

Abstract
Cardinal pathologic features of hypertensive heart disease (HHD) include not only hypertrophied cardiomyocytes and foci of scattered microscopic scarring, a footprint of prior necrosis, but also small myocytes ensnared by fibrillar collagen where disuse atrophy with protein degradation would be predicted. Whether atrophic signaling is concordant with the appearance of HHD and involves oxidative and endoplasmic reticulum (ER) stress remains unexplored. Herein, we examine these possibilities focusing on the left ventricle (LV) and cardiomyocytes harvested from hypertensive rats receiving 4 wks aldosterone/salt treatment (ALDOST) alone or together with ZnSO4, a nonvasoactive antioxidant, with the potential to attenuate atrophy and optimize hypertrophy. Compared to untreated age-/sex-/strain-matched controls, ALDOST was accompanied by: a) LV hypertrophy with preserved systolic function; b) concordant cardiomyocyte atrophy (<1000 μm2) found at sites bordering on fibrosis where they were re-expressing β-myosin heavy chain; and c) upregulation of ubiquitin ligases, MuRF1 and atrogin-1, and elevated 8-isoprostane and unfolded protein ER response with mRNA upregulation of stress markers. ZnSO4 cotreatment reduced lipid peroxidation, fibrosis and the number of atrophic myocytes, together with a further increase in cell area and width of atrophied and hypertrophied myocytes, and improved systolic function, but did not attenuate elevated blood pressure. We conclude that atrophic signaling, concordant with hypertrophy, occurs in the presence of a reparative fibrosis and induction of oxidative and ER stress at sites of scarring where myocytes are atrophied. ZnSO4 cotreatment in HHD with ALDOST attenuates the number of atrophic myocytes, optimizes size of atrophied and hypertrophied myocytes, and improves systolic function.
Keywords: hypertrophy, atrophy, oxidative stress, ER stress, cardiomyocytes, aldosteronism
INTRODUCTION
The concentric left ventricular (LV) hypertrophy found in human hypertensive heart disease (HHD) is considered a risk factor for adverse cardiovascular events, including heart failure (1–5). HHD includes not only hypertrophied cardiomyocytes, but also widely scattered foci of microscopic scarring, a footprint of myocyte necrosis, and perivascular fibrosis of intramyocardial coronary arteries and arterioles that extends into the contiguous interstitial space (6–11). As Bernard Swynghedauw has suggested, fibrosis is the “crucial determinant of myocardial heterogeneity” (12).
A more detailed morphometric analysis of cardiomyocytes in HHD reveals heterogeneity in size consisting of large cells, hypertrophied in response to the pressure overload placed on the left ventricle by arterial hypertension, and small atrophic cells (6–8, 10). These atrophic myocytes are found bordering on and within microscopic scars and perivascular/interstitial fibrosis. At these sites of fibrosis, atrophic myocytes are ensnared by fibrillar collagen that serves to anchor fibrous tissue within this hollow muscular organ and thereby preserving its structural integrity. In turn, this architectural alignment reduces myocyte work with ensuing disuse atrophy. It is therefore likely that hypertrophic and atrophic cardiomyocyte signaling are operative concurrently in HHD in response to disparate loading conditions: one which stems from the increased hemodynamic burden placed on the LV; and the other arises from unloading that prevails locally within microdomains of fibrosis. The re-expression of beta-myosin heavy chain, formerly considered a marker of increased myocyte work and hypertrophy (13), occurs instead in these smaller myocytes distributed in clusters within discrete foci of fibrosis and where it is now instead considered a marker of fibrosis (14, 15). A similar correlation exists with the re-expression of atrial natriuretic peptide at sites of perivascular fibrosis and microscopic scarring (16–22).
