

Abstract
Recent clinical and pre-clinical studies suggest that both active and passive immunization strategies targeting Aβ amyloid may have clinical benefit in Alzheimer’s disease. Here, we demonstrate that vaccination of APPswePSEN1dE9 mice with SDPM1, an engineered non-native Aβ amyloid-specific binding peptide, lowers brain Aβ amyloid plaque burden and brain Aβ1-40 and Aβ1-42 peptide levels, improves cognitive learning and memory in Morris Water maze tests and increases the expression of synaptic brain proteins. This was the case in young mice immunized prior to development of significant brain amyloid burden, and in older mice, where brain amyloid was already present. Active immunization was optimized using ALUM as an adjuvant to stimulate production of anti-SDPM1 and anti-Aβ amyloid antibodies. Intracerebral injection of P4D6, an SDPM1 peptide-mimotope antibody, also lowered brain amyloid plaque burden in APPswePSEN1dE9 mice. Additionally, P4D6 inhibited Aβ amyloid-mediated toxicity in cultured neuronal cells. The protein sequence of the variable domain within the P4D6 heavy chain was found to mimic a multimer of the SDPM1 peptide motif. These data demonstrate the efficacy of active and passive vaccine strategies to target specific Aβ amyloid oligomers using an engineered peptide-mimotope strategy.
Keywords: Alzheimer’s disease, vaccine, peptide mimotope, amyloid, neurodegeneration, learning, memory, synaptophysin, PSD95, synapse
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia in the elderly(Bachman et al., 1992). AD is triggered, at least in part, by the formation of Aβ amyloid arising from generation of Aβ1-40 and Aβ1-42 peptides(Hardy and Higgins, 1992; Selkoe, 2001a; Selkoe, 2001b), and low molecular weight Aβ peptide oligomers impact synaptic function, synaptic structure, cognition and neuronal cell death(Barghorn et al., 2005; Cleary et al., 2005; Hsieh et al., 2006; Kim et al., 2001; Lambert et al., 1998; Lesne et al., 2006; Shankar et al., 2007; Shankar et al., 2008; Walsh et al., 2002; Wei et al., 2010). Additional pathologic findings in AD brain include hyperphosphorylation of the microtubule binding protein tau, formation of neurofibrillary tangles, synaptic dysfunction, loss of synaptic density and neurodegeneration(DeKosky and Scheff, 1990; Lace et al., 2007; Marcello et al., 2012; Masliah et al., 1991; Pham et al., 2010; Selkoe, 2001a; Terry, 1996; Terry et al., 1991). Pathological findings, in turn, correlate with alterations in cognitive functions, such as learning and memory(DeKosky and Scheff, 1990; Masliah et al., 1991; Nelson et al., 2012; Terry et al., 1991). Bi-transgenic AD mouse models that overexpress early onset mutations in APP and PSEN1 mutants (APP/PS) in neurons reiterate some, but not all, of these important aspects of AD(Borchelt et al., 1997; Morrissette et al., 2009).
Both active and passive vaccine strategies based on immunization with the Aβ peptide or its variants have been shown, in some cases, to have clinically meaningful benefits in Alzheimer’s disease patients(Delrieu et al., 2012). Schenck and colleagues first showed that immunization with Aβ1-42 peptide could lower brain Aβ amyloid plaque burden in an AD mouse model(Schenk et al., 1999). This led to a phase 1 and phase2a AD clinical trials by ELAN/Wyeth (AN1792). This trial was interrupted in phase2a due to the finding of meningoencephalitis with T cell infiltrates in a subset of the vaccinated patients(Orgogozo et al., 2003; Pride et al., 2008). Despite the interruption of the trial, some patients did develop serum antibody titers to the Aβ peptide and showed cognitive improvement(Ghochikyan et al., 2006; Gilman et al., 2005; Hock et al., 2003). Analysis of patients in the AN1792 trial that have come to autopsy has documented reductions in brain Aβ amyloid plaque burden(Nicoll et al., 2006). A host of additional active vaccine trials are currently ongoing that focus on utilizing shorter Aβ peptides that are devoid of T cell activation epitopes(Cribbs, 2010; Delrieu et al., 2012; Lemere, 2009; Schneeberger et al., 2009; Tabira, 2010). Passive vaccine therapies utilizing antibodies that target different regions of the Aβ1-42 peptide or Aβ oligomers are also currently being tested (Bard et al., 2003; Carty et al., 2006; Delrieu et al., 2012; DeMattos et al., 2001; Laskowitz and Kolls, 2010; Levites et al., 2006a; Levites et al., 2006b; Ostrowitzki et al., 2012; Panza et al., 2012; Pride et al., 2008; Wilcock et al., 2003; Yamada et al., 2009).
Using a phage display method, we have identified several cysteine-bounded 20 amino acid peptides that can bind specifically to low molecular weight oligomers of Aβ1-40 and Aβ1-42 amyloid and can block subsequent Aβ oligomerization(Kang et al., 2003; Wang et al., 2010). One of these peptides is SDPM1. Some antibodies that bind SDPM1 can display peptide-mimotope activity, having the same Aβ amyloid binding and blocking activities as SDPM1 itself has(Wang et al., 2010). We have previously shown that immunization of APP/PS mice with SDPM1 to generate such peptide-mimotope antibodies lowers brain Aβ amyloid plaque burden and improves learning and memory without causing increased brain inflammation(Wang et al., 2010). While effective, this previous approach was not easily translatable to the clinic. Here we have altered our approach to immunize APP/PS mice with SDPM1 bound to ALUM, a commonly used adjuvant in clinical vaccines(Schijns and Lavelle, 2011). In addition, we assess passive vaccination with P4D6, an SDPM1-peptide mimotope antibody(Wang et al., 2010), and report its sequence.
MATERIALS AND METHODS
Animals
APPswePSEN1dE9 transgenic mice (JAX strain B6C3-Tg(APPswePSEN1dE9)85Dbo/Mmjax, stock# 004462) and non-transgenic littermate controls were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were bred and genotyped as previously described(Borchelt et al., 1997). All experiments were done with approval of the Institutional Animal Care and Use Committee at Nationwide Children’s Hospital. Transgenes were maintained as heterozygous alleles for all experiments. Mice had access to food and water ad libitum and were kept on a 12:12hr light dark cycle. All animals were housed in a clean barrier facility and were housed individually prior to Morris Water Maze and Open Field assessments. For each measure in each experimental group, half of the animals analyzed were male and half of the animals analyzed were female.
Vaccination protocols
SDPM1-4E peptide (EEEEAECDWGKGGRWRLWPGASGKTEACGP) was synthesized and purified to >95% purity by AmbioPharm (North Augusta, SC). Peptide purity was analyzed by HPLC and mass spectrometry. SDPM1-4E was bound to Alhydrogel (Al(OH)3,) (Accurate Chemical and Scientific Corp.; Westbury, NY from Brenntag; Fredrickssund, Denmark). 1mL 2% Alhydrogel (hereby called ALUM, formulated with 10.3mg/ml aluminum) was washed 3 times with 10mL sterile phospho-buffered saline (PBS, pH 7.4) prior to peptide addition.
For vaccine optimization experiments, 25μg, 50μg or 100μg of SDPM1-4E was added to 25μg of ALUM in 100μL PBS. Total amounts of each solution made were scaled according to the number of injections to be performed. The solution was mixed by gentle rocking overnight at 4°C. After overnight conjugation, centrifugation of the ALUM-SDPM1-4E-conjugate showed no unbound peptide remaining in the supernatant, as measured by Bradford assay. Controls of 100μg SDPM1-4E alone or 25μg ALUM alone were also prepared. Mice were bled one day prior to first immunization to assess baseline serum antibody titers. Adult wild type mice (C57Bl/6) were then injected subcutaneously using a 0.3mL insulin syringe with 100μL vaccine solution in the dorsal neck area. Mice were injected once every two weeks for a total of five injections. Relative to the time of the first injection, mice were bled at 0, 6, 12 and 24 weeks and serum antibody titers analyzed.
