

Abstract
Multiple natural killer (NK) cell-based anticancer therapies are currently under development. Here, we compare the efficiency of genetically modified NK-92 cells expressing chimeric antigen receptors (CARs) at killing NK cell-resistant B-lymphoid leukemia cells to the antibody-dependent cell-mediated cytotoxicity (ADCC) of NK-92 cells expressing a high affinity variant of the IgG Fc receptor (FcγRIII). First, we compared in vitro the abilities of NK-92 cells expressing CD20-targeting CARs to kill primary chronic lymphocytic leukemia (CLL) cells derived from 9 patients with active, untreated disease to the cytotoxicity of NK-92 cells expressing FcγRIII combined with either of the anti-CD20 monoclonal antibodies (mAbs) rituximab or ofatumumab. We found that CAR-expressing NK-92 cells effectively kill NK cell-resistant primary CLL cells and that such a cytotoxic response is significantly stronger than that resulting from ADCC. For studying CAR-expressing NK cell-based immunotherapy in vivo, we established xenograft mouse models of residual leukemia using the human BCR-ABL1+ cell lines SUP-B15 (CD19+CD20−) and TMD-5 (CD19+CD20+), two acute lymphoblastic leukemia (ALL) lines that are resistant to parental NK-92 cells. Intravenous injection of NK-92 cells expressing CD19-targeting CARs eliminated SUP-B15 cells, whereas they had no such effect on TMD-5 cells. However, the intrafemoral injection of NK-92 cells expressing CD19-targeting CAR resulted in the depletion of TMD-5 cells from the bone marrow environment. Comparative studies in which NK-92 cells expressing either CD19- or CD20-targeting CARs were directly injected into subcutaneous CD19+CD20+ Daudi lymphoma xenografts revealed that CD20-targeting CAR is superior to its CD19-specific counterpart in controlling local tumor growth. In summary, we show here that CAR-expressing NK-92 cells can be functionally superior to ADCC (as mediated by anti-CD20 mAbs) in the elimination of primary CLL cells. Moreover, we provide data demonstrating that the systemic administration of CAR-expressing NK-92 cells can control lymphoblastic leukemia in immunocompromised mice. Our results also suggest that the direct injection of CAR-expressing NK-92 cells to neoplastic lesions could be an effective treatment modality against lymphoma.
Keywords: CAR, NK cells, NK-92, lentiviral vector, lymphoid malignancies
Introduction
Anticancer treatments based on monoclonal antibodies (mAbs) administered in combination with chemotherapy have significantly improved clinical outcome in patients with non-Hodgkin lymphoma (NHL). Nevertheless, about 40% of all NHL patients ultimately relapse1,2 and subsequent stem cell transplantation can rescue only a small percentage of these individuals.3 Thus, cell-based approaches are being increasingly considered as a therapeutic strategy to overcome the resistance to chemotherapy of leukemic cells and attack the malignant stem cell clone through an alternative cytotoxic modality. Recent reports on the therapeutic use of chimeric antigen receptor (CAR)-expressing autologous T lymphocytes to treat chemotherapy-resistant lymphoid malignancies substantiate the potential clinical benefits of cellular therapy.4,5 As an alternative to T cells, natural killer (NK) cells expressing CARs directed against CD19 or CD20 (2 markers of the B-cell lineage) can also be used as cytotoxic effector cells for cell-based immunotherapy. However, current NK cell-based therapies are constrained by the necessity to isolate sufficient numbers of NK cells from donors as well as by the need to achieve acceptable transfection efficiencies.6,7 The human NK cell line NK-928,9 presents an attractive alternative to donor NK cells, as it can be propagated and expanded in vitro.10 Phase I clinical trials testing NK-92 cells have been successfully completed, confirming their clinical safety.11,12 Furthermore, NK-92 cells are amenable to transfection with vectors coding for proteins of interest.7 Preclinical studies performed so far have explored the genetic engineering of NK-92 cells with CARs targeting multiple tumor-associated antigens, including v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2, also known as HER-2/neu),13 CD19,14 CD20,15 ganglioside GD2,16 epithelial cell adhesion molecule (EPCAM),17 and Epstein-Barr virus (EBV) nuclear antigen 3C (EBNA3C).18 On average, a 50% transduction has been achieved by using fresh NK-92 cells and a lentiviral construct, and the percent purity of transduced cells could be increased to 100% upon cell sorting.7 Therefore, NK-92 cells can provide an “off the shelf,” CAR-customized NK-cell product for anticancer immunotherapy.
NK cells, by virtue of expressing the IgG Fc receptor FcγRIII are also major effectors of antibody-dependent cell-mediated cytotoxicity (ADCC).19,20 Although not all monoclonal antibodies kill target cells through ADCC, in some instances this is their primary killing mechanism.19 In support of this notion, it has previously been shown that patients whose lymphocytes express a high affinity FcγRIII polymorphic variant achieve a better outcome in response to mAbs.21 Unfortunately, only about 10% of the population actually harbors the allele coding for the high affinity FcγRIII variant (V/V), with the majority of individuals expressing the intermediate (F/V) or low affinity (F/F) variants of the receptor.22
Hence, the cytotoxic effects of some mAbs may be augmented by simultaneously infusing NK cells selected for expression of high FcγRIII, as previously demonstrated by in vitro studies.23 The objective of the study presented herein was to compare the cytotoxicity of NK-92 cells expressing CD20-targeting CARs to that of ADCC, as mediated by the anti-CD20 mAbs drugs rituximab and ofatumumab, against a panel of primary NK cell-resistant chronic lymphocytic leukemia (CLL) cells. We further examined whether NK-92 cells expressing CD19- or CD20-targeting CARs exert antitumor effects in xenograft models of human B-lymphoblastic leukemia and lymphoma.