Diffuse cardiomyocyte atrophy is known to accompany ventricular hemodynamic unloading associated with heterotopic transplantation (23, 24), right ventricular failure with underfilling of the left ventricle (25), dietary caloric restriction (26) or taurine deficiency (27), dexamethasone treatment (28) and cardiac sympathetic neuron ablation (29). Localized atrophy is seen with the cardiac fibrosis associated with arterial hypertension (6–10) and Trypanosoma cruzi infestation (30). Whether atrophic signaling is intrinsically coordinated with hypertrophy remains to be elucidated. Redox signaling and endoplasmic reticulum (ER) stress are common to disuse atrophy in skeletal muscle (31–34) and oxidative stress is an integral pathophysiologic feature of hypertension (35). Atrophic remodeling is a coordinated interaction between redox signaling and FoxO (Forkhead box-containing protein, O subfamily) transcription factors-dependent activation of the redox-sensitive proteolytic ubiquitin-proteasome system (UPS) with its E3 ligases, MuRF1 and atrogin-1 (28, 29, 36, 37).
Herein, we tested our hypothesis whether atrophic signaling is coupled to oxidative/ER stress in the myocardium and its cardiomyocytes harvested from rats with HHD in response to 4 wks chronic aldosterone/salt treatment (ALDOST) (38, 39). We compared observed iterations to those found in untreated age-/sex-/strain-matched controls. In ALDOST rats, plasma aldosterone levels are raised (inappropriately for 1% dietary Na+ intake) to those seen in human primary or secondary aldosteronism, together with suppressed plasma renin activity and angiotensin II, and is accompanied by a gradual rise in arterial pressure and appearance of concentric LVH. A pathologic structural remodeling of myocardium, resembling its clinical counterpart (40), first appears at 4 wks ALDOST (38, 39). We further sought to identify potential targets for intervention that could attenuate atrophy and optimize hypertrophy. In this context, we explored the relevance of oxidative stress and fibrosis in regulating cardiomyocyte size (vis-à-vis hypertension) using cotreatment with ZnSO4, a non-vasoactive antioxidant and Zn2+ donor, which together with upregulated expression of its binding protein, metallothionein, accounts for increased tissue Zn2+ at sites of cardiac injury to provide cardioprotection (41–44).
METHODS
Animal Model
Eight-week-old male Sprague-Dawley rats were used throughout this series of experiments approved by the Animal Care and Use Committee of our institution. As reported previously and following uninephrectomy, an osmotic minipump containing ALDO was implanted subcutaneously to raise circulating ALDO levels to those commonly found in human CHF (45). Drinking water was fortified with 1% NaCl and with 0.4% KCl to prevent hypokalemia. A separate group of rats received ALDOST plus zinc sulfate (40 mg/day by gavage) as cotreatment. Unoperated, untreated age-/sex-/strain-matched rats served as controls. Each group consisted of 6 rats. Animals were killed at week 4 of each regimen.
Transthoracic Echocardiography
Echocardiography was performed with 7.0 MHz pediatric transducer in anesthetized rats. Parasternal short axis 2-D and M-mode views were acquired at the level of papillary muscles. Fractional shortening was calculated from the M-mode view. Relative wall thickness was determined using the ratio of (septum thickness + posterior wall thickness) ÷ end-diastolic diameter.
Blood Pressure Measurement
Blood pressure (BP) was measured at wk 4 ALDOST using the tail-cuff method. Animals were acclimated for three days before BP was taken.
Collagen Volume Fraction
The presence of cardiac pathology based on the extent of fibrosis was assessed by collagen-specific picrosirius red staining in coronal sections (6 μm) of the ventricles and observed by light microscopy with polarized light as previously reported (39). Collagen volume fraction was determined separately for microscopic scars and perivascular/interstitial fibrosis found in each section using a computer image analysis system (NIH Image software, 1.60), as previously reported (39).
Cardiac Myocyte Isolation
Cardiomyocytes were isolated by retrograde perfusion of the crystalloid-perfused heart as we previously reported (42).
Cardiomyocyte Size Planimetry
For discriminating cell size, freshly isolated cardiac myocytes were spread on glass slides, air-dried and fixed for 15 min in 10% formalin. Cells were stained with hematoxylin-eosin and photographed using a Kodak digital camera. Cell area and length were measured using NIH Image software. Average cell width was calculated as area/length. One hundred or more cardiomyocytes were measured from each rat heart isolate.