For AD vaccine therapy experiments, APPswePSEN1dE9 (APP/PS) mice were injected using one of two paradigms. In the first paradigm, YOUNG (6 month-old) APP/PS mice were injected subcutaneously once every two weeks for a total of four injections with 100μg SDPM1-4E peptide conjugated to 25μg ALUM. Control mice were similarly injected with 25μg ALUM or PBS alone. In the second paradigm, OLD (12 month-old) APP/PS mice were injected subcutaneously once every two weeks for a total of four injections with 100μg SDPM1-4E peptide conjugated to 25μg ALUM (again with ALUM or PBS as controls). Mice may have varied by as much as 3 weeks in age below the 6- or 12-month time point for YOUNG or OLD mice, respectively, at the beginning of the experiment, but in each instance analysis was done after a treatment period of exactly 6 months. Mice were analyzed for learning and memory and bled to assess serum antibody titers at 24 weeks after the first immunization. They were then euthanized and their brains harvested for further analysis.
Serum ELISA assays of SDPM1 and Aβ amyloid antibody titers
100–200μL of blood was isolated from the facial vein and allowed to clot for 30 minutes at 37°C in non-heparinized tubes, after which samples were centrifuged at 3000g for 5 minutes to collect serum. SDPM1-specific antibody titers and Aβ1-42 amyloid-specific antibody titers were assayed on 96-well ELISA plates (NUNC #449824) as previously described (Wang et al., 2010). Addition of secondary antibody alone to SDPM1- or Aβ1-42-immobilized wells yielded a background signal that never exceeded 10% of the maximal primary antibody signal. Signals from SDPM2 background were subtracted from SDPM1-positive signals to generate anti-SDPM1 antibody titers. To determine antibody subtypes, the protocol was repeated, only goat anti-mouse antibodies specific for mouse IgM, IgG1, IgG2a, IgG2b, IgG3 or IgA (Southern Biotech: 5300–05) were used.
Immunohistochemistry and immunofluorescence microscopy
Mice were exsanguinated by perfusion in PBS under anesthetic with ketamine and xylazine. Mouse brains were removed and bisected at the midsagittal plane. Half of the brain was snap frozen in liquid nitrogen to use in biochemical experiments. The other half was fixed in 4% paraformaldehyde overnight. Brains were then washed in PBS and fixative quenched in 0.1M glycine, sunk in 10–30% sucrose gradient overnight, mounted in OCT (Optimal Cutting Temperature) and frozen in dry ice-cooled isopentane. 10μm sagittal sections of brain were cut on a cryostat. Slides were stained in Thioflavin S for 5 minutes, then sequentially washed in 70% ethanol and doubly distilled (dd)H2O. For Aβ1-42 staining, slides were blocked in PBS with 10% goat serum, incubated with biotinylated mouse anti-Aβ1-42 (6E10) antibody, washed in PBS and incubated with Cy3-conjugated streptavidin. A similar approach was used to stain for astrocytes (anti-GFAP, Zymed, 1800063), microglia (anti-Iba1, Wako 019-19741), activated microglia (anti-CD68, AbD Serotec MCA1957GA), T cells (anti-CD4, BDPharmingen, 550278; anti-CD8, BDPharmingen, 550281) or B cells (anti-B220, BDPharmingen 550286), only Cy3-conjugated goat anti-rabbit (for Iba1, GFAP) or goat anti-rat (for CD68, B220, CD4, CD8) secondary antibodies were used. Synaptophysin (Ab9272, Upstate Biotechnology, Lake Placid, NY) staining was done to identify relative synaptic density in the frontal region of the cortex and in the outer molecular layer (ML) of the hippocampus. All fluorophore-, biotin-, or HRP-conjugated secondary antibodies or streptavidin were purchased from Jackson ImmunoResearch (Seattle, WA).
Image analysis
10μm serial sagittal sections were collected beginning 720μm laterally from the midplane by cryostat sectioning and stained with Thioflavin S to visualize fibrillar dense core Aβ amyloid plaques. 8–12 sections, each spaced 80μm apart, were used to image amyloid plaque burden, plaque number and plaque size in the hippocampus and the cortex. For cortex, equivalent numbers of images were averaged from frontal, medial and posterior cortical regions. Stained images were recorded using a Zeiss Axiophot epifluorescence microscope and Zeiss AxioVision LE 4.1 software. 10x images were recorded to analyze amyloid plaque burden, while 20x images were used to record cells stained for GFAP, Iba1, or CD68. Synaptic density was indirectly assessed by immunostaining with synaptophysin in the molecular layer of the hippocampus and in the frontal neocortex. For comparisons of staining intensity, all images were collected using identical exposure settings using the same illumination intensity and filters. Images were then converted to a greyscale TIFF format and analyzed using NIH Image J (1.42) software as previously described(Wang et al., 2010). Sections were blinded with respected to the investigator prior to analysis, and multiple investigators participated in the quantification of each measure, with similar results. Each datum reflects summation of a 708×528μm image for quantification of Aβ plaque burden, number and size, and a 354×265μm image for quantification of GFAP-, Iba1-or CD68-positive cell density and synaptophysin staining intensity. 8–12 sections per animal were used to calculate the number of cells immunostained with antibodies to GFAP, Iba1 and CD68 and to quantify synaptophysin immunostaining.
Quantification of brain Aβ1-40 and Aβ1-42 peptides
Unfixed snap-frozen brain was weighed and solubilized in 2% SDS denaturation buffer as previously described(Wang et al., 2010). After precipitation of insoluble material at 16000g for 20 minutes, extracted brain proteins (SDS-soluble) were assayed using ELISA kits for Aβ1-40 (Invitrogen, KHB3482) and Aβ1-42 peptide (Invitrogen, KHB3442) following the manufacturer’s instructions. Precipitated (SDS-insoluble) pellets were extracted in formic acid, as before(Wang et al., 2010), centrifuged at 100000g, and supernatant neutralized in 1M Tris-base/0.5M NaH2PO4. The neutralized insoluble protein fraction was then assayed as above. Plates were measured for absorbance at 450nm on a SpectraMax M2 plate reader and concentrations of Aβ1-40 or Aβ1-42 normalized to weight of brain sample used.
Western Blotting
Brain hemispheres were solubilized by boiling in 2% SDS and 40μg (or 70μg) of extracted protein was separated on 10–20% Tris-Tricine gradient polyacrylamide gels (Life Technologies, Grand Island, NY) and immunoblotted with anti-Aβ1-42 (biotinylated 6E10, Covance SIG-39340-200) or GAPDH (Millipore, MAB374) as previously described(Wang et al., 2010). For PSD95 (Ab 12093, Abcam), synaptophysin (Ab 9272, Upstate Biotechnology) and β-actin (49507, Cell Signaling) immunoblots, brain plasma membranes (cortex+hippocampus) were prepared and extracted using buffer A as previously described by Masliah and colleagues(Pham et al., 2010). Blots were developed using ECL chemiluminescence (Amersham, Piscataway, NJ) and quantified as previously described(Jayasinha et al., 2003).
