreate bona fide features of congenital DM1.
Mouse models of DM2
Given the dissociation between foci and missplicing in transfected cells [71] and in a DM1 mouse model [52], alternative pathways of disease pathogenesis were investigated, such as inactivation of CNBP/ZNF9 in DM2. Zfn9+/− mice exhibited myotonia, muscle wasting, defective walking, cardiac conduction defects and ocular cataracts in the absence of repeat expansion and missplicing [72], suggesting a pathogenic role for CNBP/ZNF9 haploinsufficiency. However, contradictory reports on the effects of CCTG expansions on CNBP/ZNF9 expression in DM2 patients challenge the relevance of Cnbp-deficient mice in recreating a true disease scenario [73,74].
Analogous to HSALR mice, DM2-HSAtg mice express an intronic (CCTG)121 expansion. DM2-HSAtg mice display CCUG-containing ribonuclear inclusions, CELF1 upregulation in liver and recapitulate aspects of DM2 muscle pathology, but no missplicing [14] (R. Krahe, unpublished data), in further support of the dissociation between foci accumulation and splicing defects [52,71]. Transgenic lines will be crucial to gain insight into specific aspects of DM2 and to test new therapeutic strategies.
From molecular pathogenesis to rational therapeutic strategies
At present there is no effective treatment for DM1 or DM2. The dissection of the molecular mechanism has opened new avenues for rational therapeutic targeting of multiple disease intermediates: the DNA expansion, toxic transcripts, RNA-interacting proteins, splicing regulators and misspliced transcripts (Figure 1). As our understanding of disease mechanism improves, therapeutic targeting of pathological events could replace supportive care.
Modulation of trinucleotide repeat dynamics
Slowing down repeat expansion may delay disease onset and symptom severity, whereas contraction to the normal repeat size range is predicted to be curative [69]. Indeed, suppression of somatic mosaicism in a mouse model of Huntington disease resulted in delayed histopathology [75]. In addition, small compounds capable of reducing the expansion rate of the DM1 repeat were identified in cell culture [76,77]. The impact of modified repeat dynamics on DM1 manifestations can be assessed in mouse models exhibiting both somatic instability and disease phenotypes (e.g. the DMSXL model). Deciphering repeat expansion mechanisms will guide us towards rational testing of small molecules predicted to interfere with specific steps of the expansion process [69]. Importantly, whereas targeting of the downstream pathogenesis is most likely to be disease-specific, control of repeat dynamics might present general benefits for repeat expansion disorders.
Neutralisation or elimination of expanded CUG-containing transcripts
The reversible phenotype of GFP-DMPK-(CTG)5 mice on transgene silencing [52] supports neutralisation of toxic RNA as an attractive therapeutic strategy. Antisense oligonucleotides targeting expanded transcripts resulted in diffusion of CUG-containing transcripts and MBNL1, splicing correction in HSALR and DM300/DMSXL mice and reduced myotonia in HSALR animals [78,79]. Alternatively, pharmacological inhibition of the interaction between toxic CUG-containing transcripts and MBNL1 also reduced foci accumulation, redistributed MBNL1 in cell models, and partially reversed missplicing in HSALR skeletal muscle [80]. Because cytoplasmic aggregates seem to be innocuous [81], chemicals that increase DMPK transcript export out of the nucleus might be beneficial. Many of the transgenic mice described here offer a unique resource for preclinical assessment of anti-sense strategies and small molecules capable of modifying RNA toxicity in DM and other toxic RNA diseases.
Upregulation of MBNL1 activity
Therapeutic MBNL1 overexpression is predicted to be beneficial in DM1 and DM2. MBNL1 overexpression in HSALR skeletal muscle by adeno-associated virus (AAV) transduction overcame MBNL1 sequestration, corrected missplicing and reversed myotonia, but failed to rescue myofibre structure [82]. Efficient systemic AAV delivery in neuromuscular disorders is currently being optimised [83].
