

Abstract
Vascular endothelial growth factor (VEGF) and reelin are two major signaling pathways involved in many neuronal functions including neurogenesis and neuronal migration. Both VEGF and reelin have been shown to regulate NMDA type glutamate receptor (NMDAR) activity via independent mechanisms. However, it is not known whether the above signaling pathways influence each other on NMDAR regulation. We demonstrate that Disabled 1 (Dab1), a downstream signaling molecule of reelin pathway mediates VEGF-induced regulation of NMDAR subunit NR2B. Furthermore, VEGF treatment led to the association of VEGF receptor-2 (Flk1) and reelin receptor (apolipoprotein E receptor 2, ApoER2), and Dab1 as well as NR2B activation were Flk1-dependent. Moreover, VEGF treatment could significantly rescue the deficits in phospho-Dab1 levels in reeler (Reln−/−) neurons. Our results suggest a major role of VEGF in the regulation of reelin signaling, and Dab1 as a key molecule in the cross talk between reelin and VEGF signaling pathways.
Keywords: reelin, Dab1, VEGF, NR2B, neurons
Introduction
Vascular endothelial growth factor (VEGF) was originally identified as an endothelial growth factor stimulating angiogenesis and vascular permeability [1]. Recent studies have shown the role of VEGF in neuronal functions such as neurogenesis and cell survival [2]. In particular, VEGF regulates NMDAR2B (NR2B; the predominant form of NMDA receptors in embryonic neurons) activity in neurons via Src family kinase (SFK) pathway [3]. However, the mechanism(s) of regulation of NR2B phosphorylation following VEGF treatment remains unknown.
Disabled-1(Dab1) is an SRC substrate involved in neuronal migration during mammalian brain development [4]. Dab-1 acts downstream of reelin as an intracellular, adaptor protein that is phosphorylated by SFKs when the reelin ligand binds to very low density lipoprotein receptor and Apolipoprotein E Receptor-2 (VLDLR and ApoER2) [5–6]. Moreover, reelin-induced tyrosine phosphorylation of NMDA receptors was found significantly inhibited in Dab1 knockout neurons indicating that Dab1 is necessary for SFK mediated NMDAR activity [7].
Because both VEGF and Reelin regulate NMDAR activity and Dab1 is a downstream target of SFK, we hypothesized that Dab1 signaling pathway is involved in VEGF regulation of NMDA receptor activity.
Materials and Methods
Ethic statement
All experiments in the present study were conducted with the approval of the Georgia Regents University, Committee on Animal Use for Research.
Animals
Timed pregnant CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA, USA). Heterozygous reeler mice were obtained from Jackson Laboratories (Bar Habor, ME, USA) and the colony was maintained in our animal housing facility at the Georgia Regents University. Reeler embryos were genotyped using the DNA extraction HOT SHOT method. The following Reelin primers used: Wild-type Reelin RP 5′ACA GTT GAC ATA CCT TAA TC 3′; Reelin FP 5′TAA TCT GTC CTC ACT CTG CC 3′; Reelin RP 5′TGC ATT AAT GTG CAG TGT TG 3′. Samples were analyzed by PCR and ran on a 2% agarose gel for UV determination of bands using ethidium bromide.
Cerebral Cortical Neuronal Cultures
Mouse cortical neurons were cultured as described previously [8]. Briefly, cerebral cortices from murine embryos (E16) were aseptically dissected and plated at 2.5 × 105 cells per well on polyethyleneimine-coated 6-well plates. Neurons were cultured in Neurobasal medium supplemented with B27, 2 mM L-glutamine, and antibiotics (Invitrogen). On the third day in vitro (DIV3), media was replaced with Neurobasal supplemented with B27 minus antioxidants, glutamine, and antibiotics. Purified neuronal cultures were routinely >97% neurons, as assessed by MAP-2 immunostaining. Neurons were used for treatments between DIV 5 and 7. Following treatments in culture, cells were washed in Phosphate Buffered Saline (PBS) and collected in ice-cold radioimmununoprecipitation assay (RIPA) buffer.