Morris water maze tests
Groups of 12 month-old (YOUNG) mock -or SDPM1-immunized APP/PS mice, or 18 month-old (OLD) mock -or SDPM1-immunized APP/PS mice, all time-matched to exactly 6 months after immunizations began, were tested in the Morris water maze, along with non-immunized, age-matched, wild type mouse controls. All mice were blinded to the experimenter with respect to vaccination status and genotype. The tasks were conducted in a large pool (diameter 122cm) filled with 24°C water. Water was made opaque using non-toxic white paint. A hidden square platform (10cm2) was placed 1cm beneath the surface of the water in one quadrant of the pool. Visual cues were mounted on a screen surrounding the pool in fixed positions. The mice were pre-trained for 1 day to find and climb on to the hidden platform within 60 seconds of being placed in the water. If a mouse was unable to find the platform, it was placed there manually for 10 seconds. Mice were given up to three times to find the platform. Mice that were unable to find the platform during these training sessions were eliminated from the experiment. One or no animals per group were eliminated due to this criterion. Mice were then tested for 6 successive days, with four 60-second trials per day, and escape time to platform (escape latency), swim speed and distance swum were recorded as before(Wang et al., 2010). A probe test to measure memory of the learned skill was done 12 hours after the final day of training as before(Wang et al., 2010).
Open field tests
Ambulatory movements were measured using a Photobeam Activity System (PAS, San Diego Instruments) open field test. All measurements were done in a dedicated mouse behavior room at the same time of day with defined temperature, ambient noise and ambient light. Each mouse was placed in the center of a 16- by 16-inch clear acrylic chamber with photobeams that run along its base at 1 inch spacing along the X- and Y-axis. Ambulatory activity was recorded every time an animal crossed each one square-inch region of the box into another region. Movements were recorded in 4 5-minute sessions for a total of 20 minutes. Total recorded events were summed for each independent measure (fine motor, ambulatory, rearing) as before(Wang et al., 2010).
Sequencing of P4D6 Antibody
P4D6 heavy and light chains were sequenced from antibody chain mRNA transcripts amplified by RT-PCR from P4D6-expressing hybridoma cell clones by GenScript (Piscataway, NJ). Ten clones were sequenced from two different P4D6 expressing hybridoma cell variants, with all yielding the same single heavy chain sequence and the same single light chain sequence.
Intracerebral P4D6 antibody injection
15 month-old APP/PS mice were anaesthetized using isofluorane and kept under deep anesthesia for the remainder of the procedure. Surgery was performed using a stereotaxic apparatus (David Kopf Instruments; Tujunga, CA). The cranium was exposed by making an incision through the skin along the median sagittal plane. Four holes were drilled through the cranium of each animal, one each over the right and left frontal cortex injection site and one each over the right and left hippocampus injection site. Brain coordinates from brain surface were: +1.5 anterior-posterior, −1.75/+1.75 mediolateral (right/left), −1.5 dorsoventral (frontal cortex) and −2.7 anterior-posterior, −2.5/+2.5 mediolateral (right/left) and −2.0 dorsoventral (hippocampus). 2 μL of purified P4D6 antibody (2μg total antibody) was dispensed on right side of the frontal cortex and hippocampus, each over a 4-minute period using a 10μL syringe, while 2μL of sterile PBS was similarly dispensed on the left side in each area as a control. The incision was then cleaned and closed with 3M Vetbond tissue adhesive and animals allowed to return to the colony after waking. 1 day or 14 days after injection, mice were anesthetized in ketamine and xylazine and perfused, via intracardiac perfusion, with PBS followed by 4% paraformaldehyde. Brains were removed and fixed further in 4% paraformaldehyde for 16 hours, after which they were washed in PBS, sunk in 10–30% sucrose gradient, mounted in OCT, frozen in dry-ice cooled isopentane and sectioned for analysis.
Cell culture assays of cell death
SH-SY5Y cells (ATCC, Manassas, VA) were grown in a 1:1 mixture of Ham’s F12 nutrient and Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50μg/mL penicillin, 50U/mL streptomycin, 2mM glutamine and non-essential amino acids (Gibco, Gaithersburg, MD). Cells were incubated in a 95% air 5% CO2 humidified incubator. For differentiation, cells were seeded in 96-well tissue culture plates at 30,000 cells per well for 1 day, after which medium was replaced with retinoic acid (RA) and 1% N2 supplement (Invitrogen, Carlsbad, CA). One half of medium was replaced every other day for the ensuing 7 days, after which cell death experiments were performed.
For cell death experiments, Aβ1-42 was first diluted (from a DMSO stock) to make an aqueous 100μM Aβ1-42 stock solution in PBS, with or without 5μM P4D6. Aβ1-42 amyloid (+/-P4D6) was added to RA-differentiated SH-SY5Y cells for 48 hours in media in 2% fetal bovine serum. Cells were washed and assayed for Caspase 3/7 activity, cellular dye uptake or chromatin condensation. Caspase 3/7 assays were done using a fluorometric substrate-based assay (Promega, Apo-ONE G7790) according to the manufacturer’s instructions. To assay dye uptake and chromatin condensation, 10μg/mL propidium iodide (Sigma, P-4864) and 10μM Hoechst 33342 (Invitrogen, H21492) were added in serum-free media to cells for 20 minutes. After removal of staining solution, cells were quickly washed in PBS and fixed in 4% paraformaldehyde. Cells positive for propidium iodide or condensed chromatin Hoechst staining were visualized and images quantified as previously described(Kelly et al., 2003; Vaisid et al., 2008). A total of 600 cells were counted from each well, using at least 3 different, non-overlapping, 20x visual fields per well.
SDPM14 production and binding experiments
A cDNA encoding SDPM14 protein (AECDWGKGGRWRLWPGASGKTEACGPGAECDWGKGGRWRLWPGASGKTEACGPGAE CDWGKGGRWRLWPGASGKTEACGPGAECDWGKGGRWRLWPGASGKTEACGP) was subcloned into the HindIII and EcoR1 sites in the polylinker of pCVM1-FLAG vector to encode secreted, N-terminal FLAG epitope-tagged, protein. The plasmid was transfected into HEK293T cells and SDPM14 protein was purified from the cell supernatant using M2 (anti-FLAG)-agarose as previously described(Yoon et al., 2009). Eluted purified protein was quantified and used to determine relative binding to immobilized P4D6, Aβ1-40 amyloid and Aβ1-42 amyloid, as previously described(Wang et al., 2010).
Statistics
For comparison between only two groups, significance was determined using a two-tailed unpaired Student’s t test. For comparisons with more than two groups, significance was determined by ANOVA with post-hoc t test.
RESULTS
Immunization with SDPM1-ALUM vaccine reduces amyloid plaque burden in APPswePSEN1dE9 mouse brain
To increase the translational potential of our experiments, we utilized ALUM as the adjuvant for our immunization protocol, as ALUM has been used extensively in clinical vaccines(Schijns and Lavelle, 2011). We first developed and optimized a vaccination method using SDPM1-ALUM as the immunogen to induce anti-SDPM1 and anti-Aβ amyloid specific antibodies in wild type mice (Supplemental Figure 1). We next determined if our optimized immunization protocol could be used to affect brain Aβ amyloid burden, learning and memory, and the density of synaptic brain proteins in the APPswePSEN1dE9 (APP/PS) bitransgenic mouse model of Alzheimer’s Disease (AD). We employed two vaccine paradigms, one aimed at understanding AD prevention and one aimed at understanding AD treatment. For the AD prevention paradigm, we immunized 6 month-old APP/PS mice (termed YOUNG), a time at which brain Aβ amyloid burden is first becoming evident(Borchelt et al., 1997; Jankowsky et al., 2001; Savonenko et al., 2005). For the AD treatment paradigm, we immunized 12 month-old APP/PS mice (termed OLD), a time at which brain Aβ amyloid burden is already significant(Borchelt et al., 1997; Jankowsky et al., 2001; Savonenko et al., 2005). In both instances, mice were immunized once every two weeks with 100μg SDPM1-ALUM for a total of four immunizations. Control animals included age-matched non-immunized wild type mice and APP/PS mice immunized with ALUM alone. All measures were performed 24 weeks, or 6 months, after the first immunization was given. No morbidity or mortality was noticed during either treatment regimen, and total serum IgG and IgE levels showed no treatment-induced elevation (not shown).