Results
The cytotoxic activity of NK-92 cells expressing CD20-targeting CAR against primary CLL cells is superior to ADCC induced by anti-CD20 monoclonal antibodies
A number of mAbs rely on NK cells as cytotoxic effectors to mediate ADCC.19,20 Here, we compared the abilities of two FDA-approved anti-CD20 mAbs, namely, rituximab and ofatumumab, to elicit ADCC with the cytotoxicity of NK-92 cells transduced with a lentiviral construct for the expression of CD20-targeting CAR. Primary CLL cells from a total of 9 patients with active, untreated disease were tested as targets (Fig. 1). The mean cytotoxicity of NK-92 cells expressing CD20-targeting CAR (αCD20-CAR) was significantly greater than ADCC as mediated by either rituximab or ofatumumab (40.2% ± 2.6 for αCD20-CAR NK-92 cells as compared with 25.1% ± 2.1 and 30.5% ± 3.0 for rituximab and ofatumumab, respectively; p = 0.001 and p = 0.044, respectively).

Figure 1. Cytotoxic potential of ADCC vs. CAR-expressing NK-92. Antibody-dependent cell-mediated cytotoxicity (ADCC) against primary chronic lymphocytic leukemia (CLL) cells (n = 9) as triggered by the anti-CD20 antibodies rituximab (gray, full), and ofatumumab (white, full) as compared with CLL cell killing mediated by NK-92 cells engineered to express a CD20-specific chimeric antigen receptor (αCD20-CAR) (black, full). The cytotoxic response to parental NK-92 cells alone (checkered, control for CAR-dependent cytotoxicity) and parental NK-92 cells with mAbs (stripes, control for ADCC) is presented for each sample. In all experiments, the effector to target cell ratio was 10:1.
Systemic administration of NK-92 cells expressing CD19-targeting CAR controls the growth of metastatic SUP-B15 ALL cells transplanted in immunocompromised mice
Having shown that CAR-expressing NK-92 cells can effectively kill NK cell-resistant primary CLL cells in vitro, we developed a xenograft mouse model to test whether the intravenous infusion of NK-92 cells engineered to express CD19-specific CAR (αCD19-CAR) can eliminate human leukemia in immunocompromised NOD scid gamma (NSG) mice. SUP-B15 is an aggressive Philadelphia chromosome positive (Ph+) CD19+CD20− B-cell acute lymphoblastic leukemia (ALL) line that has been previously shown to be resistant to killing by parental NK-92 cells, yet sensitive to killing by NK-92 cells expressing αCD19-CAR in vitro.7 When injected intravenously with luciferase- (Luc-) tagged SUP-B15 cells, NSG mice succumbed within 20 to 40 d, depending on the size of the inoculum. The distribution of leukemic cells in these mice faithfully recapitulated the distribution of ALL cells observed in humans, including infiltrations into the bone marrow and the central nervous system (cranium and spine; Figure 2A–C). Mice developed signs of illness (hunched posture, scruffiness) and a reduction in mobility that eventually turned into a complete paralysis of the lower body and hind limbs. Cytofluorometric analyses at the time of sacrifice showed extensive infiltration of CD19+CD45+ SUP-B15 blast cells in the bone marrow (femur) and spleen (86.7% ± 3.8 and 50.7% ± 19.5, respectively), as well as in the peripheral blood (14.7% ± 7.7).
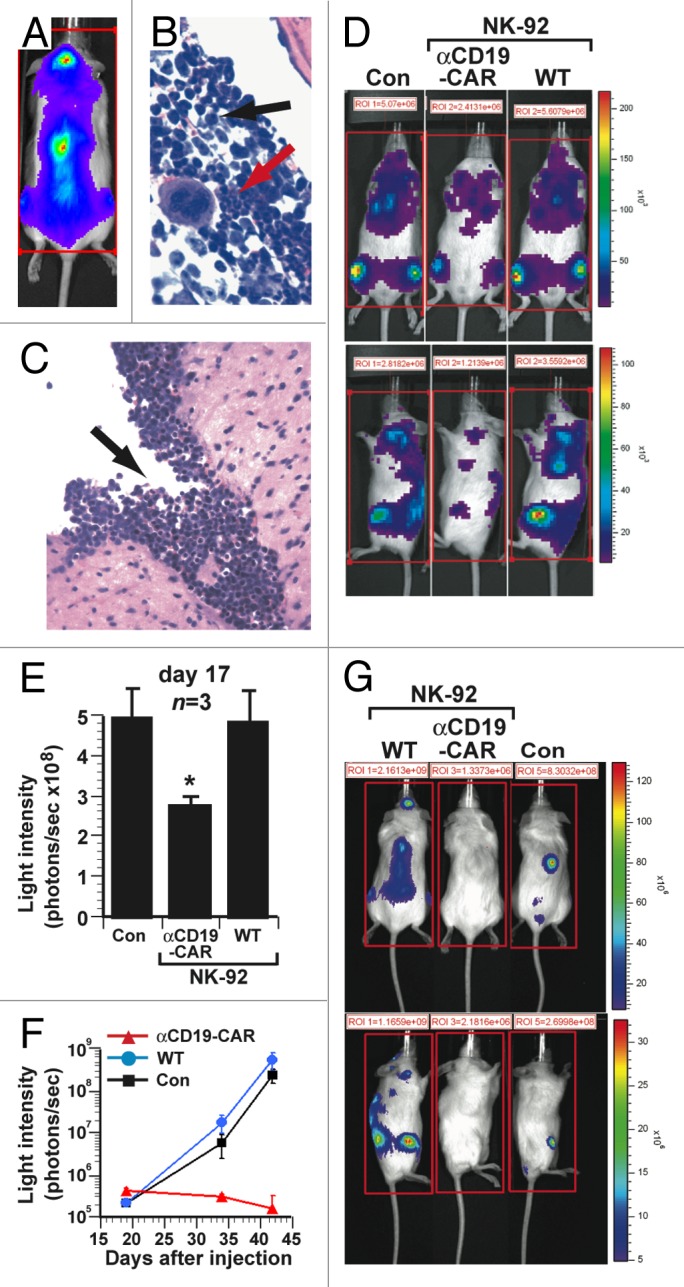