Reagents and antibodies
Recombinant VEGF protein (CYT-241) was purchased from ProSpec (Israel). SU5416 and PP2 were purchased from Tocris Biosciences (Ellisville, MO). Antibodies used in the study were obtained from commercial sources as follows: anti- rabbit Flk1 and anti-rabbit Dab1 (Santa Cruz Biotechnology); anti-rabbit phosphoDab1 and anti-rabbit β-tubulin (Cell Signaling Technology); anti-rabbit phospho NR2b (Millipore), anti-rabbit NR2b (Novus Biologicals) and anti-mouse β-actin (Sigma Aldrich). Anti-rabbit ApoER2 was a gift from Dr. Joachim Herz. Dab1 siRNA (M-043867-02) and control siRNA (D-001206-13-05) were purchased from Dharmacon. Effectene Transfection Reagent wwas purchased from Qiagen. NR1A and NR2B constructs were gifted by Dr. Bo-Shiun Chen. pCAX Dab1 plasmid (30139) was purchased from Addgene [9].
Immunoblotting and Immunoprecipitation
Protein concentrations were determined by the bicinchoninic acid method (BCA Protein Assay Kit, Sigma, USA). Equal amounts of protein were resolved in SDS-polyacrylamide gels and transferred electrophoretically onto a nitrocellulose membrane (Bio-Rad). Blots were incubated with primary antibodies overnight at 4 °C. After washing with 1× PBS and blocking with 5% milk in 1× PBS, blots were incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibody (Santa Cruz Biotechnology) for 1 h, followed by developing with the ECL Plus Western Blotting Detection System (GE Healthcare). Chemiluminescence signals were captured on autoradiographic blue films (Bioexpress). Films were scanned and the densitometric values for the proteins of interest were corrected using β-actin or β-tubulin with Image J Software. For immunoprecipitation, 300 μg of proteins were pre-cleared for 1 h with 30 μl of PureProteome Protein A and G Magnetic Beads (Millipore), followed by incubation overnight at 4°C in the presence of an anti-Flk1 antibody. The immunoprecipitated proteins were subjected to immunoblotting for the detection of ApoER2 or Flk1.
Transfection
Human kidney-derived HEK 293 cells were co-transfected by Effectene Transfection Reagent with NR1A, NR2B and pCAXDab1. Cells were transfected again 38 h later by Effectene Transfection Reagent with control siRNA or siRNA specific to Dab1. Cells were used for treatment 48 h after the second transfection.
Stereotaxic administration of VEGF
CD-1 mice (p9) were anesthetized with 3% isofluorane, and the surgical plane of the anesthesia was maintained using 1.5% isofluorane. They were placed on a stereotaxic adaptor for mouse pups mounted on a stereotaxic frame integrated with a Quintessential auto-injector (Stoelting Co, Wood Dale, IL), and a low-speed micro-drill apparatus for the bone (Harvard Apparatus, Boston MA). The skin over the skull was cleaned with alcohol followed by iodine-pivodine duo swabs. The skull was exposed through a small cut on the skin. The stereotaxic coordinates for the frontal cortex (anterioposterior +0.5 mm, medio lateral +0.5 mm and dorso–ventral − 1.0 mm relative to bregma and ventral from dura were located and the site was carefully drilled slowly. Through a skull hole, a 30 gauge needle attached to a 5 μL Hamilton syringe containing rhVEGF (150 ng/ul) solution was manually lowered according to the ventral coordinates and inserted into the brain to deliver 1 μl of the solution at the rate of 0.5 uL/ min.
Statistical Analysis
Unless otherwise indicated, experiments were performed in triplicate and repeated at least twice. Data were analyzed using GraphPad Prism and statistical significance was assessed by means one way ANOVA or Student’s t-test (*P<0.05).