Transgenic mouse models demonstrated that MBNL1 depletion and CELF1 upregulation have overlapping, as well as independent, downstream effects [11,84]. Therefore, targeting MBNL1 alone may not fully address all DM1 symptoms.
Downregulation of CELF1
CELF downregulation provides a window for therapeutic intervention. EpA960 mice have pointed to PKC as a potential therapeutic target [9,85]. Administration of PKC inhibitors to EpA960 transgenic mice expressing toxic expanded repeats in heart tissue reduced steady-state CELF1 protein levels, corrected CELF1-mediated splicing defects, improved mouse survival and ameliorated the cardiac phenotype [85]. Pharmacological or genetic inhibition of PKC activation might be envisaged as a cardiac therapy in DM1. The involvement of PKC-mediated CELF1 upregulation in other tissues and in DM2 is less clear. Thus, therapies based on modulation of CELF1 phosphorylation and protein levels might be limited to specific tissues and to DM1 in particular. Animal models will provide further insight.
Reverse of spliceopathy by exon skipping
Antisense oligonucleotides can be designed to induce skipping of abnormally included exons. This highly specific strategy was used to selectively correct Clcn1 missplicing in HSALR and Mbnl1Δ3/Δ3 transgenic mice [86]. The sustained suppression of myotonia suggests that additional aspects of the disease might be reversed by similar strategies. To this end, different antisense oligonucleotides could be combined in a cocktail to target various misspliced transcripts. Strategies to enhance systemic delivery and uptake of antisense oligonucleotides are under development [87].
Critical comparison of DM1 mouse models: towards an assessment of tissue-specific therapies
Given their intrinsic molecular features and phenotypes, some mouse models show greater potential than others for preclinical assessment of therapies that target specific organ systems. In the following subsections we discuss the potential of DM1 transgenic lines for therapy development in skeletal muscle, heart, CNS and other organ systems (Table 1).
Table 1. Preclinical assessment of therapeutic strategies in DM1 mouse models.
| Mouse modela | Therapeutic strategiesb | Quantifiable phenotypes in target tissues | Limitations | Refs | |||
|---|---|---|---|---|---|---|---|
| Skeletal muscle | Heart | CNS | Others | ||||
| HSALR | Elimination of toxic RNA, disruption of RNA–MBNL protein interactions, MBNL upregulation, restoration of missplicing | Myotonia, myopathy, foci accumulation, MBNL1 sequestration, missplicing events | Not applicable | Not applicable | Not applicable | No muscle weakness, normal CELF1 levels | [43] |
| EpA960 | Elimination of toxic RNA, disruption of RNA–MBNL protein interactions, MBNL upregulation, CELF1 downregulation, PKC inactivation | Myotonia, motor function, muscle wasting, myopathy, foci accumulation, MBNL1 sequestration, missplicing events | Cardiac conduction defects, cardiomyopathy, foci accumulation, MBNL1 sequestration, missplicing events | Not reported | Not reported | Leakage of transgene expression in skeletal muscle, high mortality due to cardiac complications | [9,53,54] |
| DM300 and/or DMSXL | Elimination of toxic RNA, disruption of RNA–MBNL protein interactions, MBNL upregulation, restoration of missplicing, modification of triplet repeat dynamics | Myotonia, myopathy, foci accumulation, muscle strength, missplicing events | Foci accumulation, missplicing events | Foci accumulation, missplicing events, tauopathy | Foci accumulation, missplicing events | Mild splicing abnormalities, interindividual variability, time-consuming breeding strategies | [47,49,51, 98,99] |
| DMPK-GFP-(CTG)5 | Elimination of toxic RNA, NKX2-5 downregulation | Myotonia, myopathy, missplicing events. | Cardiac conduction defects | Not reported | Not reported | No muscle weakness, no RNA foci, short repeat sequence, high mortality | [52] |
| Mbnl1Δ3/Δ3 | MBNL upregulation or replacement | Myotonia, myopathy, missplicing events | Cardiomyopathy | Motivation and apathy deficits | Not reported | No muscle weakness, normal CELF1 levels | [59,61] |
| TRECUGBP1 | CELF1 downregulation, restoration of missplicing | Motor function, myopathy, muscle weight, missplicing events | Cardiac conduction defects, cardiomyopathy | Not reported | Not reported | High mortality | [11,12] |
Different mouse lines have different advantages and limitations in assessing the benefits and efficacy of new therapeutic schemes in specific tissues and organ systems.