Figure 2. Therapeutic efficacy of NK-92 cells expressing CD19-specific CAR in a xenograft model of aggressive human B-cell acute lymphoblastic leukemia. (A–C) Immunocompromised NOD scid gamma (NSG) mice were inoculated i.v. with 1 × 106 luciferase (Luc)-expressing SUP-B15 acute lymphoblastic leukemia (ALL) cells. (A) Bioluminescence imaging (dorsal view) of mice 17 d after the injection of ALL cells, showing localization to the spine and calvarium. (B) Infiltration of bone marrow by SUP-B15 leukemia cells (black arrow) adjacent to area of normal hematopoiesis (red arrow). (C) Leptomeningeal SUP-B15 leukemic infiltrate. (D–E) NSG mice (n = 3) were inoculated i.v. with 2 × 105 Luc-expressing SUP-B15 cells and subsequently with 1 × 107 parental NK-92 cells (WT), 1 × 107 NK-92 cells expressing CD19-targeting chimeric antigen receptors (αCD19-CAR) or PBS control (Con), on days 1 to 5 post-inoculation. (D) Bioluminescence imaging of representative mice 17 d after the inoculation of ALL cells. (E) Quantification of leukemic burden from the experiment in (D) on day 17. The total bioluminescence (average of 4 sides measurements) in mice receiving NK-92 cells expressing the CD19-targeting CAR was significantly lower than in animals treated with parental NK-92 cells (*p = 0.02). (F-G) NSG mice (n = 3) were inoculated i.v. with 1 × 103 Luc-expressing SUP-B15 cells and then treated with 1 × 107 parental NK-92 cells (WT), 1 × 107 NK-92 cells expressing CD19-targeting CAR (αCD19-CAR) or PBS control (Con), on days 4, 5, and 6. (F) Mean serial bioluminescence of cohorts of leukemic mice. (G) Bioluminescence images of representative mice from cohorts in (F) at day 42. Statistical analyses were performed by unpaired Student t-tests.
Parental NK-92 cells or NK-92 cells engineered to express αCD19-CAR were injected intravenously (1 × 107 cells/dose) through the tail vein on 5 consecutive days, starting 24 h after the intravenous inoculation of 2 × 105 Luc-expressing SUP-B15 cells. Seventeen days after tumor cell injection, bioluminescence assessments showed that parental NK-92 cells had no effect on tumor burden, in sharp contrast to NK-92 cells expressing αCD19-CAR, which significantly reduced, but did not completely eliminate, tumor burden (3.12 × 108 ± 0.33 photons/sec vs. 5.46 × 108 ± 1.16 photons/sec in PBS-treated control mice, p = 0.02; Figure 2D-E). Since cellular therapy is surmised to be more efficacious in biological settings with reduced tumor burden, we titrated the limiting number of tumor-initiating cells by injecting decreasing numbers of Luc-expressing SUP-B15 cells (1 × 104, 1 × 103 and 1 × 102 cells) into mice, determining that 1 × 103 cells was the lowest dose sufficient to induce systemic leukemia. Mice injected with 1 × 103 Luc-expressing SUP-B15 cells were subsequently given intravenous injections of 1 × 107 NK-92 cells on days 4, 5, and 6. As shown in Figure 2F and G, after 6 weeks (42 d), the tumor burden in mice treated with NK-92 cells expressing αCD19-CAR had almost completely disappeared and no malignant cells were detected by flow cytometry in the femur, spleen, or peripheral blood when mice were sacrificed at 4 mo (data not shown). On the contrary, mice injected with 1 × 103 Luc-expressing SUP-B15 cells and treated with parental NK-92 cells exhibited progressive leukemia similar to control mice (Fig. 2F and G).
Therapeutic efficacy of CAR-expressing NK-92 cells against slow-growing TMD-5 ALL xenografts in immunocompromised mice
In addition to rapidly-growing SUP-B15 leukemia cells, we tested a less aggressive, slow-growing Ph+ CD19+CD20+ B-cell ALL cell line, namely TMD-5 cells. Similar to SUP-B15 cells, TMD-5 cells are resistant to parental NK-92 cells in cytotoxicity assays in vitro.7 When administered intravenously into sub-lethally irradiated NSG mice, Luc-expressing TMD-5 cells distributed in a manner similar to SUP-B15 cells, exhibiting a robust accumulation in the bone marrow and central nervous system (Fig. 3A). The relatively slow progression of the disease caused by TMD-5 cells and their distribution to the bone marrow, spleen and liver closely resembled a setting of residual leukemia.