Flow Cytometry
Flow cytometry was used to sort cells producing nitric oxide (NO), reactive oxygen species (ROS), and intracellular α and β smooth muscle myosin in isolated rat cardiomyocytes obtained by retrograde collagenase perfusion from the various experimental groups. CellROX® and DAF-FM diacetate (4-amino-5-methylamino- 2',7'-difluorofluorescein diacetate) (Life Technologies, Grand Island, NY) were used to detect NO and ROS, respectively, in viable, unfixed cardiomyocytes. Cardiomyocytes were positively identified with a rabbit antibody to β adrenergic receptor 1 (Abcam Biochemicals, Cambridge, MA) followed by APC-goat anti-rabbit IgG. Propidium iodide (PI) was used for dead cell discrimination. For gating and analysis, live cardiomyocytes were identified as APC+ PI− cells. Separate samples of cardiomyocyte labeled with β adrenergic receptor 1 were paraformaldehyde fixed and saponin permeabilized by Perm/Wash™ (BD Biosciences, San Jose, CA). Mouse monoclonal antibodies (mAbs) BA-G5, IgG2b and NOQ7.5.4, IgG1 were used to detect intracellular α- and β-smooth muscle myosin, respectively (15). Goat anti-mouse IgG2b-PE-Cy7 and goat anti-mouse IgG1-PE were used to detect the BA-G5 and NOQ7.5.4 mAbs, respectively. For gating and analysis, cardiomyocytes were identified as APC+ PE-Cy7+ that are either producing β-smooth muscle myosin, PE+, or not, PE−. Cardiomyocytes so identified were sorted into distinct, isolated subpopulations for downstream RNA expression analyses. Flow cytometric analyses were performed on either a BD Biosciences LSR II or FACSAria II. Sorting was performed on the FACSAria II.
Cardiac 8-Isoprostane
Cardiac tissue total 8-isoprostane (free and esterified) was measured using a competitive enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI, USA) as reported previously (46).
Western Blotting
For immunoblotting, cardiac myocytes were lysed with SDS-urea buffer (40 mM Hepes, 4 M urea, 1% SDS, pH 7.4). Protein content was measured with bicinchoninic acid assay method (Pierce Biotechnology, Rockford, IL). From each sample aliquots containing 20 μg of total protein were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes in accordance with standard procedures. Membranes were blocked in 5% non-fat milk in TBST (0.25% Tween 20) for 1 hr. Incubation with the primary antibodies to rabbit anti-protein disulfide isomerase (anti-PDI; 1:1000; Cell Signaling Technology, Danvers, MA), and mouse monoclonal to KDEL (1:1000; Stressgen Biotechnologies, San Diego, CA) and GADPH (1:10000) was performed overnight at 4°C. Immunodetection was achieved using the horseradish peroxidase (HRP) conjugated anti-mouse and anti-rabbit IgG (1:10000; Sigma-Aldrich, St. Louis, MO) and bands visualized with the ECL system (Pierce Biotechnology, Rockford, IL). Protein loading was normalized using GADPH as a housekeeping protein.
Immunofluorescence and Immunohistochemistry
For immunofluorescence and immunohistochemical staining, 6 μm thick frozen sections or air-dried cardiac myocytes on slides were fixed in 10% formalin, blocked with 3% BSA and incubated with primary antibodies to myosin (1:200, MAB 1628, Millipore, Billerica, MA) or KDEL (1:100, 10C3, Millipore) overnight at 4°C. Anti-Mouse IgG (Fab-specific)–FITC (for myosin, F8771, Sigma, St. Louis, MO), and Dako LSAB2 HRP kit (for KDEL, Dako North America, Carpintiria, CA) was used for the immunofluorescence or immunohistochemistry detection, respectively. The slides were counterstained with DAPI or hematoxylin, respectively.
