

Summary
Background
Plasminogen (Plg) binding to cell surface Plg receptors (Plg-Rs) on the surface of macrophages facilitates Plg activation and migration of these cells. Histone H2B (H2B) acts as a Plg-R and its cell surface expression is upregulated when monocytes are differentiated to macrophages via a pathway dependent on L-type Ca2+ channels and intracellular Ca2+.
Objectives
We sought to investigate the mechanism by which H2B, a protein without a transmembrane domain, is retained on themacrophage surface.
Methods
THP-1 monocytoid cells were induced to differentiate with interferon gamma + Vitamin D3 or to undergo apoptosis by treatment with camptothecin. Flow cytometry and cell surface biotinylation followed by Western blotting were used to measure the interrelationship between Plg binding, cell surface expression of H2B and outermembrane exposure of phosphatidylserine (PS).
Results
H2B interacted directly with PS via an electrostatic interaction. Anti-PS or PS binding proteins, annexin V and protein S, diminished H2B interaction with PS on the surface of differentiated or apoptotic cells and these same reagents inhibited Plg binding to these cells. L-type Ca2+ channels played a significant role in PS exposure, H2B surface expression and Plg binding induced either by differentiation or apoptosis.
Conclusions
These data suggest that H2B tethers to the surface of cells by interacting with PS on differentiated or apoptotic monocytoid cells. L-type Ca2+ channels regulate PS exposure on the surface of these cells. The exposed PS interacts directly with H2B and hence provides sites for Plg to bind to.
Keywords: histone H2B, phosphatidylserine, plasminogen, plasminogen receptor
Introduction
Plasmin, the active serine protease formed from plasminogen (Plg), is essential for efficient clearance of blood clots via its fibrinolytic activity and also facilitates cell migration via its degradation of extracellular matrix constituents. Plg activation on cell surfaces depends on its binding to a heterogeneous group of molecules bearing C-terminal lysines or mimics of this residue, which are collectively referred to as plasminogen receptors (Plg-Rs) [1,2]. Many Plg-Rs are atypical membrane proteins as they bear no signal sequence or transmembrane domains. Examples of such Plg-Rs are the glycolytic enzyme α-enolase [3] and the nuclear DNA binding protein, histone H2B (H2B) [4]. While these proteins are found primarily in the intracellular compartment, their expression at the cell surfaces is well documented and, importantly, cell-surface expression of these Plg-Rs can be modulated by changes in the activation [5,6] and adhesive status of cells [7], thereby providing a mechanism to regulate plasmin-mediated cell migration. Studies conducted by our group and others have detected cell surface expression of H2B when monocytes differentiate into macrophages [6,8], when lymphocytes are activated to lymphoblasts [9], or when cells are undergoing apoptosis [10]. On macrophages, H2B contributes significantly to Plg binding, plasmin generation and migration of these cells to sites of inflammation in vivo [6].
In investigating the mechanism for translocation of H2B to the macrophage surface, we demonstrated a major role for L-type Ca2+ channels (LTCC) using both pharmacological and genetic inhibitors [8]. The LTCC controlled elevation of intracellular Ca2+ in activated monocytes, which, in turn, controlled movement of H2B as well as other Plg-Rs, including α-enolase, to the macrophage surface [8]. However, the mechanism by which translocated H2B tethers to the cell surface is unknown.
Previous studies [11,12] have shown that histone proteins can interact with anionic phospholipids, such as phosphatidylserine (PS), when immobilized on microtiter plates. PS typically constitutes 8–15% of the total phospholipid content of the plasma membrane of mammalian cells, and is normally restricted to the inner leaflet, whereas the outer leaflet is composed mainly of the neutral phospholipid, phosphatidylcholine (PC). PS exposure on the surface of apoptotic cells serves as a ligand for macrophages that express PS binding proteins such as CD14 and CD36. On the other hand, PS expression also occurs when monocytes differentiate into macrophages, where they contribute to the phagocytic functions of these cells [13]. Differentiation of the monocytoid U937 cell line, as well as human primary monocytes, into macrophages, is associated with surface expression of PS [14]. This phenomenon is unaffected by caspase inhibitors, indicating that differentiation-induced PS exposure may follow a pathway distinct from apoptosis. In the present study, we have begun to assemble the pathway for enhanced Plg binding to macrophages by demonstrating that H2B localizes to the cell surface by interacting with PS and that Ca2+ mobilization via LTCC regulates PS exposure on macrophages.
Methods
Monocyte cell culture and differentiation
Human monocytoid THP-1 cells were obtained from the ATCC (American type culture collection, Manassas, VA, USA) and cultured in RPMI 1640 with 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g L−1 glucose, 1.5 g L−1 sodium bicarbonate, 0.05 mM 2-mercaptoethanol and 10% heat inactivated fetal bovine serum (FBS). THP-1 cells were stimulated to either differentiate with the combination of 250 U mL−1 IFNγ(eBioscience, Sandiego, CA, USA) and Vitamin D3 (1a, 25-dihydroxy, 100 nM; EMD Biosciences, San Diego, CA, USA) for 0–2 days or to induce apoptosis with camptothecin (5 μM; EMD Biosciences) for 0–1 day in complete medium. For human monocytes, human leukocytes were isolated from peripheral blood of healthy donors using Ficol Hypaque Plus (GE Healthcare Bioscience, Piscataway, NJ, USA). A portion of the leukocytes was used for FACS staining where the monocyte population was identified by PE-conjugated anti-human CD14 (eBioscience, San Diego, CA, USA) staining [15]. The remainder of the leukocytes was allowed to adhere onto fibronectin-coated plastic plates (BD Biosciences, Bedford, MA, USA) for 2 h at 37 °C. After washing, the adherent cells were either induced to differentiate by culturing them for an additional 5 days or induced into apoptosis by treatment with camptothecin (5 μM) for 24 h in RPMI-1640 with 10% human AB serum (Lonza Walkersville, Walkersville, MD, USA).
Plg binding
Plg binding was measured as described previously [7]. Details of this method are described in Data S1.