Both treatment paradigms led to 6-8-fold increases in serum antibodies specific for SDPM1 (Fig. 1A) and Aβ1-42 amyloid (Fig. 1B). We repeated SDPM1 titers using subtype-specific secondary antibodies to determine the species of antibodies induced by SDPM1-ALUM vaccination (Fig. 1C). About half of all SDPM1 antibody, both in YOUNG and OLD, APP/PS mice, was IgM. Most of the remaining antibody was IgG2b (about 30%) or IgA (about 10%).
Figure 1. Immunization of YOUNG and OLD APPswePSEN1dE9 (APP/PS) mice with SDPM1 conjugated to ALUM.
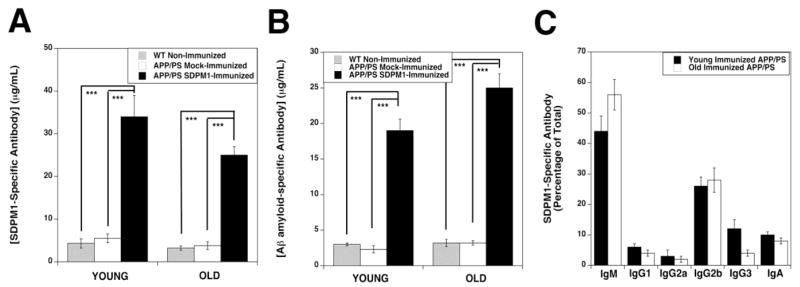
SDPM1-immunized mice were given four subcutaneous immunizations, one every two weeks, of 100μg SDPM1-4E peptide conjugated to ALUM. Mock-immunized APP/PS mice were similarly vaccinated with ALUM alone. Wild type (WT) mice were not immunized. YOUNG APP/PS mice were immunized beginning at 6 months of age, while OLD APP/PS mice were immunized beginning at 12 months of age. All mice were bled and serum antibody titers to SDPM1 (A) and Aβ1-42 amyloid (B) assayed at 6 months after immunizations had begun. Relative amounts mouse SDPM1 antibody subtypes were also determined in SDPM1-immunized APP/PS animals (C). Errors are SEM for n=10–12 animals per condition, assayed in duplicate.
We next assayed total brain Aβ1-40 and Aβ1-42 peptide levels (Fig. 2). Aβ1-40 and Aβ1-42 brain peptide levels were reduced by 50% or more in YOUNG SDPM1-immunized APP/PS mice compared to mock-immunized APP/PS animals (Fig. 2A and C). Similar results were obtained in OLD APP/PS animals. Such reductions were significant in all instances, both in SDS-soluble (Fig. 2A) and SDS-insoluble fractions (Fig. 2C). Anti-Aβ1-42 Western blots of whole brain SDS protein lysates, using the 6E10 antibody (Fig. 2B), showed a reduction in low molecular weight Aβ -containing peptides in SDPM1-immunized APP/PS brain relative to mock-immunized APP/PS controls. The APPswe transgenic protein and the endogenous brain APP protein also contain the Aβ1-42 peptide recognized by 6E10 (120kDa band in Fig. 2B). SDPM1 immunization did not significantly lower transgenic overexpression of APPswe protein in APP/PS brain.
Figure 2. Aβ1-40 and Aβ1-42 brain peptide levels are reduced in SDPM1-immunized APP/PS mice.

Aβ1-40 and Aβ1-42 peptide levels were measured in the SDS-soluble (A) and SDS-insoluble (C) fractions of YOUNG and OLD mock-immunized and SDPM1-immunized APP/PS mice. Errors are SEM for n=10–12 animals per condition, with measures assayed in duplicate. *P<0.05, **P<0.01, ***P<0.001 (B) Low molecular weight, SDS-soluble, Aβ amyloid oligomers were separated on gradient Tris/Tricine gels and blotted with an antibody to Aβ1-42 (6E10). Two samples per condition are shown. Blots were stripped and reprobed with antibody to GAPDH as a control for protein loading and transfer.
The reduction in overall brain Aβ peptide levels was further supported by staining SDPM1- and mock-immunized APP/PS brains with Thioflavin S, which stains dense core Aβ amyloid aggregates (Fig. 3A). Amyloid plaque burden, as a percentage of brain area, was reduced by 50% or more in both YOUNG and OLD SDPM1-immunized hippocampus and cortex (Fig. 3B). This was similarly correlated with a reduction in amyloid plaque number (Fig. 3C) and the average size of individual amyloid plaques in most instances (Fig. 3D). All such analyses were quantified using samples blinded to the investigators as to treatment and genotype.
Figure 3. Reduced amyloid plaque burden in APP/PS mice immunized with SDPM1.

Brains were serially sectioned and stained for dense core amyloid plaques with Thioflavin S (A). Amyloid plaque burden (B), numbers of amyloid plaque profiles (C) and average amyloid plaque size (D) were quantified from sections of cortex and hippocampus. Errors are SEM for n-10-12 animals per condition. Bar in A is 200μm. *P<0.05, **P<0.01, ***P<0.001
Immunization with SDPM1-ALUM improves learning and memory in APP/PS mice
Aside from brain amyloid plaque burden, the best outcome measure to judge clinical efficacy in APP/PS mice is the ability to improve measures of learning and memory. Prior to sacrifice, we subjected YOUNG and OLD SDPM1-immunized and mock-immunized APP/PS mice to such tests in the Morris water maze, comparing them also to non-immunized, age-matched, wild type animals (Fig. 4). By 12 months of age (the time point at which YOUNG immunized APP/PS mice were analyzed), APP/PS mice show significant deficits in learning relative to wild type animals (Fig. 4A). This was reflected in their inability to learn with repeated training to escape swimming by climbing onto a spatially fixed, but invisible, platform using spatially fixed visual cues. Both APP/PS and wild type mice, prior to training, took 40–50 seconds to complete this task. With repeated training, wild type mice learned to perform this task in half the amount of time (by days 5–6), while mock-immunized APP/PS mice did not (Fig. 4A). Improved learning, showing a significant difference compared to mock-immunized APP/PS animals, occurred in both YOUNG and OLD SDPM1-immunized APP/PS mice by 5 and 6 days of training (Fig. 4A and 4D).
Figure 4. SDPM1 immunization improves learning and memory in YOUNG and OLD APP/PS mice.

SDPM1-immunized YOUNG (A–C) and OLD (D–F) APP/PS mice showed improvement in Morris Water maze assays of learning (A, D) and memory (B, E). Mice were compared age-matched mock-immunized APP/PS mice and non-immunized wild type (WT) mice. Overall swim speed was not significantly changed between any two groups in either experiment (C, F). Errors are SEM for n=10–12 animals per condition. *P<0.05, **P<0.01, ***P<0.001
After training was completed, mice were given 12 hours without training and then subjected to a probe test for memory of the learned skill. SDPM1-immunized APP/PS mice approached memory levels observed in wild type animals, and this differed significantly from mock-immunized APP/PS animals (Fig. 4B and 4E). This was the case in both YOUNG and in OLD SDPM1-immunized APP/PS mice. Analysis of all three groups at the two different ages showed no significant difference in swim speed between any two groups (Fig. 4C and 4F). Thus, differences between groups did not result from an altered ability to swim. Open field tests of ambulation, fine motor activity, and rearing also showed no significant difference between any groups (data not shown).