Real-Time PCR
For real-time PCR, total mRNA was isolated and purified with the DNA-free™ kit (Ambion, Austin, TX) from cardiac myocytes lysed in Trizol. Quantitative real-time PCR was carried out on an LC480 thermocycler using FastStart TaqMan Probe Master (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. PCR primers were:
-
Glucose-regulated protein 78 (GRP78)
forward – 5′CGTAACAATCAAGGTCTACGA,
reverse – 5′AGGTGACTTCAATCTGGGGTA;
-
Metallothionein 1 (MT1)
forward – 5′CACCAGATCTCGGAATGGAC,
reverse – 5′TGGAGCAGGTGCAGGAG (antisense);
-
Homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 (HERPUD1)
forward – 5′TTACGGGAAAGGGAAGTCCT,
reverse – 5′CTTGGAAAGTCTGCTGGAC;
-
Protein disulfide isomerase (PDI)
forward – 5′GAAGAGCAACTTCGCAGAGG,
reverse – 5′CACACCATGGGGCATAG;
-
X-box binding protein 1 (XBP-1)
forward – 5′TGCCCTGGTTACTGAAGAGG,
reverse – 5′CACCAGCCTTACTCCATTC;
-
Myosin heavy chain, α
forward – 5′AACTGAAAACGGCAAGACG,
reverse – 5′TGGCCATGTCCTCGATCT;
-
Myosin heavy chain, β
forward – 5′ATCAAGGAGCTCACCTACCA,
reverse – 5′TCCTGCAGTCGCAGTAGGTT;
-
Pro-atrial natriuretic peptide (pro-ANP) precursor
forward - 5′ACAGATCTGATGGATTTCAAGA,
reverse – 5′CTCATCTTCTACCGGCATC;
-
Atrogin-1
forward – 5′GAAGACCGGCTACTGTGGAA,
reverse – 5′TCAATCGCTTGCGGATCT;
-
Muscle RING-finger protein-1 (MuRF1)
forward – 5′GGACTCCTGCCGAGTGAC,
reverse – 5′TTGTGGCTCAGTTCCTCCTT;
-
Connective tissue growth factor (CTGF)
forward – 5′GCTGACCTAGAGGAAAACATTAAGA,
reverse – 5′GCCCGGTAGGTCTTCACAC.
XBP-1 Splicing
Alternative splicing of XBP-1 was assessed with RT-PCR using primers according to Thuerauf DF et al. (47), which spans both non-spliced and spliced region of mRNA. PCR products were resolved electrophoretically on 2% agarose gel and visualized with ethidium bromide staining.
Statistical Analysis
Data were expressed as mean±SEM. Comparisons between groups were performed with one-way ANOVA using Scheffé's post-hoc analysis. Frequency distributions in cell size were analyzed using Fisher's exact test. P values less than 0.05 were considered statistically significant.
RESULTS
Arterial Hypertension in ALDOST
Four wks ALDOST was accompanied by arterial hypertension with a significant (p<0.05) elevation in systolic and diastolic blood pressures (see Table 1). Cotreatment with ZnSO4, a non-vasoactive antioxidant, did not attenuate this elevation in arterial pressure that accompanies ALDOST.
Table 1.
Hemodynamic and echocardiographic data.
| Control | ALDOST | ALDOST+ZnSO4 | |
|---|---|---|---|
| Body weight, g | 335.3±3.7 | 247.2±3.3* | 255.6±5.4‡ |
| SBP, mm Hg | 110.5±3.1 | 181.8±3.4* | 185.5±5.1‡ |
| DBP, mm Hg | 89±1.6 | 150.4±6.5* | 149.2±3.7‡ |
| EDD, mm | 0.71±0.02 | 0.63±0.02* | 0.66±0.01 |
| ESD, mm | 0.38±0.02 | 0.36±0.03 | 0.31±0.01‡ |
| FS, % | 46.2±1.5 | 42.3±4.5 | 52.9±2.0†‡ |
| RWT | 0.40±0.01 | 0.55±0.03* | 0.49±0.02‡ |
Data presented as mean±SEM
p<0.05 Control vs. ALDOST
p<0.05 ALDOST vs. ALDOST+ZnSO4
p<0.05 Control vs. ALDOST+ZnSO4
Concentric Hypertrophy, Myocardial Fibrosis and Ventricular Function
Hypertrophy
Consistent with the concentric pattern of hypertrophy in ALDOST, echocardiography demonstrated a significant (p<0.05) reduction in LV end diastolic dimension (EDD) without any change in end systolic dimension (ESD) in keeping with the observed increase in relative wall thickness (see Table 1).