Results
Dab1 is required for VEGF-induced NR2B activation
Primary cortical neurons were treated with recombinant VEGF and activation of NR2B and Dab1 were examined at 0, 5, 10, 15 min following treatment. The phospho NR2B (pNR2B) and phospho Dab1 (pDab1) levels (Western blot) were increased within the first 5 min of VEGF exposure [Fig 1a and b; 0.799±0.088 (mean ± SE) vs 1.284±0.165 for pNR2B/NR2B ratio and 1.105±0.113 vs 1.62±0.147 for pDab1/Dab1 ratio]. After administration of VEGF in the frontal cortex to postnatal day 9 (P9) mice, we observed increased phosphorylation of Dab1 in forebrain lysates (Fig 1c; 0.653±0.0613 vs 0.877±0.01 or 1.182±0.073 for 5 and 10 min respectively). Given that the Dab1 was activated following VEGF exposure, we examined whether Dab1 signaling would play a major role in VEGF-induced NR2B activation. To our surprise, this VEGF-induced NR2B activation in neurons was significantly blocked by co-treatment of VEGF with the Dab1 inhibitor PP2 (1 μM), when PP2 was added into the incubation for 30 min prior to VEGF treatment (Fig 1d; 0.926±0.04 vs 1.131±0.03 or 0.98±0.036 for VEGF and PP2+VEGF respectively). The role of Dab1 in VEGF-induced NR2B activation was further examined using Dab1 siRNA in HEK cells. The Dab1 expression level was reduced in those cells transfected with the Dab1 siRNA pool when examined at 48 h after the transfection (Fig 1e; 1.12±0.094 vs 0.64±0.008). The siRNA knockdown of Dab1 successfully abolished VEGF-induced NR2B activation as compared with the control siRNA transfected cells (Fig 1f; 0.95±0.043 vs 0.77±0.042). The effect of Dab1 siRNA knockdown on the prevention of NR2B activation was quantitatively similar to that after co-treatment of VEGF with the Dab1 inhibitor PP2. Taken together, these results indicate that Dab1 played a critical role in this VEGF-induced NR2B activation.
Figure 1.
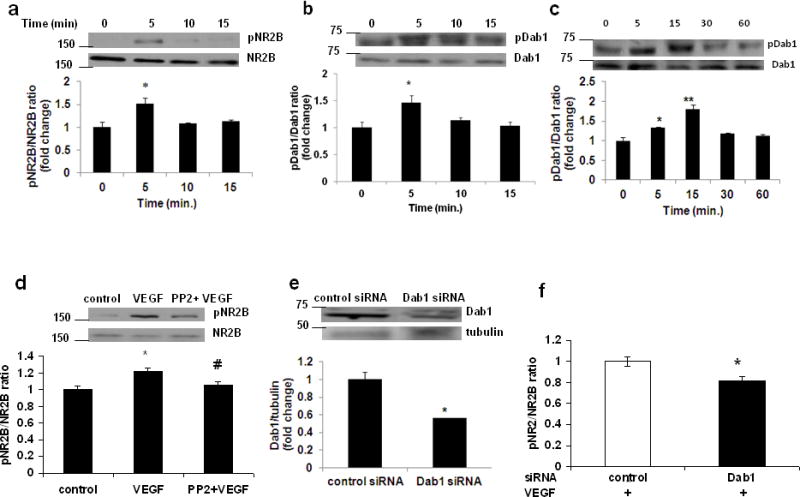
Dab1 is required for VEGF-induced NR2B activation. Representative western blot analyses (upper panel) and fold change (lower panel) in (a) pNR2B/NR2B and (b) pDab1/Dab1 ratio in primary cortical neurons treated with recombinant human VEGF protein (rhVEGF; 50 ng/ml) for 0 to 15 min. Experiments were repeated and performed in triplicate. *p<0.05 vs value at 0 min. (c) p9 mice were administered rhVEGF (100 ng/μl) in the frontal cortex for 0 to 60 min. The upper panel shows representative autoradiogram of pDab1 and Dab1, and the lower panel represents fold change in pDab1/Dab1 ratio. N=3 for 0, 