Possible therapeutic approaches are presented for mouse models that replicate key events in the biochemical cascade of RNA toxicity and develop relevant DM1 phenotypes.
Skeletal muscle pathogenesis
Therapies targeting skeletal muscle might be primarily assessed in three mouse models: the muscle-specific HSALR model [43], the inducible EpA960 line [54] and ubiquitously expressing DMSXL mice [47,51]. Myotonic discharges, histological myopathy, foci accumulation, MBNL1 sequestration and splicing abnormalities can be monitored in HSALR, inducible Ep960 and to some extent DMSXL mice to assess the efficiency of therapeutic approaches designed to revert the muscle phenotype. Progressive muscle weakness can be used as a quantitative indicator of muscle pathology in Ep960 mice. Strategies aimed at (i) destroying toxic transcripts, (ii) disrupting the interaction between CUG-containing RNA and MBNL proteins, (iii) increasing the levels of functional MBNL1 and (iv) correcting RNA splicing can be evaluated in skeletal muscle of the three transgenic models. Means to reduce CELF1 levels can be tested in EpA960 mice. The absence of repeat expansion and accumulation of RNA foci in GFP-DMPK-(CTG)5 mice, as well as the severity of their phenotype, limits their application in therapeutic assays. Nonetheless, they offer the possibility to test means of escaping RNA toxicity and NKX2-5 activation. Mbnl1Δ3/Δ3 mice display relevant, consistent and measurable myotonia and myopathic features, and can be used for testing of therapeutic strategies to circumvent the pathological consequences of MBNL1 depletion. Monitoring of motor, histopathological and molecular phenotypes in TRECUGBP1 skeletal muscle offer additional means to assess the benefits of targeting CELF1 upregulation and missplicing.
Cardiac defects
The cardiac phenotype of EpA960 mice can be monitored by ECG recordings of systolic and diastolic dysfunction, arrhythmia and AV block. RNA foci accumulation, MBNL1 sequestration, PKC activation, CELF phosphorylation and upregulation and missplicing can be investigated by molecular methods following therapeutic intervention. Whereas both TRECUGBP1 and GFP-DMPK-(CTG)5 mice provide additional means to test the cardiac response to CELF1 downregulation, only GFP-DMPK-(CTG)5 mice offer the opportunity to test the benefits of NKX2-5 down-regulation. The severe phenotype and mortality of EpA960 and GFP-DMPK-(CTG)5 mice represent limitations of these models. Characterisation of DMSXL and Mbnl1Δ3/Δ3 cardiac function is required for future therapeutic experiments.
CNS dysfunction
The efficacy of CNS therapeutic schemes can be assessed through analysis of nuclear RNA aggregation, missplicing and abnormal MAPT/tau protein profiles in DMSXL animals. Alternatively, amelioration of Mbnl1Δ3/Δ3 motivation deficits can be monitored by behavioural phenotyping. Further characterisation of molecular and behavioural CNS readouts in transgenic mice is required to investigate the impact of future therapies on DM1 neurological manifestations. Both inducible EpA960 and GFP-DMPK-(CTG)5 mice offer the possibility to express toxic RNA transcripts in the CNS.
Other tissues and organ systems
Virtually all tissues and organ systems are affected by DM1 [88]. Therefore, the benefits of therapeutic approaches should be investigated throughout the body. Both DMSXL and Mbnl1Δ3/Δ3 mice recreate multisystemic phenotypes. Further phenotypic characterisation might reveal additional DM1 features in both lines. DMSXL transgene expression under the control of the human promoter and Mbnl1 inactivation will prove valuable in evaluating the impact of systemic therapies in different body systems. The multisystem impact of new therapies can be evaluated through analysis of DMSXL and Mbnl1Δ3/Δ3 splicing profiles or accumulation of ribonuclear foci in DMSXL mice. Tissue-specific transgene expression in conditional DM1 models might further contribute to the development of therapeutic strategies that target additional tissues.