Figure 3. Therapeutic efficacy of NK-92 cells expressing CD19-specific CAR in a xenograft model of slow-growing B-cell acute lymphoblastic leukemia. (A) Bioluminescence imaging of tumor-bearing immunocompromised NOD scid gamma (NSG) mice 70 d after the intravenous injection of 5 × 106 luciferase (Luc)-expressing TMD-5 cells, showing predominant localization to the spine, long bones (bone marrow) and calvarium. (B) Quantification of leukemic burden (average of 4 sides measurements) from mice (n = 2) inoculated intravenously with 5 × 106 Luc-expressing TMD-5 cells followed by the injection of 1 × 107 parental NK-92 cells (W.T.), 1 × 107 NK-92 cells expressing CD19-targeting chimeric antigen receptors (αCD19-CAR), 1 × 107 NK-92 cells expressing CD20-targeting CAR (αCD20-CAR), or PBS control (Con), on days 7, 9, and 11 after the inoculation of leukemic cells. (C) Two NSG mice bearing TMD-5 leukemia were injected with 3 × 106 NK-92 cells expressing CD19-targeting CAR in 50 μL PBS in their right femur (NK-92) and PBS alone in the contralateral one (PBS). Forty-eight h later, mice were re-imaged using the same settings. Note the eradication of disease foci within each femur injected with CAR-expressing NK-92 cells (black arrowheads).
Cytofluorometric analyses of various murine tissues confirmed that TMD-5 cells infiltrate the bone marrow and, after an initial period of slow growth, reach > 90% of the total bone marrow cell number by week 20 (data not shown). Approximately 15 weeks after the inoculation of TMD-5 cells, mice began to show signs of progressive disease including hunched posture, scruffy fur, reduced mobility, and splenomegaly. Death typically occurred around 6 months after the injection of cancer cells. At the time of sacrifice (6 mo), CD19+/CD45+ TMD-5 cells represented > 90% of the total cell number in the bone marrow and spleen (94.2% ± 1.7 and 92.0% ± 1.6, respectively), and 50.6% ± 5.1 of the circulating cells. No hind limb paralysis was observed.
Sub-lethally irradiated NSG mice were injected intravenously with 5 × 106 Luc-expressing TMD-5 cells and subsequently treated with intravenous injections of parental NK-92 cells or NK-92 cells expressing either αCD19-CAR or αCD20-CAR (1 × 107 cells/dose) at days 7, 9, and 11 post-inoculation. Bioluminescence assessments showed that all immunotherapeutic regimens failed to control disease burden (Fig. 3B). Considering that TMD-5 cells are predominantly found in the bone marrow compartment early in the course of disease and, as such, represent a model of minimal residual leukemia, we sought to confirm whether the lack of antileukemic effects observed after the intravenous administration of NK-92 cells could be causally related to issues with the homing of NK-92 cells to disease sites in the bone marrow. To test this hypothesis, we treated NSG mice that had received 5 × 106 Luc-expressing TMD-5 cells with NK-92 cells expressing αCD19-CAR, applied as a single intrafemoral injection 100 d after cancer-cell inoculation. Specifically, 3 × 106 NK-92 cells expressing αCD19-CAR were injected into the right femur, while the left femur was injected with PBS (as a control). CAR-expressing NK-92 cells mediated a robust antitumor effect and leukemia cells were undetectable in the femur receiving NK-92 cells expressing αCD19-CAR in 48 h after treatment, whereas no marked effect could be documented in the contralateral femur (injected with PBS; Figure 3C). At sacrifice, i.e. 12 weeks after the intrafemoral injection of NK-92 cells, leukemia had recurred in the bone marrow (97% CD19+CD45+ Luc-expressing TMD-5 cells), but no disease was detected in the blood or spleen. Furthermore, we documented an expansion of CD56+/GFP+ NK-92 cells (CAR-expressing NK-92 cells) in vivo, especially in the peripheral blood (25% of cells) and in spleen (5%), whereas no NK-92 cells could be detected in the bone marrow (data not shown).
Intratumoral injection of CAR-expressing NK-92 cells controls the local growth of lymphoma
To further investigate the local antitumor effects mediated by CAR-modified NK-92 cells, we induced the formation of a solid lymphoma tumor by subcutaneously injecting 2.5 × 105 Luc-expressing CD19+CD20+ DaudiNKR cells into NOD/SCID mice, followed by the intratumoral injection of parental NK-92 cells or NK-92 cells expressing either αCD19-CAR or αCD20-CAR (5 × 106 per dose) on days 4, 5, and 6 after cancer cell inoculation. As shown in Figure 4, NK-92 cells expressing αCD20-CAR reduced tumor growth as compared with the PBS-treated control cohort, as demonstrated by an increase in bioluminescence of 1.79 ± 0.77 fold vs. 7.58 ± 1.19 fold, respectively (p = 0.001), between days 3 and 7 post-inoculation. In contrast, parental NK-92 did not affect tumor growth, nor did NK-92 cells expressing αCD19-CAR. Furthermore, at sacrifice (25 d), in the three mice of the cohort treated with NK-92 cells expressing αCD19CAR where the increase in bioluminescence was < 1.0 the size of lymphomas had remained stable over the 2 weeks period between imaging and sacrifice (0.10 ± 0.06 g, compared with 0.75 ± 0.10 g average tumor weight at sacrifice in control mice).