Cell surface biotinylation and Western blotting
THP-1 cells were differentiated with IFNγ + VD3 for 0–2 days. The cells were then surface labeled with sulfo-NHS-biotin (Thermo Fisher Scientific, Rockford, IL, USA), and biotinylated proteins were isolated by binding and elution from streptavidin beads as previously described [8]. Alternatively, cells were lysed in SDS, and the lysates were subjected to Western blotting using rabbit anti-peptide antibodies against α-enolase or H2B [6]. CD14 expression was detected in Western blots with rabbit anti-human CD14 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The intensities of Western blot bands were measured using Kodak ID 3.6 software (Eastman Kodak, Rochester, NY, USA), assigning the intensity of each selected protein in the unstimulated cells a value of 1.0; the fold change of a particular protein was then calculated relative to its intensity in the unstimulated cells.
KCl washing of cells
IFNγ+ VD3 THP-1-treated (48 h) or untreated cells were suspended in 0.5 or 1 MKCl in HBSS, pH 7.4, for 5 min. The cells were then washed three times with HBSS and used to analyze Plg binding or Plg-R expression by cell surface biotinylation as described above.
Binding of histones to phospholipids
Phospholipids were immobilized onto the wells of polystyrene microtiter plates as described [11]. Briefly, wells were coated with either PS (50 μg mL−1) in chloroform/methanol (4:1) or PC (50 μg mL−1; Avanti polar lipids, Alabaster, AL, USA) in ethanol overnight at 4 °C. Wells were blocked with 0.3% gelatin in PBS and allowed to bind either purified H2B (5 μg mL−1; New England Biolabs, Ipswich, MA, USA) or α-enolase (MorphoSys, Kidlington, UK) in 0.3% gelatin in PBS for 1 h at 22 °C. Wells were washed with PBS or varying concentrations of KCl. Bound H2B or α-enolase was detected with rabbit anti-H2B or rabbit anti-enolase followed by Alexa-488 labeled anti-rabbit Ig.
Flow cytometry
To detect PS exposure on differentiated or apoptotic cells, THP-1 cells were pretreated with either zVAD-fmk (Calbiochem), amlodipine (Sigma-Aldrich, St. Louis, MO, USA) or verapamil (Sigma-Aldrich) for 1 h and then treated with either IFNγ + VD3 for 48 h to induce differentiation or camptothecin (Calbiochem) for 24 h to induce apoptosis. PS exposure was measured by FITC-annexin V (BD Biosciences) staining according to the manufacturer’s protocol for FACS, and the CellQuest (BD Biosciences) was used for data analyses. To show the requirement for PS in H2B cell surface localization, we used two different mouse anti-PS monoclonal antibodies. One was obtained from Millipore (Billerica, MA, USA) and is an IgG, and the other was kindly provided by N.S. Rote from Case Western Reserve University, Cleveland, and is an IgM antibody. This latter antibody has been extensively characterized, including its ability to block PS-dependent functions [16,17]. THP-1 cells were differentiated with IFNγ+ VD3, in the presence of either anti-PS antibodies or its mouse IgG (Sigma) or IgM (BD Pharmingen) isotype controls. Cells were washed, preblocked with human FcR blocking reagent (Miltenyl Biotec, Auburn, CA, USA), incubated with anti-H2B and further incubated with Alexa-488 labeled goat anti-rabbit IgG in 0.1% BSA in HBSS. The extent of antibody binding was measured by FACS. To show that apoptosis induced H2B surface expression and Plg binding, camptothecin- treated cells were labeled for FACS with either anti-H2B followed by Alexa-488 anti-rabbit Ig or with Alexa-488 Glu-Plg.
Plasmin generation
THP-1 cells were either untreated or treated with IFNγ+ VD3 in the absence or presence of anti-PS IgM or its isotype IgM control. Cells were washed in HBSS buffer and used to measure plasmin generation using S-2251 as a substrate as described previously [5].
Statistical analysis
Values were expressed as means ± SD. For multiple group comparisons, a one-way ANOVA test of SigmaPlot version 10.0 (Systat Software, San Jose, CA, USA) was used and for two group comparisons, a paired Student’s t-test was applied. Results were considered statistically significant with P values of < 0.05.
Results
H2B binds to phosphatidylserine via an ionic interaction
While our prior studies demonstrated that H2B is a major contributor to Plg binding of both murine and human macrophages [6] and that expression of H2B during monocyte to macrophage differentiation is dependent on intracellular calcium mobilization and driven by LTCC [8], the mechanism by which H2B tethers to the cell surface is undetermined. Histones are known to bind to anionic phospholipids [11,12], and we considered whether H2B binding to PS, an anionic phospholipid that becomes surface expressed during monocyte to macrophage differentiation, might serve as an H2B binding site. Microtiter wells were coated with PS or PC, a neutral lipid control, and their ability to bind purified human recombinant H2B was assessed using anti-H2B followed by binding of Alexa-488 anti-rabbit Ig. As shown in Fig. 1(A), H2B does bind DNA but not BSA, which serves as a positive and a negative control for these experiments. PS also binds H2B, whereas PC does not. H2B interaction with PS was only detected at the 5 μg mL−1 input concentration but not at 0.4 and 1 μg mL−1 H2B under the assay conditions used. However, when the incubation time was extended, interaction at the lower concentrations of H2B was also detected. Thus, compared with DNA, the binding of H2B to PS appears to be weaker.
Fig. 1.

Binding of human H2B to phosphatidylserine (PS). (A) Recombinant H2B was allowed to bind to microtiter wells coated with BSA, PS, PC or DNA. Recombinant H2B (0.4–5 μg mL−1) was added to the wells for 2 min at 22 °C. After washing thoroughly with PBS, anti-H2B, followed by Alexa-488 anti-rabbit IgG, was added to detect bound H2B. H2B bound to DNA (positive control) and PS but not to BSA or PC. *P<0.001 for PS and DNA vs. BSA as analyzed by a one-way ANOVA test. (B) Binding of H2B (5 μg mL−1) to immobilized PS was inhibited by KCl, indicative of an electrostatic interaction. Data are the means ± SD from triplicates. (C) H2B or α-enolase was added to BSA or PS-coated wells, and anti-H2B or anti-enolase was used for detection. Binding anti-enolase to enolase-coated wells is a control to validate the detection system. *P = 0.01 for PS vs. BSA. #P = 0.02 for enolase vs. BSA as analyzed by Student’s t-test.