15, 30 and 60 min; N=2 for 5 min. *p<0.05 and **p<0.01 vs value at 0 min. (d) Primary cortical neurons were preincubated for 30 min with PP2 (1 μM), and then exposed to rhVEGF (50 ng/ml) for 5 min. The upper panel shows representative autoradiogram of pNR2B and NR2B, and the lower panel represents fold change in pNR2B/NR2B ratio normalized to vehicle-treated controls. Values are mean ± SE. Experiments were repeated and performed in triplicate. *p<0.05 vs control and #p<0.05 vs VEGF. (e) Representative western blot analyses (upper panel) and fold change (lower panel) in Dab1/β-tubulin ratio in human kidney-derived HEK 293 cells transfected with control or Dab1 siRNA. Experiments were performed in triplicate. *p<0.05 vs control siRNA. (f) HEK 293 cells were co-transfected by Effectene Transfection Reagent with NR1, NR2B and pCAX Dab1. Cells were transfected again 38h later with control siRNA or siRNA specific to Dab1. Cells were treated 48 h later with rhVEGF (50 ng/ml) for 5 min before lysis. Fold change in pNR2B/NR2B ratio normalized to vehicle-treated controls. Values are mean ± SE. Experiments were repeated and performed in triplicate. *p<0.05 vs control.
VEGFR2 phosphorylation is required for VEGF-induced Dab1 activation
Although VEGFs signal through the high affinity receptor tyrosine kinases VEGFR-1, VEGFR-2 (Flk1), and VEGFR-3, Flk1 mediates VEGF-induced functions in neurons [10–11]. The observed increase of Dab1 phosphorylation following VEGF suggested that Dab1 activation may be dependent on the activation status of Flk1. We therefore performed rhVEGF-dependent Dab1 phosphorylation experiments in the presence of the VEGF receptor blocker SU5416. SU5416 is a potent and specific inhibitor of the Flk-1 tyrosine kinase [12]. Treatment of primary cortical neurons with SU5416 prior to stimulation with rhVEGF led to a decrease in Dab1 as well as NR2B phosphorylation (Fig. 2), which indicates that Flk1 phosphorylation is required for VEGF-induced activation of Dab1 and NR2B.
Figure 2.
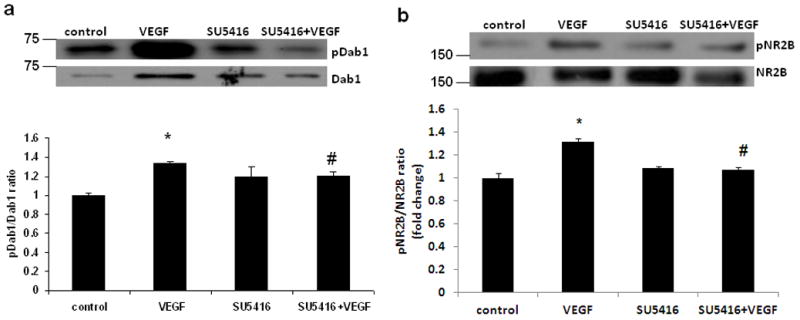
VEGFR2 phosphorylation is required for VEGF-induced Dab1 and NR2B activation. Primary cortical neurons were preincubated for 1 h with SU5416 (10μg/mL), and then exposed to rhVEGF (50 ng/ml) for 5 min. Representative western blot analyses (upper panel) and fold change (lower panel) in (a) pDab1/Dab1 and (b) pNR2B/NR2B ratio are shown. Values are mean ± SE. Experiments were repeated and performed in triplicate. *p<0.05 vs control and #p<0.05 vs VEGF.