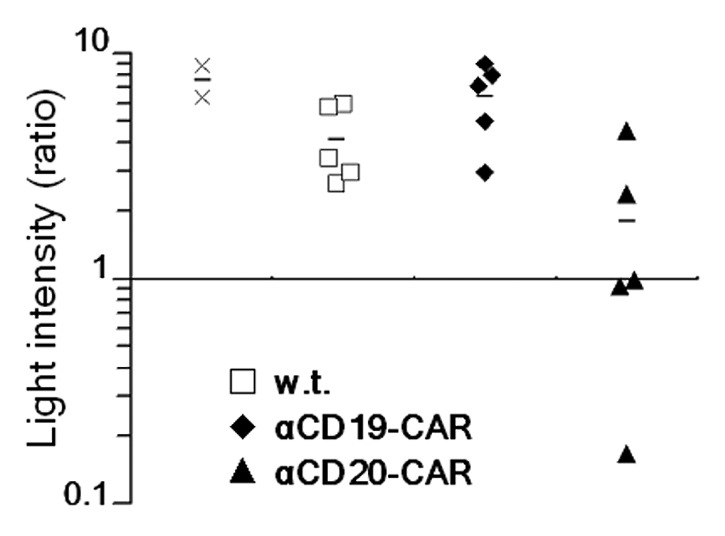
Figure 4. Therapeutic effects of intratumoral injection of CAR-expressing NK-92 cells against subcutaneous Daudi lymphomas. NOD/SCID mice (n = 5) were injected subcutaneously with 2.5 × 105 natural killer (NK) cell-resistant luciferase (Luc)-expressing DaudiNKR cells in PBS. When the tumor had grown to about 0.5 cm3, mice were treated with intratumoral injections of 5 × 106 parental NK-92 cells (w.t.), 5 × 106 NK-92 cells expressing CD19-targeting chimeric antigen receptors (αCD19-CAR), or 5 × 106 NK-92 cells expressing CD20-targeting CAR (αCD20-CAR), on days 4, 5, 6. Results are expressed as fold change in bioluminescence before and after the injection of NK-92 cells. Data relative to each mouse are shown for cohorts of animals treated with NK-92 cells expressing CD19- (black diamonds) or CD20-targeting (black triangles) CARs, treated with parental NK-92 cells (white squares), or treated with PBS (x marks). Mean values (black lines) are also shown for each cohort.
Discussion
Despite recent advances in chemotherapy and stem cell transplantation, a significant proportion of patients with lymphoid leukemia succumb to their disease. The elimination of the clonogenic cancer stem cell requires a multi-modality treatment approach involving cooperative components that should not counteract each other. Cellular therapy is considered one of those treatments and the infusion of autologous T lymphocytes expanded in culture and engineered to express CD19-targeting CARs has recently been shown to control advanced B-lymphoid malignancies.4,5
Much of the focus of cellular immunotherapy so far has been on bulk peripheral T cells, EBV-specific T cells, or lymphoid progenitor cells (reviewed in ref. 24). However, autologous cytotoxic cells are frequently dysfunctional due to prior chemotherapy and/or exposure to the immunosuppressive tumor microenvironment.25 Additionally, the absent or aberrant expression of MHC molecules by malignant cells possibly precludes their recognition by autologous MHC-restricted T cells. Furthermore, allogeneic T lymphocytes can cause acute graft-vs.-host disease (GvHD), a pathological condition that can be debilitating and, in extreme cases, fatal.26 Conversely, NK cells do not induce GvHD and necessitate only the presence of “foreign” epitopes, often mutated MHC antigens, to become fully activated (reviewed in ref. 27). NK cells have been found to be suitable effectors for CAR-dependent immunotherapy, with the caveat that their isolation and expansion from the peripheral blood is labor intensive and costly (reviewed in Refs. 6,28). There is also a significant donor-to-donor variability with respect to NK cells yield and cytotoxic activity.29
As an immortalized NK cell line that can be easily and predictably expanded in culture while maintaining a broad anticancer activity, NK-92 cells provide a potential solution to these issues.8,9 Phase I clinical trials testing NK-92 cells have been completed establishing an acceptable safety profile.11,12 Importantly, NK-92 cells are amenable to be genetically engineered by various methods.7 Six variants of NK-92 cells expressing unique CARs have indeed been reported to date.13-18 Furthermore, since the parental NK-92 cell line does not express endogenous Fc receptors, a NK-92 cell derivative expressing high affinity variant of FcγRIII has been developed.30 This variant has also been employed for quantifying the potential of individual mAbs to trigger ADCC.20
The principal aim of this project was to compare the ADCC triggered by 2 FDA-approved anti-CD20 mAbs against primary CLL cells to the anticancer cytotoxicity achieved by NK-92 cells engineered to express a CD20-specific CAR. Our findings indicate that NK-92 cells expressing αCD20-CAR exert a significantly higher cytotoxicity than the anti-CD20 antibodies rituximab or ofatumumab through ADCC. Although these results suggests that CAR-based immunotherapies may be intrinsically superior to mAb-based approaches, one should bear in mind that mAbs may kill their target through multiple mechanisms, including ADCC, complement-mediated cytotoxicity, and the interference with key signaling pathways. Hence, the actual antineoplastic activity of mAbs in vivo originates from a compilation of these distinct effects. Additionally, the FcγRIII employed in our assays does not possess an intracellular activation domain, and is therefore dependent on endogenous proteins for the transduction of an activatory signal, unlike CAR molecules, which contain the CD3ζ chain of the T-cell receptor (TCR) signaling complex. It should also be noted that FcR and TCR preferentially signal through different tyrosine kinases (SYK/LYN and ZAP70, respectively), which may affect the strength of the transduced signal.