Tissue-specific versus systemic approaches
The choice of model to be used in therapeutic experiments depends on the target tissue and the question being asked. HSALR and inducible EpA960 and GFP-DMPK-(CTG)5 mice recreate consistent tissue-specific phenotypes that are easily quantifiable and facilitate monitoring of the benefits and efficacy of new therapeutic strategies. Despite their milder phenotype and interindividual variability, DMSXL mice have the advantage of expressing full-length expanded DMPK transcripts in multiple tissues under the control of the endogenous human promoter. Therefore, they provide a unique tool for extending and refining primary tissue-specific therapeutic assays for multisystem evaluation of their benefits and efficiency.
Concluding remarks and future perspectives: can multiple transgenic lines replace a ‘perfect’ model?
Complementary DM1 mouse models have recreated many molecular and phenotypic disease features, pointing to an RNA-mediated disease mechanism. Although fundamental aspects of molecular pathogenesis still deserve further investigation (Box 3), our current understanding of DM1 is sufficient to design rational therapeutic strategies that target molecular events of the pathobiological cascade. The vast collection of mouse models available offers opportunities to therapeutically target specific tissues and organ systems and investigate different levels of disease severity. Independent DM1 lines make possible proof-of-principle studies by different laboratories: parallel tests on multiple models enable replication of results, data refinement and strengthening of conclusions. Protocol standardisation for phenotype monitoring will facilitate direct comparison of independently generated results. Nonetheless, large-scale therapeutic trials on multiple transgenic lines are not practical or financially feasible. Major preclinical experiments should focus on the selected mouse model that is most relevant to the question being asked and the phenotypic readouts being measured. It might be neither necessary nor reasonable to generate a ‘perfect’ transgenic mouse that recreates all DM1 (and/or DM2) features. Nonetheless, there is certainly room for improvement. Whereas muscle and heart phenotypes have been reproduced and characterised in mice, investigation of CNS manifestations is lagging behind. Characterisation of the cognitive and behavioural phenotypes of the available lines is required.
Box 3. Outstanding questions.
Molecular pathogenesis
To what extent does the DM1 (and DM2) spliceopathy explain the full disease spectrum? What is the pathogenic role of deregulated gene expression [18,19,57,96,97] and/or cell stress-induced effects on protein translation [22]?
What is the degree of molecular pathogenesis overlap between adult and congenital DM1? Do they share the same pathobiology?
It has been shown that the mouse strain affects repeat dynamics [67], but a detailed study of the effects of strain background and/or genetic drift on disease manifestation and progression has not been reported. To what extent is the DM phenotype influenced by genetic modifiers in humans and in mice?
-
Tissue-specific expression of toxic RNA in an individual tissue is sufficient to induce relevant DM1 features. However, it is conceivable that non-expressing tissues might exhibit phenotypes secondary to integral dysfunction of body physiology. What is the boundary between the primary and secondary impacts of DM1 expansion? Monitoring of secondary phenotypes in tissue-specific transgenic lines will shed light on this question, despite the confounding effects of possibly leaky transgene expression.
Therapeutic strategies
What are the most relevant phenotypic readouts for assessing the efficiency of therapeutic schemes in the CNS of transgenic mouse models?
What is the best strategy for systemic treatments that target multiple tissues and organs affected by DM?
Is it possible to develop pharmacological approaches based on non-toxic chemical compounds capable of reversing DM-associated spliceopathy?
Toxic RNA repeats are the central pathogenic agent behind a number of human diseases. The combined study of existing DM1, DM2, SCA8, FXTAS and SCA10 mouse models will help to further dissect RNA toxicity and develop novel therapeutic strategies in vivo. The progress made so far does not bring the development and analysis of mouse models to a halt. Instead, it should be a source of great optimism for future advances.