Immunization with SDPM1-ALUM increases expression of synaptic proteins in APP/PS brain
Akin to alterations in cognitive performance, APP/PS mice show altered expression of synaptic brain proteins, much as can also occur in AD(Buttini et al., 2005; DeKosky and Scheff, 1990; Masliah et al., 1991; Pham et al., 2010; Sze et al., 1997; Terry et al., 1991). Therefore, we next assessed and quantified the expression of synaptophysin, a presynaptic vesicle marker of synaptic density, by immunostaining and immunoblotting (Fig 5). We identified a significant decrease in synaptophysin staining in both the frontal cortex and the molecular layer of the hippocampus in OLD mock-immunized APP/PS mice compared to wild type mice. Synaptophysin staining density was significantly increased in both brain regions of OLD SDPM1-immunized APP/PS mice compared to mock-immunized APP/PS animals (Figs. 5A and C). We also assayed for the expression of synaptic proteins by immunoblotting of purified brain membranes isolated from cortex and hippocampus, which were pooled for each animal (Figs. 5B and D). We found a significant reduction in synaptophysin protein, and also a reduction in the postsynaptic glutamate receptor anchoring protein PSD95, in OLD mock-immunized APP/PS mice compared to wild type mice (Fig. 5B and D). Levels of both proteins were significantly increased in OLD SDPM1-immunized APP/PS mice compared to mock-immunized APP/PS animals (Fig. 5D). For both staining and blotting, similar trends were observed in YOUNG animals, though these differences were not always significant. Thus, the positive changes of SDPM1 immunization on cognitive performance correlated with increased expression of synaptic proteins, including synaptophysin and PSD95.
Figure 5. Increased expression of synaptic brain proteins after SDPM1 immunization of APP/PS mice.

(A) Regions of the frontal cortex and the outer molecular layer of the hippocampus (ML) were imaged after staining of brain sections with an antibody to synaptophysin. OLD APP/PS mice, either mock-immunized or SDPM1-immunized, were compared to age-matched wild type (WT) mice. Small inserts at the right show absence of signal with secondary antibody alone. Bar is 200μm. (B) 20μg of purified brain plasma membranes were solubilized from SDPM1- or mock-immunized YOUNG and OLD APP/PS mice and non-immunized wild type (WT) mice and separated on gradient SDS-PAGE gels. Brain lysates were immunoblotted for synaptophysin, PSD95 or βActin. Quantification of synaptophysin immunostaining (C) and PSD95 and synaptophysin immunoblotting (D) for age-matched wild type (WT), mock-immunized and SDPM1-immunized APP/PS mice. Errors are SEM for n=10–12 animals per condition. *P<0.05, **P<0.01, ***P<0.001
Reduced inflammatory markers in SDPM1-immunized APP/PS brain
To assess overall levels of inflammatory cells, we quantified positive cell staining in brain sections taken from SDPM1- and mock-immunized APP/PS mouse cortex for GFAP, a marker for astrocytes (and whose increased expression marks astroglial activation and gliosis), Iba1, a marker for brain microglia, and CD68, a marker for activated brain microglia (Fig. 6). Consistent with reduced amyloid plaque burden, we saw a significant reduction in CD68-positive cells in both YOUNG and OLD SDPM1-immunized APP/PS brain, relative to mock-immunized APP/PS controls. In addition, we did not observe any significant increase in brain Iba1 or GFAP positive cells in SDPM1-immunized APP/PS brain. No staining for T or B cells (using antibodies to CD45, CD4, CD8 or B220) was found in grey or white matter of SDPM1-immunized APP/PS brain, nor was any increased staining evident in the meninges (not shown). These data suggest that there is no increase in brain inflammation as the result of SDPM1 immunization.
Figure 6. Absence of increased inflammatory cells in SDPM1-immunized APP/PS mouse brain.
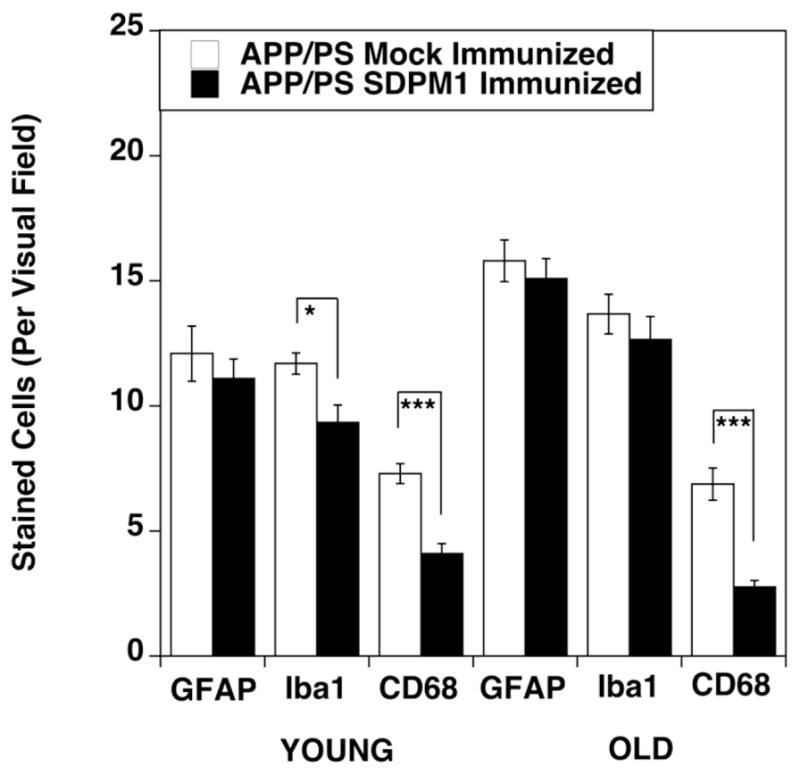
Serial brain sections of cortex from YOUNG and OLD APP/PS mice, either mock-immunized or SDPM1-immunized, were analyzed for expression GFAP, Iba1 or CD68. Errors are SEM for n=10–12 animals per condition. *P<0.05, **P<0.01
Therapeutic response of APP/PS mice to passive vaccination with P4D6, an SDPM1 peptide-mimotope antibody
We had previously shown that peptide-mimotope antibodies induced by SDPM1 immunization can, like the SDPM1 peptide itself, bind to low molecular weight Aβ amyloid oligomers and block subsequent Aβ amyloid aggregation(Wang et al., 2010). One such antibody is P4D6. Co-staining of P4D6 with Thioflavin S in 15 month-old APP/PS brain shows that P4D6 recognizes regions at the periphery of dense core amyloid plaques in sections taken from the frontal cortex (Supplemental Fig. 2).
We next determined if intracerebral injection of P4D6 would reduce Aβ amyloid plaque burden at the site of injection, much as other anti-Aβ peptide antibodies are known to do(Delrieu et al., 2012; Laskowitz and Kolls, 2010; Ostrowitzki et al., 2012; Panza et al., 2012; Roher et al., 2011). We used stereotactic brain injection methods to focally introduce P4D6 into a region of the frontal cortex and the hippocampus in 15 month-old APP/PS mice. The contralateral side of the brain was similarly injected (both cortex and hippocampus) with vehicle alone. At 1 day post-injection, we could identify diffuse, evenly distributed, staining of P4D6 antibody on the injected side of the brain, where it was concentrated at dense core amyloid plaques (co-stained with Thioflavin S) (Supplemental Fig. 3). By contrast, injected P4D6 did not co-localize with microglia (stained with Iba1) (Supplemental Fig. 3). Staining of the contralateral (control) side of the brain showed very low background in the absence of P4D6 (Supplemental Fig. 3).
By two weeks post-injection, most of the injected P4D6 antibody had been taken up by cells or cleared from the brain. Cellular staining of internalized antibody, however, was still visible in injected regions of the hippocampus (Fig. 7A) and cortex (Fig. 7B). Co-staining for P4D6 and Thioflavin S showed reduced Thioflavin S staining in P4D6-injected brain regions. Amyloid plaque burden (Fig. 7C) and plaque number (Fig. 7D) were both significantly reduced in P4D6-injected areas. Interestingly, the size of remaining plaques (Fig. 7E), most of which were present at the edges of the injection sites, was unchanged, suggesting a local therapeutic effect at the site of injection. These data demonstrate that an SDPM1 peptide-mimotope antibody can directly affect brain Aβ amyloid plaque burden.