Fibrosis
Light microscopy revealed scarring, a biomarker of earlier cardiomyocyte necrosis of the nonpressure overloaded, nonhypertrophied RV and the pressure overloaded hypertrophied LV, as well as perivascular/interstitial fibrosis involving intramyocardial coronary arteries and arterioles. Pro-atrial natriuretic peptide (ANP), a molecular marker re-expressed at sites of fibrosis, was also upregulated with 4 wks ALDOST (vida infra).
Collagen volume fraction (CVF) related to scarring was increased (p<0.05) more than two-fold with ALDOST compared to controls (4.79±0.87 vs. 2.22±0.04%), as was the perivascular/interstitial fibrosis component of CVF (p<0.05) compared to controls (4.29±0.61 vs. 2.96±0.02%). Four weeks cotreatment with antioxidant ZnSO4 attenuated necrosis as estimated by reduced microscopic scarring (3.89±0.34%), but did not modify the coronary vasculopathy with perivascular fibrosis (4.20±0.31%) or re-expression of ANP.
Ventricular Function
Fractional ventricular shortening was unchanged between controls and 4 wks ALDOST, but it was increased (p<0.05) with ZnSO4 cotreatment (see Table 1). The decline in EDD in the absence of any change in shortening implicates diastolic dysfunction with ALDOST and is consistent with the appearance of myocardial fibrosis (vide infra).
ZnSO4 cotreatment revealed a tendency toward increased EDD and reduced ESD, together with increased shortening compared to ALDOST alone, and is suggestive of improved systolic function which is also consistent with the morphologic evidence of less myocardial scarring and myocyte atrophy (see Table 1).
Heterogeneity in Cardiomyocyte Size
Hypertrophy
A heterogeneity in cardiomyocyte size is found in the normal left ventricle harvested from untreated control rats. Atrophic myocytes are seen at sites where normal fibrillar collagen encircles these cells, such as occurs at the base of the mitral valve or with their pending emergence as chordae tendineae from the papillary muscle (see Figure 1). However, there was no evidence of β-myosin heavy chain (MHC) re-expression at these sites (not shown).
Figure 1.
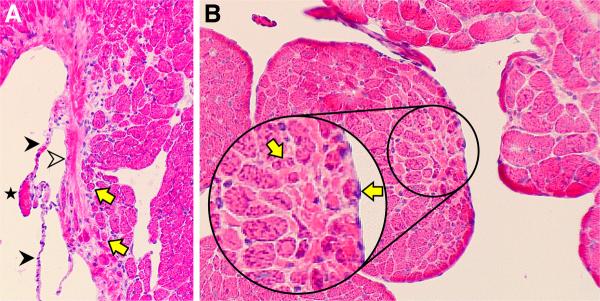
Atrophic cardiomyocytes (yellow arrows) are found in the normal rat heart where they are ensnared by fibrillar collagen. A) This includes the base of the mitral valve (open arrowhead). Constitutive chordae tendineae (solid arrowheads) and tip of sectioned papillary muscle (star) are also seen. B) Below the sectioned tip of the papillary muscle is fibrillar collagen (pink interstitial space) surrounding smaller myocytes (yellow arrows) and which will form chordae tendineae,. Hematoxylin and eosin; original magnification, 200×.