Supplementary Material
Acknowledgments
We are grateful to the Marigold Animal Model Working Group for helpful discussions and to their colleagues who contributed with personal communications prior to publication. We would like to apologise in advance to many authors and colleagues whose work was not directly discussed in this review owing to space limitations.
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.molmed. 2011.05.004.
References
- 1.Suominen T, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liquori CL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 3.Taneja KL, et al. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timchenko LT, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fardaei M, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 7.Savkur RS, et al. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 8.Philips AV, et al. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 9.Kuyumcu-Martinez NM, et al. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin X, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 11.Koshelev M, et al. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. 2010;19:1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward AJ, et al. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelletier R, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. 2009;36:181–190. doi: 10.1016/j.nbd.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 14.Salisbury E, et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol. 2009;175:748–762. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mankodi A, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 16.Lueck JD, et al. Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am J Physiol Cell Physiol. 2007;292:C1291–1297. doi: 10.1152/ajpcell.00336.2006. [DOI] [PubMed] [Google Scholar]
- 17.Charlet BN, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 18.Osborne RJ, et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18:1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du H, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timchenko NA, et al. Overexpression of CUG triplet repeatbinding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. 2004;279:13129–13139. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 21.Timchenko NA, et al. CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBPbeta mRNA and regulates translation of C/EBPbeta isoforms. Nucleic Acids Res. 1999;27:4517–4525. doi: 10.1093/nar/27.22.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huichalaf C, et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 2010;24:3706–3719. doi: 10.1096/fj.09-151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timchenko NA, et al. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/enhancer-binding protein beta by interacting with the alpha and beta subunits of eukaryotic initiation translation factor 2. J Biol Chem. 2005;280:20549–20557. doi: 10.1074/jbc.M409563200. [DOI] [PubMed] [Google Scholar]
- 24.Daughters RS, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tassone F, et al. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 26.Sellier C, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwahashi CK, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 28.White MC, et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes E, et al. SCA12: an unusual mutation leads to an unusual spinocerebellar ataxia. Brain Res Bull. 2001;56:397–403. doi: 10.1016/s0361-9230(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 30.Yu Z, et al. Altered RNA splicing contributes to skeletal muscle pathology in Kennedy disease knock-in mice. Dis Model Mech. 2009;2:500–507. doi: 10.1242/dmm.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corbeil-Girard LP, et al. PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol Dis. 2005;18:551–567. doi: 10.1016/j.nbd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Fu YH, et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science. 1993;260:235–238. doi: 10.1126/science.8469976. [DOI] [PubMed] [Google Scholar]
- 33.Klesert TR, et al. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet. 1997;16:402–406. doi: 10.1038/ng0897-402. [DOI] [PubMed] [Google Scholar]
- 34.Thornton CA, et al. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet. 1997;16:407–409. doi: 10.1038/ng0897-407. [DOI] [PubMed] [Google Scholar]
- 35.Jansen G, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13:316–324. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 36.Reddy S, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996;13:325–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 37.Sarkar PS, et al. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet. 2000;25:110–114. doi: 10.1038/75500. [DOI] [PubMed] [Google Scholar]