VEGF-induced Dab1 activation is Reelin-independent
It has been shown that Reelin regulates NMDA-type glutamate receptor activity through a mechanism that requires Dab1 [7] and phosphorylation of Dab1 in cortical neurons isolated from reeler mice could be rescued by stimulation with exogenous Reelin [13]. Given that the Dab1 was activated following VEGF exposure, we examined whether reelin would play a major role in VEGF-induced Dab1 activation. First, we determined the effect of VEGF treatment on reelin protein levels at 0, 1, 6, 12, 24 and 48 -h exposure of primary cortical neurons. The reelin level was significantly increased when examined at 1 h of VEGF exposure, which lasted up to at least 6 h after VEGF exposure (Fig 3a). Next, we examined whether reelin is necessary for VEGF-induced activation of Dab1 in neurons. Primary cortical neurons isolated from reeler (Reln−/−) and wild-type mice were treated with VEGF and phosphorylation of Dab1 was examined. We found an increase in Dab1 levels with a reduction in Dab1 phosphorylation levels in Reln−/− neurons as reported in previous studies [13, 19] (Fig 3b). Surprisingly, stimulation of Reln−/− neurons with VEGF could significantly induce Dab1 phosphorylation. However, the increase in Dab1 phosphorylation in Reln−/− neurons was not up to those levels in VEGF-treated WT neurons. The incomplete recovery of Dab1 phosphorylation following VEGF stimulation in Reln−/− neurons indicate that other mechanisms are also likely to be involved in the activation of Dab1 by VEGF.
Figure 3.
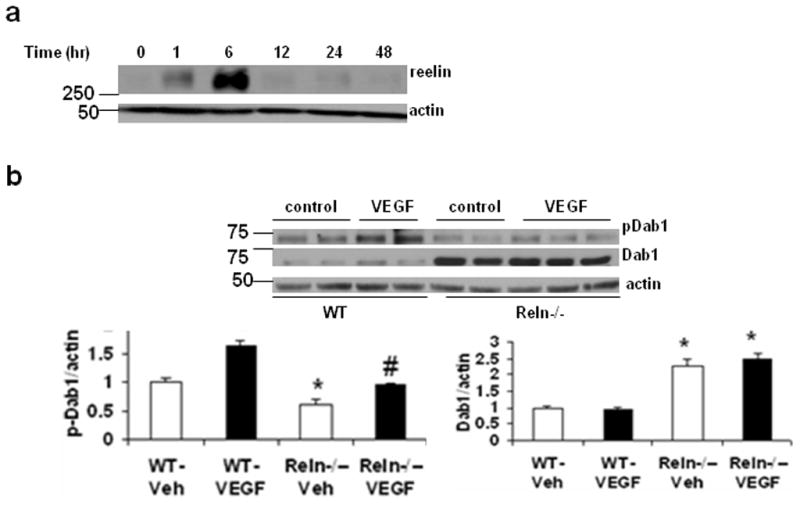
VEGF-induced Dab1 activation is Reelin-independent. (a) Representative western blot analyses of reelin and β-actin expression in primary cortical neurons treated with recombinant human VEGF protein (rhVEGF; 50 ng/ml) for 0 to 48 h. Experiments were repeated and performed in triplicate. (b) Primary cortical neurons from wild-type (WT) or reeler (Reln−/−) embryos were treated with rhVEGF (50 ng/ml) for 5 min. The upper panel shows representative autoradiogram of pDab1, Dab1 and actin, and the lower panel represents fold change in (left) pDab1/actin and (right) Dab1/actin ratio normalized to vehicle-treated WT neurons. Values are mean ± SE. Experiments were repeated and performed in triplicate. *p<0.05 vs WT-vehicle and #p<0.05 vs Reln−/−-vehicle.
VEGF increases the association between APOER2 and Flk1 in neurons
ApoER2 and VLDLR serve as receptors for Reelin activation of Dab1 signaling. To determine whether VEGF treatment promotes the association between Flk1 and ApoER2 or VLDLR, we performed co-immunoprecipitation experiments following VEGF treatment in neurons. Flk1 immunoprecipitation followed by western blot analysis revealed enhanced Flk1-ApoER2 complex formation after stimulation with VEGF (Fig 4).