Next, we assessed the antineoplastic potential of CAR-expressing NK cells in xenograft models of human BCR-ABL1+ B-lymphoblastic leukemia in NSG mice, using the B-ALL cell lines SUP-B15 (CD19+CD20−) and TMD-5 (CD19+CD20+). We showed that the intravenous delivery of NK-92 cells expressing a CD19-specific CAR can effectively control the growth of aggressive SUP-B15 cells, but not that of slow-growing TMD-5 cells. The eradication of TMD-5 cells from the bone marrow could only be achieved when the NK-92 cells expressing αCD19-CAR were injected directly into leukemia-infiltrated femurs. In this context, the clearance of leukemic cells from the bone marrow was relatively rapid (within 48 h), suggesting that TMD-5 cells are sensitive to the cytotoxic potential of NK-92 cells in vivo and that NK-92 cells are capable of eliminating a large number of targets upon engagement. It also implies that the lack of response to the intravenous delivery of NK-92 cells is caused by factors that are not directly related to their cytolytic functions. For example, the contact between NK cells and slow-growing TMD-5 cells could alter signaling pathways in NK cells that are critical for their sustained activation.31 TMD-5 cells may also secrete a different spectrum of chemokines than SUP-B15 cells, such that TMD-5 cells may be less efficient at attracting NK-92 cells to the bone marrow. The recurrence of TMD-5 cells in the bone marrow several weeks after apparent clearance could result from either the incomplete elimination of tumor cells by CAR-expressing NK-92 cells or, alternatively, from the re-colonization of the bone marrow by tumor cells coming from other parts of the body. This finding also suggests that NK-92 cells are not capable of long-term engraftment and proliferation in the bone marrow compartment.
We also sought to determine whether NK-92 cells expressing αCD19-CAR or αCD20-CAR exhibit preferential anticancer cytotoxicity. To this aim, we injected NK-92 cells expressing either the CD19- or CD20-targeting CAR into a subcutaneous Daudi lymphoma that expresses both CD19 and CD20. In this system, only NK-92 cells bearing the CD20-targeting CAR were effective, although it is unclear to what extent the relative density of CD19 and CD20 molecules on Daudi cells could have biased these results.
The superiority of CAR-expressing NK-92 cells over ADCC as triggered by anti-CD20 mAbs could be relevant for patients affected by multiple types of hematological cancers. Although we only tested primary tumor cells from patients with CLL, similar data were reported by Tassev et al., who found that the antineoplastic potential of NK-92 cells engineered to express CARs is superior to that of ADCC when the target is an EBV-transformed B-cell tumor.18. Importantly, we found that CAR-expressing NK-92 cells also controlled the growth of some neoplastic B-lymphoid xenografts in immunocompromised mice when given systemically or locally. Altogether, our findings support the initiation of clinical assays testing the antineoplastic potential of CAR-expressing NK-92 in patients with hematological malignancies.
Materials and Methods
Primary effector and target cells
Primary CLL samples from 9 patients with untreated CLL diagnosed according to the National Cancer Institute-Working Group criteria were available for testing. All patients had stage 0 or I CLL (Rai staging system). Mononuclear cells (MNCs) from all samples were obtained by density gradient centrifugation using Ficoll-Hypaque Plus (Amersham Biosciences).
Cells lines and cell culture
NK-92 cells8 were maintained in Myelocult® medium (StemCell Technologies) supplemented with 500 IU/mL Proleukin (recombinant human interleukin-2; Novartis Pharmaceuticals Corporation). The high affinity FcγRIII-expressing NK-92.26.5 cell line (“NK-92Fc”) was provided by Dr K Campbell (Fox Chase Cancer Center).30 Daudi (Burkitt’s lymphoma, CD19+CD20+, NK-92 cell-sensitive) and SUP-B15 (BCR-ABL1+ B-precursor ALL, CD19+CD20−, NK-92 cell-resistant) cell lines were purchased from American Type Culture Collection (ATCC). TMD-5 (BCR-ABL1+ B-ALL, CD19+CD20+, NK-92 resistant) cells were provided by Dr N Nara (Tokyo Medical and Dental University).32 HEK-293T packaging cells were provided by Dr C Kuperwasser (Molecular Oncology Research Institute, Tufts Medical Center). Daudi, SUP-B15, and HEK-293T cells were maintained in RPMI-1640 medium (Mediatech Inc.) supplemented with 20% fetal bovine serum (FBS), 100 µL/ml penicillin, 10 µg/mL streptomycin, and 250 µg/mL amphotericin B (all from Gibco Invitrogen), as well as with 10 µg/mL ciprofloxacin (Mediatech-Corning). TMD-5 cells were maintained in α-MEM medium (Gibco) supplemented with 10% FBS and antibiotics as described above.
Antibodies and cytofluorometry
The anti-CD20 mAb rituximab and the anti-HER2 mAb trastuzumab (also known as Herceptin) were obtained from Genentech/Roche. The anti-CD20 mAb ofatumumab was obtained from GlaxoSmithKline. For the detection of human cells from xenografts by fluorescence cytometry, phycoerythrin (PE)-conjugated anti-CD56, PE-conjugated anti-CD45, and fluorescein isothyocyanate (FITC)-conjugated anti-CD19 mAbs were purchased from BD Biosciences PharMingen. Cytofluorometric analyses were performed on a Dako CyanTM flow cytometer (Beckman-Coulter, Inc) and analyzed using the Summit software (Beckman-Coulter, Inc).
Recombinant lentiviral vectors and lentivirus production
The Luc-expressing reporter construct used in this study was provided by Dr M Rosenblatt (Tufts University School of Medicine). This construct encodes firefly luciferase (Luc), the mCherry fluorescent protein and a puromycin resistance determinant separated by 2A “self-cleaving” peptides, all cloned as a polycistronic cDNA into a pFUW lentiviral vector. The CAR constructs used in this study consist of single chain variable fragments (scFvs) from a murine antibody specific for human CD19 or CD20 linked to the CD3ζ chain of the TCR complex (first-generation CAR). The cDNAs coding for CD19- and CD20-specific CARs were subcloned from the retroviral vector pLXSN14,15 into the lentiviral vector pCL20c-IRES-GFP (provided by Dr R Childs, NHLBI). The CAR-coding constructs were transfected into HEK-293T packaging cells alongside helper plasmids, using the FugeneTM lipofection system (Roche). Culture supernatants were collected after 48 h, filtered (with 0.22 µm filters) and stored at -80°C. The supernatants consistently had a titer of > 107 infectious units/mL.