- 38.Klesert TR, et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet. 2000;25:105–109. doi: 10.1038/75490. [DOI] [PubMed] [Google Scholar]
- 39.Berul CI, et al. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest. 1999;103:R1–7. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkar PS, et al. Six5 is required for spermatogenic cell survival and spermiogenesis. Hum Mol Genet. 2004;13:1421–1431. doi: 10.1093/hmg/ddh161. [DOI] [PubMed] [Google Scholar]
- 41.Wakimoto H, et al. Characterization of cardiac conduction system abnormalities in mice with targeted disruption of Six5 gene. J Interv Card Electrophysiol. 2002;7:127–135. doi: 10.1023/a:1020881520353. [DOI] [PubMed] [Google Scholar]
- 42.O'Cochlain DF, et al. Transgenic overexpression of human DMPK accumulates into hypertrophic cardiomyopathy, myotonic myopathy and hypotension traits of myotonic dystrophy. Hum Mol Genet. 2004;13:2505–2518. doi: 10.1093/hmg/ddh266. [DOI] [PubMed] [Google Scholar]
- 43.Mankodi A, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 44.Wheeler TM, et al. Ribonuclear foci at the neuromuscularjunction in myotonic dystrophy type 1. Neuromuscul Disord. 2007;17:242–247. doi: 10.1016/j.nmd.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gourdon G, et al. Moderate intergenerational and somatic instability of a 55-CTG repeat in transgenic mice. Nat Genet. 1997;15:190–192. doi: 10.1038/ng0297-190. [DOI] [PubMed] [Google Scholar]
- 46.Seznec H, et al. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet. 2000;9:1185–1194. doi: 10.1093/hmg/9.8.1185. [DOI] [PubMed] [Google Scholar]
- 47.Seznec H, et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 48.Guiraud-Dogan C, et al. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim Biophys Acta. 2007;1772:1183–1191. doi: 10.1016/j.bbadis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Vignaud A, et al. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin–proteasome pathway. Neuromuscul Disord. 2010;20:319–325. doi: 10.1016/j.nmd.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 50.Sergeant N, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- 51.Gomes-Pereira M, et al. CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet. 2007;3:e52. doi: 10.1371/journal.pgen.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahadevan MS, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang GS, et al. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orengo JP, et al. Expanded CTG repeats within the DMPK 3′UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Musova Z, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A. 2009;149A:1365–1374. doi: 10.1002/ajmg.a.32987. [DOI] [PubMed] [Google Scholar]
- 56.Braida C, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19:1399–1412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]
- 57.Yadava RS, et al. RNA toxicity in myotonic muscular dystrophy induces NKX2-5 expression. Nat Genet. 2008;40:61–68. doi: 10.1038/ng.2007.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Storbeck CJ, et al. Inhibition of myogenesis in transgenic mice expressing the human DMPK 3′-UTR. Hum Mol Genet. 2004;13:589–600. doi: 10.1093/hmg/ddh064. [DOI] [PubMed] [Google Scholar]
- 59.Kanadia RN, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 60.Lueck JD, et al. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol. 2007;129:79–94. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matynia A, et al. Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS ONE. 2010;5:e9857. doi: 10.1371/journal.pone.0009857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hao M, et al. Muscleblind-like 2 (Mbnl2)-deficient mice as a model for myotonic dystrophy. Dev Dyn. 2008;237:403–410. doi: 10.1002/dvdy.21428. [DOI] [PubMed] [Google Scholar]
- 63.Ho TH, et al. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 64.Monckton DG, et al. Hypermutable myotonic dystrophy CTG repeats in transgenic mice. Nat Genet. 1997;15:193–196. doi: 10.1038/ng0297-193. [DOI] [PubMed] [Google Scholar]
- 65.Fortune MT, et al. Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum Mol Genet. 2000;9:439–445. doi: 10.1093/hmg/9.3.439. [DOI] [PubMed] [Google Scholar]
- 66.Lia AS, et al. Somatic instability of the CTG repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum Mol Genet. 1998;7:1285–1291. doi: 10.1093/hmg/7.8.1285. [DOI] [PubMed] [Google Scholar]
- 67.van Den Broek WJ, et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- 68.Gomes-Pereira M, et al. Transgenic mouse models of unstable trinucleotide repeats. In: Wells RD, Ashizawa T, editors. Genetic Instabilities and Neurological Diseases. 2nd. Elsevier; 2006. pp. 563–583. [Google Scholar]