To evaluate heterogeneity in cardiomyocyte size present at 4 wks ALDOST, isolated cardiomyocytes were planimetered and their area, width and length determined in cells harvested from each of the 3 experimental groups. Consistent with the concentric hypertrophy associated with LV pressure overload, cell area and width (not length) were increased (p<0.05; see Figure 2) at 4 wks ALDOST compared to controls (see Table 2). ZnSO4 cotreatment led to a further increase (p<0.05) in cell area and width above that seen with ALDOST alone (Figure 2 and Table 2).
Figure 2.
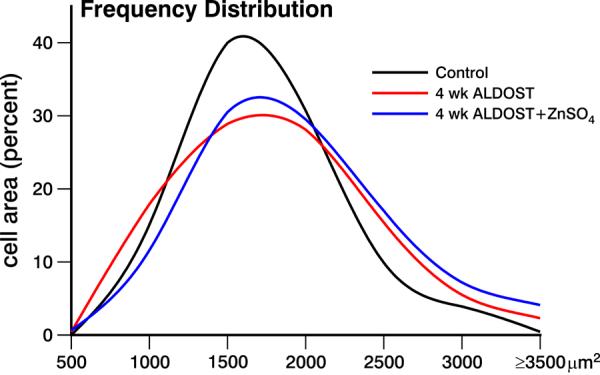
Frequency distribution of cardiomyocyte size, or percent cell area, seen in the left ventricle harvested from untreated controls, 4 wks ALDOST and 4 wks ALDOST+ZnSO4 cotreatment. An increase in atrophic cells (500–1000 μm2) and hypertrophied cells (≥2200 μm2) was seen with ALDOST. Cotreatment with ZnSO4 attenuated the number of atrophic cells while increasing the number of hypertrophied cells.
Table 2.
Cardiomyocyte morphometric data.
| 500–6000 μm2 | Control | ALDOST | ALDOST+ZnSO4 |
|---|---|---|---|
| Area, μm2 | 1498.1±18.9 | 1600.3±28.5* | 1692.1±30.0†‡ |
| Length, μm | 80.4±0.7 | 81.4±0.9 | 81.8±1.0 |
| Width, μm | 18.7±0.2 | 19.6±0.3* | 21.3±0.3†‡ |
|
| |||
| 500–1000 μm2 (atrophic myocytes) | |||
| Area | 831.7±11.7 | 782.2±13.8* | 824.9±16.8 |
| Length | 63.1±1.1 | 59.3±1.5 | 58.02±1.7 |
| Width | 13.5±0.3 | 13.8±0.3 | 14.9±0.5 |
Data presented as mean±SEM
p<0.05 Control vs. ALDOST
p<0.05 ALDOST vs. ALDOST+ZnSO4
p<0.05 Control vs. ALDOST+ZnSO4
Atrophy
Planimetry was used to examine variability in cell size. It revealed an increase in the number of atrophic cardiomyocytes (<1000 μm2) at 4 wks ALDOST (see Figure 2) compared to controls (17.9 vs. 15.1 %), and mean cell area was correspondingly reduced (p<0.05) in this model of HHD. By morphologic interrogation, atrophic cardiomyocytes were found bordering on sites of perivascular fibrosis (see panel A, Figure 3) and microscopic scars (see panel B, Figure 3), where histochemical staining revealed them to be ensnared by fibrillar collagen. In atrophic myocytes found at sites of fibrosis with scarring (panel A, Figure 4), evidence of re-expression of β-MHC was identified by both immunohistochemistry (panel B, Figure 4) and RTPCR (Table 3). A >6-fold increase in mRNA expression of slow β isoform of MHC was observed to be a molecular signal of atrophy in these smaller myocytes. Both atrophy and β-MHC re-expression were attenuated by ZnSO4 cotreatment (vida infra). The α isoform of MHC was likewise upregulated with ALDOST, but less dramatically, and ZnSO4 had no effect on the amplification of the α isoform. ZnSO4 cotreatment reduced (p<0.05) the number of atrophic cells (11.6%) and attenuated the reduction in mean cardiomyocyte area (see Table 2). We further subjected cardiomyocytes isolated at 4 wks ALDOST by flow cytometry. Smaller atrophic viable myocytes expressed biomarkers of oxidative stress and nitric oxide production as seen in top and bottom panels of Figure 5, respectively.