Figure 4.
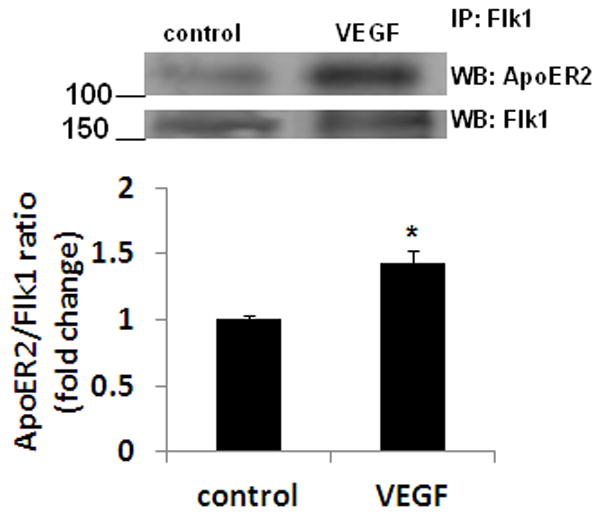
VEGF increases the association between ApoER2 and Flk1 in neurons. Primary cortical neurons were treated with rhVEGF (50 ng/ml) for 5 min. Cell extracts were immunoprecipitated (IP) with Flk1 antibody and western blotting (WB) was performed with ApoER2 or Flk1 antibody. The upper panel shows representative autoradiogram of ApoER2 and Flk1, and the lower panel represents fold change in ApoER2/Flk1 ratio. Experiments were performed in triplicate. *p < 0.05 vs control.
Discussion
NR2B regulation by VEGF and reelin has been reported by independent studies. Our data show that Dab1 functions as a key signaling molecule to mediate VEGF-induced NR2B activation. Moreover, VEGF could stimulate Dab1 phosphorylation in Reln−/− neurons.
Both VEGF and reelin play important roles in neuronal migration during brain development [14–15]. Moreover, disruption of VEGF signaling shows neuronal migration defects similar to reeler mice [16]. Our data suggest that the interactions between reelin and VEGF pathways might involve Dab1 as a common signaling molecule for their downstream effects. Dab1 is an adaptor protein with many binding partners. Dab1 acts as an anchoring protein to ApoEr2 and VLDLR, the two reelin receptors which lack intrinsic tyrosine kinase activity [17]. Dab1 has also been shown to act as a point of convergence for reelin and cdk5 pathways [18]. The present data indicate that VEGF-induced Flk1 phosphorylation is involved in Dab1 activation. Flk1 is the key receptor for VEGF-induced effects in neurons [10–11]. We found a robust increase in Flk1-ApoEr2 interaction following VEGF treatment. It is possible that Dab1 acts as a scaffold for the above receptor complex following Flk1 activation by VEGF.
Our observation that VEGF induces Dab1 phosphorylation in neurons from reeler mice suggest that Dab1 could be significantly activated by VEGF in the absence of reelin. We found a decrease in phosphoDab1, but increase in Dab1 levels in the neurons from reeler mice, which are in agreement with many previous studies [13,19]. Although VEGF increases reelin protein levels, the presence of reelin is not required for Dab1 activation in embryonic cortical neurons. Further investigations are needed to understand the signaling events including the phosphorylation site in Dab1 as well as the requirement of reelin receptors for Dab1 activation by VEGF.
Reelin, VEGF and NR2B are implicated in neuropsychiatric disorders such as schizophrenia. Decreases in reelin as well as VEGF levels have been reported in the prefrontal cortex of schizophrenia subjects [20, 21]. Our data suggest that Dab1 mediates VEGF-induced regulation of NR2B in cortical neurons. Additional studies are needed to know whether altered VEGF signaling contributes to NMDAR hypofunction in schizophrenia. It is also important to investigate whether overexpression of VEGF could normalize behavioral and neurochemical abnormalities in rodent models of schizophrenia.