To determine the nature of interaction between H2B and PS, PS-coated plates were presented with H2B in the presence of various concentrations of KCl to disrupt ionic interactions. KCl disrupted the interaction of H2B with PS in a concentration- dependent manner; 50% inhibition was observed at approximately 1.5 M (Fig. 1B). When PS- and PC-coated wells were treated with KCl first, washed and then H2B was added, H2B bound at a similar level to that observed without the KCl prewash, indicating that KCl did not simply elute PS from the plate but disrupted the PS:H2B interaction. As a control, we tested the capacity of another Plg-R, α-enolase, to bind to PS. Using anti-enolase as a disclosing reagent, α-enolase did not bind to immobilized PS or BSA; however, the anti-enolase did bind to wells coated directly with α-enolase (Fig. 1C).
H2B binds to phosphatidylserine on THP-1 cells and this interaction contributes to plasmin generation
To investigate whether H2B binds to cell surfaces via an anionic interaction, THP-1 monocytoid cells were treated with IFNγ+ VD3 for 48 h to induce differentiation towards macrophage-like cells and to up-regulate their surface expression of H2B and Plg binding [8]. The cells were then washed for 5 min with 0.5 and 1 M KCl, a concentration that did not affect cell viability as assessed by propidium iodide staining. The KCl was then removed by washing, and the cells were analyzed for Plg binding by FACS or H2B surface expression by cell surface biotinylation followed by Western blotting. A 0.5 M KCl wash of the cells led to a marked reduction of Plg binding (Fig. 2A). In parallel, KCl also reduced surface expression of H2B as assessed by Western blotting (Fig. 2B, left). By densitometry, the reduction in differentiation-induced surface H2B at 0.5 M KCl was approximately 70% (Fig. 2B, right). H2B interaction with immobilized PS was reduced by approximately 30–40% at 0.6–1.2 M (Fig. 1B). While higher concentrations of KCl (2.4 M) could completely block H2B binding to immobilized PS, the cells could not tolerate concentrations of KCl above 1 M. Indeed, 1 M KCl did induce mild apoptosis of the differentiated THP-1 cells, which may explain the slight increase in surface H2B observed at 0.5 and 1.0 M KCl (see below). In contrast to H2B, α-enolase, another Plg-R that does not bind to PS (Fig. 1C), was not eluted from the cells by KCl. Also CD14, which is an integral membrane protein and a marker of monocyte to macrophage differentiation, was unaffected by KCl concentrations up to 1 M(Fig. 2B). We have also analyzed the KCl-treated cells for their alteration in PS expression by FITC-annexin V. At 0.5 M KCl treatment, when we observed maximum H2B elution (Fig. 2), PS expression was unaffected by the KCl wash (FITC-Annexin V mean fluorescence intensity [MFI] = 138 ± 12 without KCl, MFI = 145 ± 16 with 0.5 M KCl wash). PS expression measured by FACS at 1 M KCl increased (due to some apoptosis), rather than decreased. Therefore, the decrease in H2B surface expression at both KCl concentrations used (0.5 and 1 M) to wash the differentiated THP-1 cells was not due to elution of PS.
Fig. 2.

H2B binds to the macrophage surface via PS and this interaction contributes to plasmin generation. (A) Binding of Alexa-488 Plg as detected by FACS to THP-1 cells induced to differentiated by IFNγ+ VD3 (48 h). The cells were either untreated or treated with KCl for 5 min and washed before addition of the labeled Plg.KCl reduced Plg binding to differentiated THP-1 cells compared with unwashed cells (*P ≤ 0.001, by a one-way ANOVA). (B) Effect of KCl washes (5 min) on H2B surface expression. After washing, the cells were biotinylated, and the biotinylated proteins were subjected to Western blotting (left panels). Intensities of the Western blot bands were measured by densitometry and expressed as the fold-increase in H2B, α-enolase and CD14 relative to their levels on non-stimulated THP-1 cells (right panel). Data are the means ± SD from triplicate blots. H2B, striated bars; α-enolase, gray bars; CD14, open bars. *P ≤0.002 vs. cell surface H2B on IFNγ+ VD3-treated cells as analyzed by Student’s t-test. KCl treatment eluted surface H2B but did not alter surface expression of α-enolase and CD14. (C) THP-1 cells were differentiated with IFNγ+ VD3 in the absence or presence of either anti-PS (50 μg mL−1) or its isotype control for 48 h. Cells were subjected to FACS staining by anti-H2B followed by Alexa-488 anti-rabbit IgG. Specific mean fluorescence intensity (MFI) values were quantified by subtracting the binding obtained with secondary antibody alone. Data are means ± SD from triplicate experiments. Anti-PS reduced H2B surface expression on the IFNγ+ VD3-treated cells compared with IFNγ+ VD3 and isotype control-treated cells (*P < 0.001 by ANOVA). The isotype control has no inhibitory effect on IFNγ+ VD3-mediated H2B surface expression. (D) THP-1 cells were treated with IFNγ+ VD3 in the absence or presence of anti-PS antibody (50 μg mL−1) and its isotype control (IgM). Cells were incubated with Plg (200 nM) for 1 h and then uPA(3 nM) was added with the chromogenic substrate S-2251. Plasmin generation was measured at 405 nm over 2 h. Data are representative of three experiments. Note the 60% reduction of plasmin generation at 120 min in anti-PS-treated cells compared with isotype control-treated cells.