Lentiviral transduction
Daudi, SUP-B15 and TMD-5 cells were transduced with pFUW-Luc lentivirus and selected with puromycin (Sigma-Aldrich). Daudi cells, which are normally sensitive to the cytotoxic activity of NK and NK-92 cells, lost such sensitivity upon transfection with this construct, and were subsequently referred as DaudiNKR (NK resistant).
NK-92 cells were transduced with pCL20c lentiviral particles as previously described,7 using 2 × 106 cells in 6-well plates mixed with the corresponding lentiviral supernatant and at a multiplicity of infection (MOI) of ~5. Since polybrene is toxic to NK-92 cells, only 15 µg/mL protamine sulfate (Sigma–Aldrich) was used. Transduced cells were expanded in Myelocult® medium supplemented with 1000 UI/mL Proleukin and GFP-expressing (i.e. transduced) NK-92 cells were further enriched by cell sorting to achieve > 95% purity (MoFlo, DakoCytomation). Transgene expression was confirmed by flow cytometry, measuring either mCherry or GFP expression directly, or CAR expression by using biotin-conjugated anti-scFv antibody (Jackson Immunoresearch) and allophycocyanin (APC)-conjugated streptavidin (BD Biosciences PharMingen). Of note, we always observed a strict correlation between GFP and CAR expression.
Cytotoxicity assay
Effector and target cells were co-cultured for 3 h and cytotoxicity was measured by flow cytometry, as previously published.7 ADCC assays were performed as previously described.20 Briefly, CLL cells were incubated with rituximab, ofatumumab, or trastuzumab (as a negative control) for 30 min, followed by the addition of effector cells (parental NK-92 cells or NK-92Fc cells, which expressing high affinity FcγRIII), at an effector to target cell ratio of 10:1. Heat-inactivated FBS was utilized throughout the assays to exclude any contribution of complement-mediated cytotoxicity. For NK-mediated cytolysis, CLL target cells were incubated with parental NK-92 cells or NK-92 cells expressing CD20-specific CAR as above, in the absence of mAbs. Samples were analyzed immediately by flow cytometry. ADCC was calculated by subtracting the percentage of cytotoxicity obtained with mAbs plus parental NK-92 cells (which do not express FcγRIII) from that obtained with mAbs plus NK-92fc cells. The percentage of cytotoxicity obtained with mAbs or effector cells alone was consistently lower than that detected with mAbs plus parental NK-92 cells. Trastuzumab consistently failed to induce ADCC. CAR-dependent killing was calculated by subtracting the percentage of target cell killing obtained with parental NK-92 cells from that obtained with CAR-expressing NK-92 cells.
Xenotransplantation experiments
NOD.CB17-Prkdcscid/J (NOD/SCID) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD scid gamma, or NSG) mice were purchased from the Jackson Laboratory and were housed under sterile conditions in the animal facility at Tufts University School of Medicine. This study was approved by the Tufts Institutional Animal Care and Use Committee. For the Ph+ B-acute lymphoblastic leukemia model, 6- to 10-week old female NSG mice were sub-lethally irradiated with 350 cGy 24 h before the intravenous injection of 2 × 105 or 1 × 103 Luc-expressing SUP-B15 cells, or 5 × 106 Luc-expressing TMD-5 cells (in PBS). Mice were then treated with 3 to 5 intravenous injections of 1 × 107 parental NK-92 cells, or NK-92 cells expressing CD19- or CD20-targeting CARs, every day or every 2 d as indicated. Intrafemoral injections were performed using 3 × 106 NK-92 cells expressing CD19-specififc CAR in PBS, or PBS alone (control), as previously described.33
To test the local effect of CAR-expressing NK-92 cells, non-irradiated 6- to 10-week old female NOD/SCID mice were injected subcutaneously with 2.5 × 105 Luc-expressing DaudiNKR cells in PBS. When the tumor had grown to about 0.5 cm3, mice were treated on days 4, 5 and 6 with intratumoral injections of 5 × 106 parental NK-92 cells or NK-92 cells expressing CD19- or CD20-targeting CARs, in PBS.
In vivo imaging
For the detection of tumor burden in vivo, D-luciferin (Caliper Life Sciences) was injected intraperitoneally (75 mg/Kg in PBS) 10 min prior to imaging. Mice were anesthetized using 2.5% isofluorane and imaging was performed using a Xenogen IVIS 200 Biophotonic Imager (PerkinElmer) at the Tufts small animal imaging facility. Bioluminescence quantification was performed using the Living Image® Software (PerkinElmer), and individual values were recorded as the average of measurements from the 4 sides of the whole body.
Statistical analysis
The SPSS 11.5 software (SPSS) was used to calculate statistical significance using a one-sided Student t-test (independent samples, unpaired). For small sample sizes we also used a Wilcoxon rank-sum test. All p values < 0.05 were considered statistically significant. Data are presented as means ± SEM, unless otherwise noted.
Disclosure of Potential Conflicts of Interest
HK is the Founder and employee of Conkwest Inc. The other authors do not declare any competing financial interest.
Acknowledgments
The authors thank the staff at Flow Cytometry and Small Animal Services Core Facilities of the Tufts Medical Center Cancer Center for assistance. This work was supported by grants from the National Institutes of Health (R01 CA090576 to R.A.V., R01 HL093981 to H.K. and R.A.V., and K08 CA138916 to D.S.K.)