Figure 7. Intracerebral injection of P4D6 in cortex and hippocampus lowers amyloid plaque burden at two weeks post-injection in APP/PS brain.

Brain sections of hippocampus (A) or cortex (B) were co-stained with anti-mouse IgM-rhodamine, to visualize injected P4D6, and Thioflavin S, to visualize dense core amyloid plaques, fourteen days after intracerebral injection of P4D6 antibody in the cortex or hippocampus. Individual stains were imaged on an epifluorescence microscope using rhodamine (red)- or fluorescein (green)-specific filters. Merged red and green images are shown in all four panels, with overlap in yellow. Bar is 200μm in A and B. Quantification of amyloid plaque burden (C), amyloid plaque profiles (D) and average amyloid plaque size (E) 14 days after P4D6 injection versus control. Errors are SD for n=4 animals per condition. *P<0.05, **P<0.01, ***P<0.001
P4D6 inhibits Aβ amyloid-mediated neurotoxicity
Because APP/PS mice fail to properly model the full extent of neurodegeneration that occurs in AD, we tested whether P4D6 could block Aβ-mediated neurotoxicity using neuronally differentiated SH-SY5Y cells(Lambert et al., 1994; Pahlman et al., 1984). We performed three assays to address neurotoxicity. The first was a fluorescence-activated Caspase 3/7 activity assay, which models activation of Aβ-mediated apoptosis, the second was propidium iodide uptake, which models cell death via perforation of the cell membrane, and the third was quantification of chromatin condensation via Hoechst staining of nuclei in dying cells. We performed these experiments by first making a 100μM Aβ1-42 peptide solution in PBS either in the presence or the absence of 5μM P4D6. Serial dilutions of these two stock solutions were then used for all experiments. Differing dilutions of this stock solution, (either of Aβ1-42 alone or Aβ1-42+P4D6) were added to differentiated SH-SY5Y cells for 48 hours for each assay. For all three assays, we observed blockage of Aβ1-42-mediated neurotoxicity by P4D6 (Fig. 8). All such changes were significant at 5, 10 and 20μM added Aβ1-42. These data support the notion that P4D6 can inhibit Aβ-mediated neurotoxicity and cell death.
Figure 8. P4D6 blocks Aβ1-42-mediated cytotoxicity of neuron-like cells.
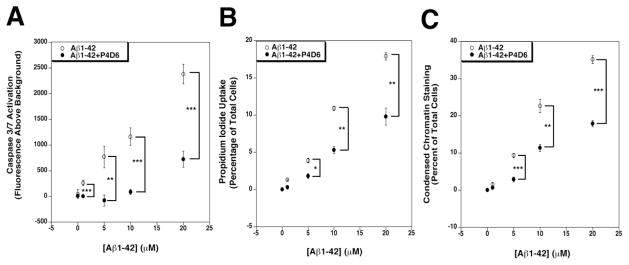
Retinoic acid differentiated SH-SY5Y cells were cultured in the presence of different concentrations of Aβ1-42 amyloid or Aβ1-42 amyloid that had been pre-incubated with (20:1 mol/mol) P4D6 antibody. Cells were assayed for activation of Caspase 3/7 (A), uptake of propidium iodide (B) or condensed chromatin after staining with Hoechst dye (C). Errors are SEM for n=10 experiments per condition. *P<0.05, **P<0.01, ***P<0.001
Sequencing of the P4D6 antibody and SDPM1 multimer binding
To identify the P4D6 antibody sequence, we used RT-PCR to subclone and sequence multiple mouse antibody heavy and light chain clones from multiple P4D6-expressing hybridoma cell clones. All heavy chain sequences identified were identical, as were all light chain sequences identified. These findings suggest a true clonal nature to the P4D6 hybridomas we had originally made (Supplemental Fig. 4). Moreover, heavy chain and light chains sequences were consistent with previous characterization of P4D6 as a mu-heavy chain/lambda-light chain antibody(Wang et al., 2010).
There are four common features in the amino acid sequence of SDPM1 and SDPM1-like peptides(Kang et al., 2003; Wang et al., 2010). First, all such peptides require a cysteine at both ends of the peptide for high affinity binding to Aβ amyloid. Second, they are all rich in hydrophobic amino acids. Third, they all have a net positive charge, and fourth, they all contain a proline within the hydrophobic-basic region. In analyzing the 73 amino acid variable domain of the P4D6 heavy chain bounded by cysteines, we identified two SDPM1-like motifs in the P4D6 heavy chain (Fig. 9A). Each SDPM1-like motif was rich in hydrophobic amino acids, had an abundance of lysine and arginine (with net positive charge of +2 per motif) and contained a proline. No SDPM1-like motifs were evident in the variable domain of the P4D6 light chain (Fig. 9A).
Figure 9. Identification of multiple SDPM1 peptide motifs in the P4D6 heavy chain variable domain and a role for SDPM1 multivalency in Aβ amyloid binding.

(A) Comparison of amino acids from the SDPM1 peptide (and a second SDPM1-like peptide) with the cysteine-bridged variable domain of the heavy and light chain of P4D6 show two SDPM1-like motifs within the P4D6 heavy chain sequence. Hydrophobic and basic amino acids are in bold, while SDPM1 motifs are underlined. (B) An epitope-tagged tetramer of the SDPM1 peptide (SDPM14) was produced, purified and studied for binding to Aβ1-40 or Aβ1-42 amyloid and the P4D6 antibody (Ab). Errors are SEM for n=6 experiments per condition.
We had previously shown that the SDPM1 peptide has a lower binding affinity for Aβ amyloid than does P4D6(Wang et al., 2010). Binding of monomeric SDPM1 to Aβ1-40 and to Aβ1-42 amyloid was saturable at 1μM, while binding of P4D6 to these same proteins was saturable at 15nM. To determine if this could be due to the multimeric presentation of SDPM1 in P4D6, which theoretically would have four SDPM1 motifs per Ig domain, we produced a recombinant tetrameric form of the SDPM1 peptide (SDPM14). SDPM14 showed high affinity binding to P4D6, and this binding was nearly identical in profile to SDPM14 binding to Aβ1-40 and Aβ1-42 amyloid (Fig. 9B). Binding of SDPM14 to all three proteins was saturable at 15nM (Fig. 9B). Thus, multimerization of the SDPM1 motif, much as may occur in P4D6, increased binding to Aβ amyloid by 1–2 logs relative to the monomeric peptide.
DISCUSSION
We have demonstrated that immunization of APPswePSEN1delE9 (APP/PS) mice with SDPM1, an Aβ-amyloid specific binding and blocking peptide(Kang et al., 2003; Wang et al., 2010), reduces brain amyloid plaque burden and brain Aβ1-40 and Aβ1-42 peptide levels, improves learning and memory, and increases expression of synaptic brain proteins. The immunization protocol was optimized for its translational relevance by utilizing ALUM as the adjuvant and only four immunizations for the vaccine paradigm. YOUNG (6 month-old) APP/PS mice, immunized just as brain Aβ amyloid plaques were developing(Borchelt et al., 1997), and OLD (12 month-old) APP/PS mice, immunized after brain amyloid burden was already significant(Borchelt et al., 1997), both showed improvements in most or all of these measures when analyzed 6 months after immunizations were begun. In addition, we found no increase in inflammatory cells in the brains of either YOUNG or OLD APP/PS mice compared to age-matched APP/PS controls. Thus, immunization with the SDPM1 peptide bound to ALUM shows potential as a vaccine therapy for Alzheimer’s disease.