To verify the role of PS in surface localization of H2B and Plg binding, THP-1 cells were differentiated with IFNγ + VD3 for 48 h in the presence of a monoclonal anti-PS or its isotype control. As shown in Fig. 2(C), differentiation enhanced H2B surface expression by 5-fold as assessed by FACS. This induction of H2B surface expression was inhibited 55% by an anti-PS IgM, whereas its isotype IgM control had no inhibitory effect. A similar extent of inhibition was observed when the anti-PS IgG was used (data not shown).
In our previous study [4], we used an antibody to H2B that blocked its binding of Plg to implicate their interaction in plasmin generation on the surface of macrophage-related cells. We now sought to test whether PS is important in plasmin generation on the differentiated THP-1 cells. THP-1 cells were treated with IFNγ+ VD3 to induce differentiation in the absence or presence of anti-PS IgM or its isotype control. Cells were then allowed to bind with Plg, and plasmin generation was measured as a function of time upon addition of uPA and a chromogenic substrate for plasmin, S-2251. As shown in Fig. 2(D), upon IFNγ + VD3 treatment, a 2.3-fold increase in plasmin generation was observed at the 2 h point compared with untreated THP-cells. In the presence of the anti-PS, uPA-mediated Plm generation was suppressed by 60% compared with isotype control. Furthermore, with PS-coated microtiter plates, we found that addition of H2B enhanced plasmin generation by 2-fold compared with the absence of H2B (not shown).
Regulation of H2B surface expression and plasminogen binding by PS binding partners
Annexin V and protein S are two high affinity PS binding proteins (Kd values estimated at 15–0.03 nM, respectively) [18,19]. To demonstrate the PS interaction with H2B on the cell surface, THP-1 cells were treated with IFNγ+ VD3 for 48 h in the presence of either annexin V or protein S. Cell surface biotinylation followed by Western blotting showed a dose-dependent inhibition of H2B surface localization by either annexin V or protein S, whereas α-enolase surface localization, which does not interact with PS (Fig. 1C), was unaffected by these two PS binding proteins (Fig. 3A). Of note, these reagents that inhibited H2B surface expression associated with differentiation did not inhibit differentiation of IFNγ+ VD3-treated cells per se as indicated by CD14 and αMβ2 integrin expression levels measured by FACS (data provided as supporting information, Fig. S1). Thus, both annexin V and protein S interfere with H2B association with the cell surface.
Fig. 3.

Inhibition of cell surface H2B expression and Plg binding by annexin V and protein S. (A) THP-1 cells were differentiated with IFNγ+ VD3 in the absence or presence of various concentrations of annexin V or protein S for 48 h. Cell surface biotinylation followed by Western blot of biotinylated proteins with anti-H2B indicated a dose-dependent inhibition of H2B localization on the cell surface. Annexin V and protein S did not alter differentiation-induced α-enolase and CD14 cell surface localization. Intensities of the Western blot bands of H2B, α-enolase and CD14 are expressed as the fold increase compared with non-stimulated THP-1 cells (lower panel). Bar represents means ± SD from three independent Western blots; H2B, striated bars; α-enolase, gray bars; CD14, open bars. (B) THP-1 cells were treated with IFNγ+ VD3 in the presence or absence of annexin V or protein S, and Alexa-488 Plg binding was analyzed by FACS. Data are presented as means ± SD of four independent experiments. Differentiation-induced Plg binding was inhibited by annexin V and protein S.*P < 0.001 for IFNγ+ VD3 vs. untreated cells, **P < 0.001 for annexin V vs. IFNγ+ VD3 alone treated cells and ***P ≤ 0.001 for protein S vs. IFNγ+ VD3 alone treated cells as analyzed by ANOVA.
To determine the consequence of loss of H2B on Plg binding, THP-1 cells were treated with IFNγ + VD3 for 48 h in the presence of either annexin V or protein S and analyzed for their capacity to bind Plg. As depicted in Fig. 3(B), differentiation enhanced Plg binding by 3.5-fold. This induced binding was inhibited in a dose-dependent manner by annexin V and protein S. A 60–70% reduction of differentiation-induced Plg binding was observed at 2 μM annexin V and protein S, and H2B binding to the cell surface was completely inhibited. These data are consistent with a previous study showing that protein S suppressed Plg binding to THP-1 cells through an undefined mechanism [20].
Comparison between differentiation and apoptosis-induced PS exposure on Plg binding and H2B cell surface expression
In addition to exposure of PS during differentiation [13,14], apoptosis of macrophages leads to surface expression of PS [21,22]. We sought to compare how these two pathways of PS exteriorization influence Plg binding and H2B surface expression. As shown in Fig. 4(A), cells induced to differentiate with IFNγ + VD3 increased their surface exposure of PS (3-fold), and this increase was insensitive to zVAD-fmk, a broad spectrum caspase inhibitor. When the THP-1 cells were treated with camptothecin to induce apoptosis, they also increased their PS exposure (7-fold), but this increase was completely blocked by zVAD-fmk (Fig. 4A). Differentiation enhanced Plg binding (Fig. 4B) and H2B (Fig. 4C) surface expression by 3.3- and 4.7-fold, respectively, compared with the undifferentiated cells, and these increases were also unaffected by zVAD-fmk. Apoptosis induced by camptothecin was associated with a 10-fold increase in Plg binding (Fig. 4B) and a 4.6-fold induction of H2B surface expression (Fig. 4C). In contrast to differentiation, apoptosis induced Plg binding (Fig. 4B) and cell surface expression of H2B (Fig. 4C) was 70–80% inhibited by z-VAD-fmk. Thus, two distinct pathways for PS exposure, differentiation and apoptosis, lead to enhanced exteriorization of H2B. To verify the independence of these pathways in inducing PS exposure, we tested whether IFNγ+ VD3 induced apoptosis and whether camptothecin induced differentiation by FACS analyses. Upon IFNγ+ VD3 treatment, THP-1 cells showed an increased CD14 and αMβ2 integrin surface expression, markers of differentiation [23] and only a very small percentage (7% of total population) were positive for annexin V and propidium, indicative of apoptosis, compared with untreated cells. On the other hand, THP-1 cells treated with camptothecin showed no significant difference in CD14 and integrin expression compared with untreated cells, even though 55% of the cells were positive for annexin V + PI staining (see Fig. S2 of supporting information).