Glossary
Abbreviations:
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ALL
acute lymphoblastic leukemia
- CAR
chimeric antigen receptor
- CLL
chronic lymphocytic leukemia
- FcγRIII
Fc receptor, IgG, low affinity III
- GvHD
graft-versus-host disease
- Luc
luciferase
- NHL
non-Hodgkin lymphoma
- NK
natural killer
- NKR
natural killer resistant
- NSG
NOD scid gamma
Citation: Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, Klingemann H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. OncoImmunology 2013; 2:e26527; 10.4161/onci.26527
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26527
References
- 1.Coiffier B. Effective immunochemotherapy for aggressive non-Hodgkin’s lymphoma. Semin Oncol. 2004;31(Suppl 2):7–11. doi: 10.1053/j.seminoncol.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Micallef IN, Maurer MJ, Wiseman GA, Nikcevich DA, Kurtin PJ, Cannon MW, Perez DG, Soori GS, Link BK, Habermann TM, et al. Epratuzumab with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated diffuse large B-cell lymphoma. Blood. 2011;118:4053–61. doi: 10.1182/blood-2011-02-336990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacher U, Klyuchnikov E, Le-Rademacher J, Carreras J, Armand P, Bishop MR, Bredeson CN, Cairo MS, Fenske TS, Freytes CO, et al. Lymphoma Working Committee of the CIBMTR Conditioning regimens for allotransplants for diffuse large B-cell lymphoma: myeloablative or reduced intensity? Blood. 2012;120:4256–62. doi: 10.1182/blood-2012-06-436725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11:855–73. doi: 10.1517/14712598.2011.573476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg M, Childs R. Ex-vivo expansion of NK cells: what is the priority--high yield or high purity? Cytotherapy. 2010;12:969–70. doi: 10.3109/14653249.2010.536216. [DOI] [PubMed] [Google Scholar]
- 7.Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, Klingemann H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53:958–65. doi: 10.3109/10428194.2011.634048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8. [PubMed] [Google Scholar]
- 9.Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 10.Tam Y, Martinson JA, Doligosa K, Klingemann HG. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. . Cytotherapy. 2003;5:259–72. doi: 10.1080/14653240310001523. [DOI] [PubMed] [Google Scholar]
- 11.Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–32. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 12.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 13.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, Wels W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100:1265–73. [PubMed] [Google Scholar]
- 14.Romanski A, Uherek C, Bug G, Müller T, Rossig C, Kampfmann M, Krossok N, Hoelzer D, Seifried E, Wels W, et al. Re-targeting of an NK cell line (NK92) with specificity for CD19 efficiently kills human B-precursor leukemia cells. Blood. 2004;104a:2747. [Google Scholar]
- 15.Müller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, Tonn T, Wels WS. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57:411–23. doi: 10.1007/s00262-007-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, Aperlo-Iffland C, Huston JS, Uherek C, Schönfeld K, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61:1451–61. doi: 10.1007/s00262-012-1212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassev DV, Cheng M, Cheung NK. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 2012;19:84–100. doi: 10.1038/cgt.2011.66. [DOI] [PubMed] [Google Scholar]
- 19.Alderson KL, Sondel PM. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J Biomed Biotechnol. 2011;2011:379123. doi: 10.1155/2011/379123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weitzman J, Betancur M, Boissel L, Rabinowitz AP, Klein A, Klingemann H. Variable contribution of monoclonal antibodies to ADCC in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:1361–8. doi: 10.1080/10428190903026500. [DOI] [PubMed] [Google Scholar]
- 21.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 22.Lehrnbecher T, Foster CB, Zhu S, Leitman SF, Goldin LR, Huppi K, Chanock SJ. Variant genotypes of the low-affinity Fcgamma receptors in two control populations and a review of low-affinity Fcgamma receptor polymorphisms in control and disease populations. Blood. 1999;94:4220–32. [PubMed] [Google Scholar]
- 23.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jewett A, Tseng HC. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J Cancer. 2011;2:443–57. doi: 10.7150/jca.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dazzi F, Goldman J. Donor lymphocyte infusions. Curr Opin Hematol. 1999;6:394–9. doi: 10.1097/00062752-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR) Cancer Biol Ther. 2009;8:2211–20. doi: 10.4161/cbt.8.23.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koepsell SA, Miller JS, McKenna DH., Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53:404–10. doi: 10.1111/j.1537-2995.2012.03724.x. [DOI] [PubMed] [Google Scholar]
- 29.Klingemann H, Grodman C, Cutler E, Duque M, Kadidlo D, Klein AK, Sprague KA, Miller KB, Comenzo RL, Kewalramani T, et al. Autologous stem cell transplant recipients tolerate haploidentical related-donor natural killer cell-enriched infusions. Transfusion. 2013;53:412–8, quiz 411. doi: 10.1111/j.1537-2995.2012.03764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. 2008;180:6392–401. doi: 10.4049/jimmunol.180.9.6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill AI, Vasey AE, Baker JB, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119:5758–68. doi: 10.1182/blood-2012-03-415364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tohda S, Sakashita C, Fukuda T, Murakami N, Nara N. Establishment of a double Philadelphia chromosome-positive acute lymphoblastic leukemia-derived cell line, TMD5: effects of cytokines and differentiation inducers on growth of the cells. Leuk Res. 1999;23:255–61. doi: 10.1016/S0145-2126(98)00172-6. [DOI] [PubMed] [Google Scholar]
- 33.Mazurier F, Doedens M, Gan OI, Dick JE. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med. 2003;9:959–63. doi: 10.1038/nm886. [DOI] [PubMed] [Google Scholar]