Figure 3.
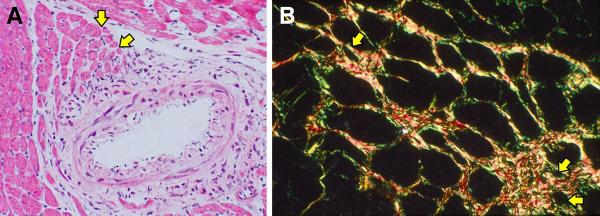
Histopathology reveals heterogeneity in myofiber size found in the rat left ventricle at 4 wks aldosterone/salt treatment (ALDOST). (A) Hematoxylin and eosin (H&E) staining. Perivascular/interstitial fibrosis involving an intramyocardial coronary artery. Yellow arrows point to atrophic myofibers bordering on fibrosis and surrounded by fibrillar collagen appearing as pink-stained interstitium (×20). (B) Picrosirius red staining with polarized light to enhance fibrillar collagen surrounding atrophic myofibers (yellow arrows) (original magnification, 100×).
Figure 4.
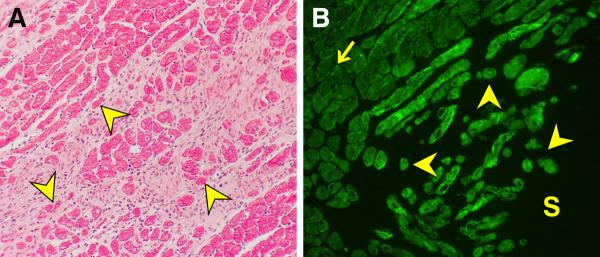
LV free wall; 4 wks ALDOST. (A) Light microscopy (H&E) with pink fibrous tissue surrounding cardiomyocytes (red) some of which are atrophic (yellow arrowhead) at the site of scarring. (B) Immunohistochemistry. Atrophic cardiomyocytes re-express β-myosin heavy chain (yellow arrowhead) at a site of scarring (S) as contrasted to its low level expression in normal-sized and hypertrophied myocytes (arrow) (original magnification, 200×).
Table 3.
mRNA expression by real-time PCR.
| mRNA | Control | ALDOST | ALDOST+ZnSO4 |
|---|---|---|---|
| MHC-β | 1.0±0.07 | 6.6±0.34* | 4.0±0.18*† |
| MHC-α | 1.0±0.11 | 1.9±0.26 | 1.8±0.60 |
| Pro-ANP | 1.0±0.21 | 3.9±0.46* | 4.3±0.28* |
| CTGF | 1.0±0.45 | 3.0±0.43* | 2.9±0.26* |
| Atrogin-1 | 1.0±0.11 | 2.2±0.21* | 1.8±0.22* |
| MuRF1 | 1.0±0.29 | 2.5±0.42* | 2.7±0.16* |
| GRP78 | 1.0±0.30 | 2.9±0.20* | 1.9±0.23 |
| XBP-1 | 1.0±0.21 | 1.9±0.23* | 1.6±0.20 |
| PDI | 1.0±0.32 | 3.2±0.22* | 2.0±0.26* |
| HERPUD1 | 1.0±0.28 | 3.8±0.16* | 2.0±0.23 |
| MT1 | 1.0±0.14 | 2.9±0.23* | 2.5±0.20* |
Data presented as mean±SEM
p<0.05 Control vs. ALDOST
p<0.05 ALDOST vs. ALDOST+ZnSO4
Figure 5.

Identification of smaller viable cardiomyocytes producing reactive oxygen species (ROS, upper panel) and nitric oxide (NO, lower panel) at 4 wks ALDOST using flow cytometry.