Previously, we had shown that immunization with biotinylated SDPM1 (conjugated to streptavidin and coupled with Freund’s incomplete adjuvant) stimulated production of SDPM1-specific antibodies that cleared brain Aβ amyloid plaque burden and improved learning and memory in APPswePSEN1(A246E) mice(Wang et al., 2010). Besides the use of a different mouse model here, there were several other important differences between the two studies. First, our previous study showed improvements in learning and memory in YOUNG, but not OLD, APP/PS mice, while here both YOUNG and OLD APP/PS SDPM1-immunized animals showed significant cognitive improvement. Use of the ALUM-SDPM1 vaccine stimulated roughly the same amounts of SDPM1-specific and Aβ amyloid-specific antibodies as was seen in the previous study, but the vaccine paradigm used here, four injections (one every two weeks) versus 6 injections previously (one every month) may have led to more rapid therapeutic antibody production. Second, the distribution of SDPM1-specific antibody species produced in the two studies was different; the biotin-SDPM1-strepavidin vaccine produced anti-SDPM1-specific antibodies that were predominantly IgG1 (65–85%) with most of the remainder being IgM (10–20%), while the SDPM1-ALUM vaccine produced predominantly IgM (45–55%) with most of the remainder being IgG2b (25–30%) or IgA (10%). Both SDPM1 vaccines were strongly biased towards a Th2 type immune response(Finkelman et al., 1990), with IgG1+IgG2b antibody levels being roughly 10-fold higher than IgG2a. The preferential production of IgG2b and IgA here perhaps suggests a TGFβ-mediated, Treg-dependent, mechanism for T cell help, as TGFβ can be produced by Treg cells and can induce IgG2b and IgA class switching(Josefowicz et al., 2012; Park et al., 2005; Tran, 2012). Third, in this study we have correlated improvements in cognitive performance here with increased expression of synaptic brain proteins (synaptophysin and PSD95). This data is consistent with previous studies showing reduced staining and blotting for synaptic proteins in both AD and APP/PS brain, suggestive of changed synaptic density and/or integrity, and increased expression after immunization with Aβ peptides (Buttini et al., 2005; Buttini et al., 2002; DeKosky and Scheff, 1990; Masliah et al., 1991; Pham et al., 2010; Scheff and Price, 2003; Shankar et al., 2009; Terry et al., 1991). Improvements in cognitive performance, however, may also reflect the decreased brain inflammation in SDPM1-vaccinated animals. Fourth, the vaccine paradigm utilized here is more translationally relevant for AD than our previous study. Formulation of a clinical vaccine as has been used in this study would only require cGMP production of the SDPM1-4E peptide and its conjugation with ALUM, an adjuvant that has been used in many clinical vaccines(Schijns and Lavelle, 2011).
The SDPM1-ALUM vaccine joins a large number of proof of concept experiments showing that immunization with various formulations of Aβ peptide can have therapeutic effects in AD mouse models(Delrieu et al., 2012; Lemere and Masliah, 2010; Nelson et al., 2012; Panza et al., 2012; Tabira, 2010). The relevant question, then, is why would SDPM1 be any better than these myriad other approaches. Several features of SDPM1 recommend its use. The first, and most obvious, is that the SDPM1 peptide is not structurally related to the Aβ1-40 or Aβ1-42 peptide. SDPM1 was isolated as binding to Aβ amyloid from a cysteine-linked random 20 amino acid phage display peptide library(Kang et al., 2003). To our knowledge, this peptide sequence does not exist in nature, showing no more than 50% identity to any protein in any database. As such, it represents a non-native protein sequence, which in turn, perhaps, also reflects a non-native immunogenic epitope. The issue of endogenous protein homology is very important for the effectiveness of an AD vaccine. Only a minority of the AD patients in the AN1792 trial, where Aβ1-42 amyloid was used as the immunogen, produced increased levels of anti-Aβ antibodies (Gilman et al., 2005; Orgogozo et al., 2003; Schenk, 2008). Such failures arise, at least in part, from the reduced responsiveness of elderly patients to vaccines(Chen et al., 2009; Grubeck-Loebenstein et al., 2009; Haynes and Swain, 2006). An additional issue, however, may be the endogenous expression of Aβ1-42 within APP and its natural release by β and γ secretase, which could tolerize patient immune responses to Aβ1-42 immunization. Second, immunization with SDPM1 induces peptide-mimotope antibodies such as P4D6 that specifically bind to and affect low molecular weight Aβ oligomers(Wang et al., 2010). We show here that P4D6 can directly clear amyloid plaque burden in APP/PS brain and can block Aβ-mediated neurotoxicity in cultured neuron-like cells. While immunization with Aβ peptides would also allow for immune responses to low molecular weight Aβ oligomers, such Aβ oligomers would likely only be present in small amounts, as Aβ1-42 oligomerization to high molecular weight forms occurs quite rapidly. Both passive and active anti-Aβ immunization vaccine strategies can have toxic side effects that can be antibody- or lymphocyte-mediated, including meningoencephalitis, vasogenic cerebral edema, and microhemmorhage(Delrieu et al., 2012; Laskowitz and Kolls, 2010; Monsonego et al., 2003; Orgogozo et al., 2003). The ability to eliminate such side effects by targeting a specific subset of Aβ oligomers could be very important. The sequencing of the P4D6 antibody also allows for the creation of humanized antibodies to test in future studies.
Other studies suggest that the SDPM1 motif may be part of a class of peptide sequences that abrogate common protein amyloid aggregation domains within pathologic proteins. For examples, O’Nuallain and colleagues used phage display to map peptides that could mimic binding of the 11-1F4 antibody to the human κ4 Bence Jones protein Len, a protein cause of non-Aβ amyloidosis(O’Nuallain et al., 2007). Like P4D6, 11-1F4 shows therapeutic efficacy at inhibiting amyloidosis in vivo and protein fibrillogenesis in vitro(Hrncic et al., 2000; O’Nuallain et al., 2007). Similar to SDPM1, they identified only proteins with a proline flanked by aromatic amino acids, suggesting a hydrophobic cis-proline-mediated β turn was involved in aberrant protein aggregation. Another study by Tanaka and colleagues showed a hydrophobic and positively charged motif in the B6-C15 cyclic peptide could block Aβ1-42 fibrillogenesis, again defining positively charged and hydrophobic amino acids (with a proline) as being important for blocking protein aggregation(Tanaka et al., 2011). Interestingly, like SDPM1 and P4D6(Wang et al., 2010), the B6-C15 peptide appeared to block Aβ aggregation at the level of Aβ tetramers(Tanaka et al., 2011). Thus, while not identical to the sequence we have identified, these studies support the notion that SDPM1-like peptide motifs can have therapeutic efficacy in AD and perhaps other forms of protein amyloidosis.
Supplementary Material
Highlights.
Vaccination with SDPM1 shows pre-clinical efficacy in lowering brain Aβ amyloid plaque burden, brain Aβ1-40 and Aβ1-42 peptide levels, and improving learning and memory and expression of synaptic proteins in the APPswePSEN1de9 mouse.
An SDPM1 peptide-mimotope antibody, P4D6, can lower brain Aβ amyloid plaque burden in the APPswePSENde9 mouse
An SDPM1 peptide-mimotope antibody, P4D6, can block Aβ-mediated neurotoxicity
The therapeutic SDPM1 peptide-mimotope antibody, P4D6, was sequenced and mimics the SDPM1 peptide motif in the variable domain of its heavy chain.
Acknowledgments
We would like to thank Leslie Turner for technical support. Leslie was supported by the NCH high school summer scientist’s internship program. This work was supported by NIH grant AR049722 to PTM and by a Technology Transfer grant from Nationwide Children’s Hospital to PTM.