Fig. 4.

PS exposure, Plg binding and H2B surface expression induced by differentiation and apoptosis. THP-1 cells were pretreated with zVAD-fmk, amlodipine and verapamil for 1 h or were untreated. The cells were either differentiated with IFNγ+ VD3 for 48 h or induced to apoptosis with camptothecin for 24 h and then labeled with either FITC-annexin V to detect PS exposure (A), Alexa-488 Plg (B) or anti-H2B followed by Alexa-488 anti-rabbit IgG (C) and analyzed by FACS. Data are presented as fold change over untreated THP-1 cells from three independent experiments. *P < 0.001 vs. untreated cell; #P ≤0.001 vs. camptothecin-treated cells as analyzed by a one-way ANOVA test.
We have previously shown that L-type Ca2+ channels (LTCC) are required for increased intracellular Ca2+ and H2B surface exposure during monocyte differentiation [8]. Here, we sought to determine the role of LTCC in PS exposure associated with differentiation and apoptosis. As shown in Fig. 5(A), 10 μM concentrations of the LTCC blockers, amlodipine and verapamil, inhibited differentiation-induced PS exposure by ≥90%. Camptothecin-induced PS exposure was reduced by approximately 50% by both amlodipine and verapamil (Fig. 5A). Because LTCC are involved in PS exposure during both differentiation and apoptosis, we compared the effects of these pathways on Plg binding and H2B surface expression. Differentiation-induced Plg binding (Fig. 5B) and H2B cell surface expression (Fig. 5C) was completely abrogated by amlodipine or verapamil. The LTCC inhibitors reduced camptothecin-induced Plg binding by 45–50% (Fig. 5B). Similarly, camptothecin-induced H2B surface expression was reduced by approximately 50% by both amlodipine and verapamil (Fig. 5C). Thus, two distinct pathways for PS exposure lead to LTCC-dependent increases in Plg binding and H2B surface expression. The increases in Plg binding and H2B expression associated with differentiation are entirely dependent on LTCC, whereas these events are partially dependent on LTCC on apoptotic cells.
Fig. 5.

Effect of the L-type calcium channel blockers, amlodipine and verapamil, on PS exposure, Plg binding and H2B surface expression. THP-1 cells were either pretreated with amlodipine or verapamil for 1 h or were untreated. The cells were then differentiated with IFNγ + VD3 for 48 h or induced to apoptosis with camptothecin for 24 h. Cells were labeled with FITC-annexin V (A), Alexa-488 Plg (B) or anti-H2B followed by Alexa-488 anti-rabbit IgG (C) and analyzed by FACS. Data are presented as fold change over untreated THP-1 cells from three independent experiments. *P ≤0.001 vs. untreated cells; #P ≤ 0.001 vs. IFNγ+ VD3 alone treated cells; **P ≤ 0.001 vs. camptothecin alone treated cells as analyzed by ANOVA test.
To extend the observations regarding PS exposure, Plg binding and H2B surface localization, we turned to primary monocytes derived from human peripheral blood. These cells were either directly stained for PS exposure, Plg binding and H2B surface expression or induced to undergo differentiation or apoptosis and then analyzed by FACS. As shown in Fig. 6, there was a 6.4- and 12.5-fold increase of PS exposure (Fig. 6A) when monocytes were induced to undergo differentiation or apoptosis, respectively. This increase of PS exposure was associated with a 7.2- and 12.4-fold increase in Plg binding (Fig. 6B) and a 7.3- and 8-fold increase of H2B surface expression (Fig. 6C) in association with differentiation and apoptosis, respectively. These results emphasize the close correlation between PS exposure, Plg binding and surface expression of H2B on both differentiated and apoptotic primary cells and recapitulates our observations with THP-1 cells (Figs 4 and 5).
Fig. 6.
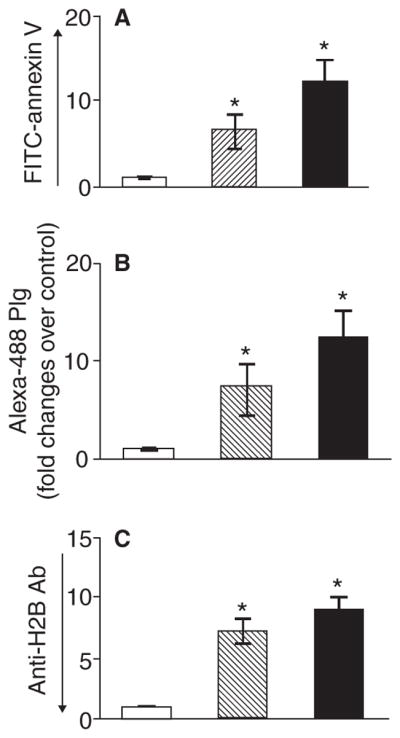
Phosphatidylserine exposure, Plg binding and H2B surface expression on human peripheral blood monocytes induced to differentiate or to undergo apoptosis. Isolated human blood monocytes (open bar) were either subjected to FACS analysis or cultured. The cultured cells were either induced to differentiate (striated bar) by continued culture for 5 days or induced to undergo apoptosis with camptothecin (black bar) for 24 h. The cells were then harvested and labeled with either FITC-annexin V to detect PS exposure (A), Alexa-488 Plg (B) or anti-H2B followed by Alexa-488 anti-rabbit IgG (C) and analyzed by FACS. Data are presented as fold change compared with freshly isolated monocytes from three independent experiments. *P < 0.001 vs. untreated cells as analyzed by one-way ANOVA test.