Highlights.
Dab1 mediates VEGF-induced regulation of NR2B in cortical neurons
VEGF treatment led to the association of VEGF receptor-2 (Flk1) and reelin receptor (apolipoprotein E receptor 2, ApoER2).
VEGF-induced Dab1 activation is Flk1-dependent.
VEGF treatment could significantly rescue the deficits in phospho-Dab1 levels in reeler (Reln−/−) neurons.
Acknowledgments
We thank Dr. Bo-Shiun Chen for NR1A and NR2B constructs, Dr. Dennis Selkoe for pCAX Dab1 plasmid and Dr. Joachim Herz for ApoER2 antibody. This work was supported by grants from NIH/NIMH to AP.
Footnotes
Competing interest
We have no conflicts of interest in presenting this manuscript.
Author contributions
AP designed the research. KRH performed the in vitro experiments and protein analyses. MNH performed the in vivo experiments. AP and KRH analyzed the data. AP wrote the manuscript. All authors read and approved the final manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zachary I. Neuroprotective role of vascular endothelial growth factor: signalling mechanisms, biological function, and therapeutic potential. Neurosignals. 2005;14:207–221. doi: 10.1159/000088637. [DOI] [PubMed] [Google Scholar]
- 3.Meissirel C, et al. VEGF modulates NMDA receptors activity in cerebellar granule cells through Src-family kinases before synapse formation. Proc Natl Acad Sci U S A. 2011;108:13782–13787. doi: 10.1073/pnas.1100341108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 5.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 6.Rice DS, et al. The reelin pathway modulates the structure and function of retinal synaptic circuitry. Neuron. 2001;31:929–941. doi: 10.1016/s0896-6273(01)00436-6. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, et al. Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howell KR, Kutiyanawalla A, Pillai A. Long-term continuous corticosterone treatment decreases VEGF receptor-2 expression in frontal cortex. PLoS One. 2011;6:e20198. doi: 10.1371/journal.pone.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young-Pearse TL, et al. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27:14459–144569. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao L, et al. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36:827–835. doi: 10.1038/ng1395. [DOI] [PubMed] [Google Scholar]
- 12.Vajkoczy P, et al. Inhibition of tumor growth, angiogenesis, and microcirculation by the novel Flk-1 inhibitor SU5416 as assessed by intravital multi-fluorescence videomicroscopy. Neoplasia. 1999;1:31–41. doi: 10.1038/sj.neo.7900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sentürk A, Pfennig S, Weiss A, Burk K, Acker-Palmer A. Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature. 2011;472:356–360. doi: 10.1038/nature09874. [DOI] [PubMed] [Google Scholar]
- 14.Huang Z. Molecular regulation of neuronal migration during neocortical development. Mol Cell Neurosci. 2009;42:11–22. doi: 10.1016/j.mcn.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Mackenzie F, Ruhrberg C. Diverse roles for VEGF-A in the nervous system. Development. 2012;139:1371–1380. doi: 10.1242/dev.072348. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz Q, et al. Vascular endothelial growth factor controls neuronal migration and cooperates with Sema3A to pattern distinct compartments of the facial nerve. Genes Dev. 2004;18:2822–2834. doi: 10.1101/gad.322904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 18.Keshvara L, Magdaleno S, Benhayon D, Curran T. Cyclin-dependent kinase 5 phosphorylates disabled 1 independently of Reelin signaling. J Neurosci. 2002;22:4869–4877. doi: 10.1523/JNEUROSCI.22-12-04869.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proceedings of the National Academy of Sciences United States of America. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulzele S, Pillai A. Decreased VEGF mRNA expression in the dorsolateral prefrontal cortex of schizophrenia subjects. Schizophrenia Research. 2009;115:372–373. doi: 10.1016/j.schres.2009.06.005. [DOI] [PubMed] [Google Scholar]