Footnotes
CONFLICT OF INTEREST
PTM is the inventor of SDPM1 peptide and P4D6, an SDPM1 peptide-mimotope antibody, both of which are protected by US patents US7745569 and US8394375. None of the other authors have any conflicts to disclose related to the publication of this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachman DL, et al. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology. 1992;42:115–9. doi: 10.1212/wnl.42.1.115. [DOI] [PubMed] [Google Scholar]
- Bard F, et al. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100:2023–8. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, et al. Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem. 2005;95:834–47. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Buttini M, et al. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9096–101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, et al. Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid beta peptides but not on plaque formation. J Neurosci. 2002;22:10539–48. doi: 10.1523/JNEUROSCI.22-24-10539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty NC, et al. Intracranial administration of deglycosylated C-terminal-specific anti-Abeta antibody efficiently clears amyloid plaques without activating microglia in amyloid-depositing transgenic mice. Journal of neuroinflammation. 2006;3:11. doi: 10.1186/1742-2094-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WH, et al. Vaccination in the elderly: an immunological perspective. Trends Immunol. 2009;30:351–9. doi: 10.1016/j.it.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cribbs DH. Abeta DNA vaccination for Alzheimer’s disease: focus on disease prevention. CNS Neurol Disord Drug Targets. 2010;9:207–16. doi: 10.2174/187152710791012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Delrieu J, et al. ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches. J Neurochem. 2012;120(Suppl 1):186–93. doi: 10.1111/j.1471-4159.2011.07458.x. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelman FD, et al. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol. 1990;8:303–33. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- Ghochikyan A, et al. Prototype Alzheimer’s disease epitope vaccine induced strong Th2-type anti-Abeta antibody response with Alum to Quil A adjuvant switch. Vaccine. 2006;24:2275–82. doi: 10.1016/j.vaccine.2005.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Grubeck-Loebenstein B, et al. Immunosenescence and vaccine failure in the elderly. Aging Clin Exp Res. 2009;21:201–9. doi: 10.1007/BF03324904. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Haynes L, Swain SL. Why aging T cells fail: implications for vaccination. Immunity. 2006;24:663–6. doi: 10.1016/j.immuni.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–54. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Hrncic R, et al. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol. 2000;157:1239–46. doi: 10.1016/S0002-9440(10)64639-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–65. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jayasinha V, et al. Inhibition of dystroglycan cleavage causes muscular dystrophy in transgenic mice. Neuromuscul Disord. 2003;13:365–75. doi: 10.1016/s0960-8966(03)00040-3. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, et al. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CK, et al. Identification of peptides that specifically bind Abeta1-40 amyloid in vitro and amyloid plaques in Alzheimer’s disease brain using phage display. Neurobiol Dis. 2003;14:146–56. doi: 10.1016/s0969-9961(03)00105-0. [DOI] [PubMed] [Google Scholar]
- Kelly KJ, et al. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol. 2003;284:C1309–18. doi: 10.1152/ajpcell.00353.2002. [DOI] [PubMed] [Google Scholar]
- Kim JH, et al. Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001;21:1327–33. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace GL, et al. A brief history of tau: the evolving view of the microtubule-associated protein tau in neurodegenerative diseases. Clin Neuropathol. 2007;26:43–58. doi: 10.5414/npp26043. [DOI] [PubMed] [Google Scholar]
- Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, et al. Beta/A4-evoked degeneration of differentiated SH-SY5Y human neuroblastoma cells. J Neurosci Res. 1994;39:377–85. doi: 10.1002/jnr.490390404. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Kolls BJ. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2010;74:2026. doi: 10.1212/WNL.0b013e3181e03844. author reply 2026–7. [DOI] [PubMed] [Google Scholar]
- Lemere CA. Developing novel immunogens for a safe and effective Alzheimer’s disease vaccine. Prog Brain Res. 2009;175:83–93. doi: 10.1016/S0079-6123(09)17506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6:108–19. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Levites Y, et al. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. The Journal of clinical investigation. 2006a;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, et al. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006b;26:11923–8. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, et al. Synaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol. 2012;970:573–601. doi: 10.1007/978-3-7091-0932-8_25. [DOI] [PubMed] [Google Scholar]
- Masliah E, et al. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am J Pathol. 1991;138:235–46. [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissette DA, et al. Relevance of transgenic mouse models to human Alzheimer disease. J Biol Chem. 2009;284:6033–7. doi: 10.1074/jbc.R800030200. [DOI] [PubMed] [Google Scholar]
- Nelson PT, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- O’Nuallain B, et al. Phage display and peptide mapping of an immunoglobulin light chain fibril-related conformational epitope. Biochemistry. 2007;46:13049–58. doi: 10.1021/bi701255m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgogozo JM, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Ostrowitzki S, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69:198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- Pahlman S, et al. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–44. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Panza F, et al. Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4:213–38. doi: 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- Park SR, et al. Analysis of transforming growth factor-beta1-induced Ig germ-line gamma2b transcription and its implication for IgA isotype switching. Eur J Immunol. 2005;35:946–56. doi: 10.1002/eji.200425848. [DOI] [PubMed] [Google Scholar]
- Pham E, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–67. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pride M, et al. Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis. 2008;5:194–6. doi: 10.1159/000113700. [DOI] [PubMed] [Google Scholar]
- Roher AE, et al. Neuropathology and amyloid-beta spectrum in a bapineuzumab immunotherapy recipient. Journal of Alzheimer’s disease: JAD. 2011;24:315–25. doi: 10.3233/JAD-2011-101809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko A, et al. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18:602–17. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Schenk D. Current challenges for the successful treatment and prevention of Alzheimer’s disease: treating the pathologies of the disease to change its clinical course. Alzheimers Dement. 2008;4:S119–21. doi: 10.1016/j.jalz.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schijns VE, Lavelle EC. Trends in vaccine adjuvants. Expert Rev Vaccines. 2011;10:539–50. doi: 10.1586/erv.11.21. [DOI] [PubMed] [Google Scholar]
- Schneeberger A, et al. Development of AFFITOPE vaccines for Alzheimer’s disease (AD)--from concept to clinical testing. J Nutr Health Aging. 2009;13:264–7. doi: 10.1007/s12603-009-0070-5. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001a;3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001b;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shankar GM, et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, et al. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, et al. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–44. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220:95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- Tanaka K, et al. A mimotope peptide of Abeta42 fibril-specific antibodies with Abeta42 fibrillation inhibitory activity induces anti-Abeta42 conformer antibody response by a displayed form on an M13 phage in mice. J Neuroimmunol. 2011;236:27–38. doi: 10.1016/j.jneuroim.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1996;55:1023–5. [PubMed] [Google Scholar]
- Terry RD, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol. 2012;4:29–37. doi: 10.1093/jmcb/mjr033. [DOI] [PubMed] [Google Scholar]
- Vaisid T, et al. Amyloid beta peptide toxicity in differentiated PC12 cells: calpain-calpastatin, caspase, and membrane damage. J Neurosci Res. 2008;86:2314–25. doi: 10.1002/jnr.21670. [DOI] [PubMed] [Google Scholar]
- Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang CM, et al. Immunization with the SDPM1 peptide lowers amyloid plaque burden and improves cognitive function in the APPswePSEN1(A246E) transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2010;39:409–22. doi: 10.1016/j.nbd.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, et al. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–6. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, et al. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:3745–51. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, et al. Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci. 2009;29:11393–8. doi: 10.1523/JNEUROSCI.2021-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, et al. The synaptic CT carbohydrate modulates binding and expression of extracellular matrix proteins in skeletal muscle: Partial dependence on utrophin. Mol Cell Neurosci. 2009;41:448–63. doi: 10.1016/j.mcn.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.