Our previous study [5] demonstrated that differentiation of THP-1 with IFNγ + VD3 induces intracellular Ca2+ mobilization. To test the effect of camptothecin on changes in intracellular Ca2+ and their correlation with PS exposure on THP-1 cells, cells were treated with camptothecin in the absence or presence of amlodipine and verapamil for 24 h. Cells were then loaded with Fluo-4AM as Ca2+ indicator followed by staining with PE conjugated annexin V. Cells were then analyzed by FACS. As shown in Fig. S3 of supporting information, camptothecin increased intracellular Ca2+ levels by 1.4-fold and this induction was inhibited by both amlodipine and verapamil by approximately 50%. The increased intracellular Ca2+ by camptothecin was associated with a 7-fold increase in PS levels, and this induction of PS expression was inhibited by both amlodipine and verapamil by 48–50% (Fig. S3 of supporting information).
Phosphatidylserine provides spare receptors for H2B and plasminogen
Because the increase in H2B surface expression was similar on differentiated and apoptotic cells (4.7- and 4.6-fold, respectively), but yet PS exposure was substantially more extensive on apoptotic cells, we tested whether camptothecin-treated cells might have spare receptors for H2B binding. Cells were either induced to differentiate with IFNγ + VD3 or apoptosis with camptothecin, and exogenous H2B was added. After 1 h at 22 °C, the cells were washed and H2B surface expression was measured by FACS. The exogenously added H2B led to a 2.5-fold increase on differentiated cells and 3.9-fold increase on apoptotic cells of H2B surface expression (Fig. 7A). These increments in H2B were associated with further increases (1.6- fold on differentiated and 2-fold on apoptosis relative to the differentiated or apoptotic cells, respectively) in Plg binding (Fig. 7B). These data indicated that both differentiated and apoptosis cells have the ability to bind exogenous H2B but camptothecin-treated cells bound more H2B than IFNγ + VD3 treated cells, reflecting the difference in amount of PS on the surface of these two populations of cells. Anti-PS IgM inhibited 80% and 70% of exogenous H2B binding to differentiated cells and apoptotic cells compared with the isotype control IgM.
Fig. 7.

Role of PS in apoptosis-induced Plg binding and H2B surface expression. IFNγ+ VD3 and camptothecin-treated (24 h) cells were either untreated or pretreated with anti-PS or its isotype control IgM. Cells were washed and allowed to bind exogenously added recombinant H2B (5 μg mL−1). Cells were then washed and subjected to FACS staining with either anti-H2B (A) or Alexa-488 labeled Plg (B). Specific mean fluorescence intensities are presented. Data are the means ± SD from triplicate experiments. *P < 0.001 vs. untreated; #P < 0.001 vs. IFNγ+ VD3 treated; ##P < 0.001 vs. IFNγ+ VD3 + ExoH2B treated and IFNγ+ VD3 + ExoH2B+isotype IgM treated; §P < 0.001 vs. camptothecin treated; §§P < 0.001 vs. camptothecin + ExoH2B treated and camptothecin + ExoH2B + isotype IgM treated, as analyzed by one-way ANOVA test.
Discussion
Histone H2B is a major receptor for Plg on the surface of macrophages, where it enhances Plg activation and contributes to Plg-dependent recruitment of these cells during inflammatory responses in vivo [6]. It is clear that H2B is not the only Plg-R that contributes to these responses, but it is very prominent among them. In previous studies, we demonstrated that translocation of H2B to the cell surface during differentiation of monocytes into macrophages depends upon Ca2+ mobilization and LTCC. The present study was undertaken to define the mechanism by which H2B is retained on the cell surface as it becomes exteriorized. Our data indicate that, once mobilized to the cell surface through a LTCC-dependent pathway, a major pathway for H2B retention on cell surfaces depends on it binding to PS via an ionic interaction.
Several lines of evidence have been developed to indicate that PS is critical for H2B retention at the surface of the macrophage and consequently for Plg binding. First, disruption of ionic interactions with KCl not only inhibited the direct interaction of H2B with PS in vitro but also eluted H2B from the cell surface. The reduction of H2B by KCl wash of macrophages was 73% and also led to a concomitant decrease in Plg binding but occurred without loss of PS surface expression. Second, an anti-PS antibody inhibited surface expression of H2B by 55% and plasmin generation by 60%. Third, known binding partners of PS, annexin V and Protein S, inhibited surface expression of H2B and Plg binding. Of note, the inhibition of H2B surface expression by annexin V and protein S was not associated with an inhibition of differentiation of IFNγ + VD3-treated cells per se, as indicated by CD14 and αMβ2 integrin expression levels measured by FACS. This was also the case with LTCC blockers, which inhibited H2B surface expression but not differentiation (data provided in Fig. S1). Protein S and annexin V have been shown to inhibit migration of monocytes and tumor cells [20,24], and this activity might at least in part reflect their interference with the Plg-H2B interaction. Recent data suggest that histones H3 and H4 released from macrophages contribute to septic shock [25]. In addition to these two histones, H2B, as well as other histones, binds to extracellular DNA nets during infections [26]. Unlike H2B, H3 and H4 lack C-terminal lysines and would not promote Plg activation and fibrinolysis. Thus, the histones that decorate DNA nets may have distinct functions in hemostasis.
Monocyte differentiation to macrophages is known to be associated with PS exposure [13,14]. Our data indicate that this pathway for PS exposure is insensitive to caspase inhibition. PS exposure also occurs in cells undergoing apoptosis, and apoptosis strongly correlates with increased Plg binding [27,28]. Our data show interdependence between Plg binding, H2B surface expression and PS exposure on cells induced into apoptosis with camptothecin on THP-1 cells. We also observed these same interrelationships with primary human monocytes undergoing differentiation or apoptosis. Furthermore, these data are consistent with previous results showing that ionomycin- induced apoptosis of T lymphocytes led to H2B exteriorization on the cell surface [10]. However, the extent of H2B induction (4.7-fold) on camptothecin-treated cells was substantially less than the increase in Plg binding (11-fold), suggesting that other Plg-Rs contribute to the Plg binding capacity of apoptotic cells. Annexin 2, another Plg-R expressed on the surface of macrophages, also interacts with PS [29] and could contribute to the extent of Plg binding to apoptotic cells. Indeed, we saw a 2.5-fold increase of annexin 2 expression on camptothecin-treated THP-1 cells and 56% of the annexin 2 surface expression was inhibited upon addition of the anti-PS antibody to apoptotic THP-1 cells.