- 69.Gomes-Pereira M, Monckton DG. Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res. 2006;598:15–34. doi: 10.1016/j.mrfmmm.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 70.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ho TH, et al. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 72.Chen W, et al. Haploinsuffciency for Znf9 in Znf9+/− mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol. 2007;368:8–17. doi: 10.1016/j.jmb.2007.01.088. [DOI] [PubMed] [Google Scholar]
- 73.Margolis JM, et al. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet. 2006;15:1808–1815. doi: 10.1093/hmg/ddl103. [DOI] [PubMed] [Google Scholar]
- 74.Raheem O, et al. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 in myotonic dystrophy type 2. Am J Pathol. 2010;177:3025–3036. doi: 10.2353/ajpath.2010.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wheeler VC, et al. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- 76.Gomes-Pereira M, Monckton DG. Chemically induced increases and decreases in the rate of expansion of a CAG•CTG triplet repeat. Nucleic Acids Res. 2004;32:2865–2872. doi: 10.1093/nar/gkh612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashem VI, et al. Chemotherapeutic deletion of CTG repeats in lymphoblast cells from DM1 patients. Nucleic Acids Res. 2004;32:6334–6346. doi: 10.1093/nar/gkh976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wheeler TM, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mulders SA, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci USA. 2009;106:13915–13920. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warf MB, et al. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci USA. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dansithong W, et al. Cytoplasmic CUG RNA foci are insufficient to elicit key DM1 features. PLoS ONE. 2008;3:e3968. doi: 10.1371/journal.pone.0003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kanadia RN, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 84.Kalsotra A, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang GS, et al. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest. 2009;119:3797–3806. doi: 10.1172/JCI37976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wheeler TM, et al. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.White PJ, et al. Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev Mol Med. 2009;11:e10. doi: 10.1017/S1462399409001021. [DOI] [PubMed] [Google Scholar]
- 88.Harper PS. Myotonic Dystrophy. WB Saunders; 2001. [Google Scholar]
- 89.Meola G, Sansone V, et al. Cerebral involvement in myotonic dystrophies. Muscle Nerve. 2007;36:294–306. doi: 10.1002/mus.20800. [DOI] [PubMed] [Google Scholar]
- 90.Weber YG, et al. Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology. 2010;74:1108–1117. doi: 10.1212/WNL.0b013e3181d8c35f. [DOI] [PubMed] [Google Scholar]
- 91.Romeo V, et al. Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol. 2010;257:1246–1255. doi: 10.1007/s00415-010-5498-3. [DOI] [PubMed] [Google Scholar]
- 92.Brook JD, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69:385. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- 93.Mahadevan M, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 94.Monckton DG, et al. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8. doi: 10.1093/hmg/4.1.1. [DOI] [PubMed] [Google Scholar]
- 95.Harper PS, et al. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet. 1992;51:10–16. [PMC free article] [PubMed] [Google Scholar]
- 96.Ebralidze A, et al. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science. 2004;303:383–387. doi: 10.1126/science.1088679. [DOI] [PubMed] [Google Scholar]
- 97.Botta A, et al. Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr. 2007;13:339–351. doi: 10.3727/000000006781510705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gourdon G, et al. The DMSXL transgenic mice carrying very large expansions exhibit molecular and physiological defects: an animal model for gene therapy experiments? 2009 [Google Scholar]
- 99.Panaite PA, et al. Myotonic dystrophy transgenic mice exhibit pathologic abnormalities in diaphragm neuromuscular junctions and phrenic nerves. J Neuropath Exp Neur. 2008;67:763–772. doi: 10.1097/NEN.0b013e318180ec64. [DOI] [PubMed] [Google Scholar]
- 100.Boucher CA, et al. A novel homeodomain-encoding gene is associated with a large CpG island interrupted by the myotonic dystrophy unstable (CTG)n repeat. Hum Mol Genet. 1995;4:1919–1925. doi: 10.1093/hmg/4.10.1919. [DOI] [PubMed] [Google Scholar]
- 101.Shaw DJ, et al. Genomic organization and transcriptional units at the myotonic dystrophy locus. Genomics. 1993;18:673–679. doi: 10.1016/s0888-7543(05)80372-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.