Differentiation-induced Plg binding, H2B exteriorization and PS exposure were all completely inhibited by amlodipine and verapamil, two distinct classes of LTCC inhibitors. These three events were also closely interrelated and dependent upon LTCC on apoptotic cells; Plg binding, H2B surface expression and PS exposure were reduced by 45–50% by amlodipine and verapamil. A need for elevated intracellular Ca2+ in apoptosis-induced PS exposure is well documented in various cell types [30,31]. However, in our study the inhibition of all three measured events, including intracellular Ca2+ by the LTCC blockers, was only partial. Such partial inhibition may reflect that the 10 μM concentration, as high a concentration that could be used without compromising cell viability, was insufficient to exert a full effect. Alternatively, another type of Ca2+ channel or a pathway independent of Ca2+ may contribute to apoptosis-induced PS exposure. PS exposed during differentiation and apoptosis provides a bed to tether H2B to the cell surface. While α-enolase export to the surface of differentiated cells also is regulated by Ca2+ and LTCC, it does not tether to the cell surface via PS. Thus, PS exposure is essential for H2B expression but is not the only pathway for Plg binding to cells.
We also found an increment of Plg binding when exogenous H2B was added to either IFNγ+ VD3 or camptothecin-treated cells and PS accounted for 70–80% of H2B binding to these cells. These data indicate that H2B can accumulate at the cell surface as a protein derived from other cells, as a consequence of release from necrotic or apoptotic cells, as suggested by the studies of Dejouvencel et al. [32]. Previous reports have indicated that histone proteins can bind to negatively charged cell surface molecules such as heparan sulfate proteoglycans, perlecan and glypican [33,34]. However, when we treated differentiated THP-1 cells with heparitinase I (0.5 U/ml to 2 U/ml) we did not see any reduction in H2B cell surface expression (data not shown). The same concentration of heparitinase 1 treatment showed reduced glypican 1 expression, a membrane bound heparan proteoglycan on THP-1 cells [35], we did not see any reduction in H2B cell surface expression (data not shown). PS accounted for approximately 55% of the endogenous H2B binding to differentiated THP-1 cells, and we do not exclude the involvement of other negatively charged interactions in the retention of H2B on cell surfaces. Nevertheless, our data do indicate that PS is particularly important in H2B surface localization.
In summary, Ca2+ drives exposure of PS during monocyte differentiation to macrophages and as monocytes undergo apoptosis. PS provides sites to directly interact with H2B, which ultimately contributes to an increase in Plg binding on the responding cells.
Supplementary Material
Acknowledgments
We thank N. S. Rote, Case Western Reserve University, Cleveland, OH, for his generosity in providing us with anti-phosphatidylserine antibody. We thank E. Poptic, Hybridoma Core of LRI, for measuring the IgM concentration of supernatant. We also thank T. Burke for proofreading the manuscript. This work was supported by NIH Grant HL17964 to E. F. Plow and AHA Fellowship 0825638D to R. Das.
Footnotes
Addendum
R. Das designed and performed the research, analyzed the data and wrote the manuscript. E. F. Plow designed the research, analyzed the data and wrote the manuscript.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Additional Supporting Information may be found in the online version of this article:
Data S1. Materials and Methods.
Fig. S1. Effect of anti-PS antibody, annexin V, protein S, amlodipine and verapamil on IFNγ+ VD3-mediated THP-1 cell differentiation.
Fig. S2. Effect of IFNγ+ VD3 on camptothecin-induced apoptosis and of camptothecin on differentiation of THP-1 cells.
Fig. S3. Camptothecin treatment of THP-1 cells enhances intracellular Ca2+ levels and PS surface expression.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J. 1995;9:939–45. doi: 10.1096/fasebj.9.10.7615163. [DOI] [PubMed] [Google Scholar]
- 2.Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005;10:1754–62. doi: 10.2741/1658. [DOI] [PubMed] [Google Scholar]
- 3.Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30:1682–91. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- 4.Herren T, Burke TA, Das R, Plow EF. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry. 2006;45:9463–74. doi: 10.1021/bi060756w. [DOI] [PubMed] [Google Scholar]
- 5.Wygrecka M, Marsh LM, Morty RE, Henneke I, Guenther A, Lohmeyer J, Markart P, Preissner KT. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood. 2009;113:5588–98. doi: 10.1182/blood-2008-08-170837. [DOI] [PubMed] [Google Scholar]
- 6.Das R, Burke T, Plow EF. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood. 2007;110:3763–72. doi: 10.1182/blood-2007-03-079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herren T, Burke TA, Jardi M, Felez J, Plow EF. Regulation of plasminogen binding to neutrophils. Blood. 2001;97:1070–8. doi: 10.1182/blood.v97.4.1070. [DOI] [PubMed] [Google Scholar]
- 8.Das R, Burke T, Van Wagoner DR, Plow EF. L-type calcium channel blockers exert an antiinflammatory effect by suppressing expression of plasminogen receptors on macrophages. Circ Res. 2009;105:167–75. doi: 10.1161/CIRCRESAHA.109.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holers VM, Kotzin BL. Human peripheral blood monocytes display surface antigens recognized by monoclonal antinuclear antibodies. J Clin Invest. 1985;76:991–8. doi: 10.1172/JCI112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zunino SJ, Singh MK, Bass J, Picker LJ. Immunodetection of histone epitopes correlates with early stages of apoptosis in activated human peripheral T lymphocytes. Am J Pathol. 1996;149:653–63. [PMC free article] [PubMed] [Google Scholar]
- 11.Pereira LF, Marco FM, Boimorto R, Caturla A, Bustos A, De la Concha EG, Subiza JL. Histones interact with anionic phospholipids with high avidity; its relevance for the binding of histone-antihistone immune complexes. Clin Exp Immunol. 1994;97:175–80. doi: 10.1111/j.1365-2249.1994.tb06064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furnrohr BG, Groer GJ, Sehnert B, Herrmann M, Voll RE. Interaction of histones with phospholipids – implications for the exposure of histones on apoptotic cells. Autoimmunity. 2007;40:322–6. doi: 10.1080/08916930701356457. [DOI] [PubMed] [Google Scholar]
- 13.Marguet D, Luciani MF, Moynault A, Williamson P, Chimini G. Engulfment of apoptotic cells involves the redistribution of membrane phosphatidylserine on phagocyte and prey. Nat Cell Biol. 1999;1:454–6. doi: 10.1038/15690. [DOI] [PubMed] [Google Scholar]
- 14.Callahan MK, Halleck MS, Krahling S, Henderson AJ, Williamson P, Schlegel RA. Phosphatidylserine expression and phagocytosis of apoptotic thymocytes during differentiation of monocytic cells. J Leukoc Biol. 2003;74:846–56. doi: 10.1189/jlb.0902433. [DOI] [PubMed] [Google Scholar]
- 15.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 16.Vogt E, Ng AK, Rote NS. Antiphosphatidylserine antibody removes annexin-V and facilitates the binding of prothrombin at the surface of a choriocarcinoma model of trophoblast differentiation. Am J Obstet Gynecol. 1997;177:964–72. doi: 10.1016/s0002-9378(97)70302-8. [DOI] [PubMed] [Google Scholar]
- 17.Katsuragawa H, Kanzaki H, Inoue T, Hirano T, Mori T, Rote NS. Monoclonal antibody against phosphatidylserine inhibits in vitro human trophoblastic hormone production and invasion. Biol Reprod. 1997;56:50–8. doi: 10.1095/biolreprod56.1.50. [DOI] [PubMed] [Google Scholar]
- 18.Appelt U, Sheriff A, Gaipl US, Kalden JR, Voll RE, Herrmann M. Viable, apoptotic and necrotic monocytes expose phosphatidylserine: cooperative binding of the ligand Annexin V to dying but not viable cells and implications for PS-dependent clearance. Cell Death Differ. 2005;12:194–6. doi: 10.1038/sj.cdd.4401527. [DOI] [PubMed] [Google Scholar]
- 19.Rezende SM, Simmonds RE, Lane DA. Coagulation, inflammation, and apoptosis: different roles for protein S and the protein S-C4b binding protein complex. Blood. 2004;103:1192–201. doi: 10.1182/blood-2003-05-1551. [DOI] [PubMed] [Google Scholar]
- 20.Hryszko T, Suzuki Y, Mogami H, Urano T. Protein S attenuates the invasive potential of THP-1 cells by interfering with plasminogen binding on cell surface via a protein C-independent mechanism. FEBS Lett. 2005;579:6023–6. doi: 10.1016/j.febslet.2005.09.080. [DOI] [PubMed] [Google Scholar]
- 21.Duvall E, Wyllie AH, Morris RG. Macrophage recognition of cells undergoing programmed cell death (apoptosis) Immunology. 1985;56:351–8. [PMC free article] [PubMed] [Google Scholar]
- 22.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16. [PubMed] [Google Scholar]
- 23.Schwende H, Fitzke E, Ambs P, Dieter P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J Leukoc Biol. 1996;59:555–61. [PubMed] [Google Scholar]
- 24.Balch C, Dedman JR. Annexins II and V inhibit cell migration. Exp Cell Res. 1997;237:259–63. doi: 10.1006/excr.1997.3817. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupa F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 27.O’Mullane MJ, Baker MS. Elevated plasminogen receptor expression occurs as a degradative phase event in cellular apoptosis. Immunol Cell Biol. 1999;77:249–55. doi: 10.1046/j.1440-1711.1999.00823.x. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell JW, Baik N, Castellino FJ, Miles LA. Plasminogen inhibits TNFα-induced apoptosis in monocytes. Blood. 2006;107:4383–90. doi: 10.1182/blood-2005-07-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross M, Gerke V, Steinem C. Membrane composition affects the reversibility of annexin A2t binding to solid supported membranes: a QCM study. Biochemistry. 2003;42:3131–41. doi: 10.1021/bi027069z. [DOI] [PubMed] [Google Scholar]
- 30.Williamson P, Kulick A, Zachowski A, Schlegel RA, Devaux PF. Ca2+ induces transbilayer redistribution of all major phospholipids in human erythrocytes. Biochemistry. 1992;31:6355–60. doi: 10.1021/bi00142a027. [DOI] [PubMed] [Google Scholar]
- 31.Hampton MB, Vanags DM, Pörn-Ares MI, Orrenius S. Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett. 1996;399:277–82. doi: 10.1016/s0014-5793(96)01341-5. [DOI] [PubMed] [Google Scholar]
- 32.Dejouvencel T, Doeuvre L, Lacroix R, Plawinski L, gnat-George F, Lijnen HR, Angles-Cano E. Fibrinolytic cross-talk: a new mechanism for plasmin formation. Blood. 2010;115:2048–56. doi: 10.1182/blood-2009-06-228817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henriquez JP, Casar JC, Fuentealba L, Carey DJ, Brandan E. Extracellular matrix histone H1 binds to perlecan, is present in regenerating skeletal muscle and stimulates myoblast proliferation. J Cell Sci. 2002;115:2041–51. doi: 10.1242/jcs.115.10.2041. [DOI] [PubMed] [Google Scholar]
- 34.Watson K, Gooderham NJ, Davies DS, Edwards RJ. Nucleosomes bind to cell surface proteoglycans. J Biol Chem. 1999;274:21707–13. doi: 10.1074/jbc.274.31.21707. [DOI] [PubMed] [Google Scholar]
- 35.Makatsori E, Lamari FN, Theocharis AD, Anagnostides S, Hjerpe A, Tsegenidis T, Karamanos NK. Large matrix proteoglycans, versican and perlecan, are expressed and secreted by human leukemic monocytes. Anticancer Res. 2003;23:3303–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.