Atrophic Signaling
The atrophic signaling network includes E3 ubiquitin ligases, atrogin-1 and MuRF1, of the redox-sensitive UPS that participates in the degradation of sarcomeric proteins and contributes to regulation of myocyte size. Expression of these ligase genes was upregulated (p<0.05) in hearts harvested at 4 wks ALDOST (see Table 3). ZnSO4 cotreatment attenuated the induction of atrogin-1.
Oxidative and Endoplasmic Reticulum Stress
Oxidative Stress
Cardiomyocyte overloading of cytosolic and mitochondrial Ca2+ is an integral feature of chronic aldosteronism and is accompanied by mitochondria-derived oxidative stress in affected cardiomyocytes (43, 46). The oxidative stress compromises the integrity of mitochondria by increasing the opening potential of the mitochondria permeability transition pore and promotes misfolding and degradation of intracellular proteins. The rate of ROS generation in cardiomyocytes with ALDOST exceeded their rate of detoxification by endogenous Zn2+-based antioxidant defenses. Myocardial tissue levels of 8-isoprostane, a biomarker for lipid peroxidation, were increased 10-fold at 4 wks ALDOST compared to controls (390.1±86.0 vs. 34.7±6.4 pg/mg; p<0.05), and were markedly attenuated by cotreatment with the antioxidant, ZnSO4 (24.2±3.5 pg/mg).
ER Stress
The unfolded protein response (UPR) invoked by the endoplasmic reticulum (ER) stress is induced by the misfolded and degraded proteins. The UPR increases expression of various molecular chaperones, such as glucose-regulated proteins (GRP)78 and GRP94; a ubiquitously expressed transcription factor that undergoes alternative splicing termed splicing of X-box protein (XBP)-1; and protein-disulfide isomerase (PDI) that promotes refolding of misfolded proteins or their degradation by local ER-associated protein degradation system (ERAD). Under conditions of ER-stress, the XBP-1 mRNA is converted to active form by activated endoribonuclease IRE1. IRE1 catalyses excision of a 26 nucleotide unconventional intron from XBP-1 mRNA. Removal of this intron causes a frame shift in XBP-1 coding sequence resulting in the translation of active XBP-1 isoform (48). ERAD and UPR reduce cell death induced by oxidative stress; however, excessive ER stress leads to necrosis.
We examined the expression of ER stress biomarkers (GRP78, XBP-1, PDI and homocysteine-inducible, ER stress-inducible, ubiquitin-like domain member-1 (HERPUD1)) by real time PCR. Compared to controls, 4 wks ALDOST was accompanied by the induction of these stress markers, including GRP78, XBP-1, HERPUD1 and PDI. ZnSO4 cotreatment attenuated the induced expression of each of these genes (see Table 3). Zn2+ may directly mediate protection of thiol groups in protein or induce metallothionein (MT)-1, a thiol-rich protein induced by increased cytosolic free [Zn2+]i via its intracellular sensor, metal-responsive transcription factor (MTF)-1. MT1 mRNA was upregulated nearly 3-fold with ALDOST (see Table 3). ZnSO4 attenuation of the ALDOST-induced increase in GRP78 and the active isoform of XBP-1 was confirmed by immunoblotting (Figure 6A and B, respectively).
Figure 6.

Untreated controls, 4 wks ALDOST, and 4 wks ALDOST+ZnSO4. (A) Western blotting for glucose-related protein (GRP)78, and protein disulfide isomerase (PDI) with GADPH as a housekeeping protein. (B) Transcription factor with alternative splicing, termed splicing of X-box protein (XBP)-1, assessed by RT-PCR using primers spanning both unspliced (290 bp) and spliced (264 bp) regions of mRNA. (C) Immunostaining with anti-KDEL antibody reacting with GRP78 and GRP94. In contrast to controls in which only weak staining was seen confined to perinuclear area, at 4 wks ALDOST intense staining was evident in cardiomyocytes and nonmyocyte cells surrounding orange-colored fibrous tissue, whereas ZnSO4 cotreatment downregulated their expression as shown at a site of perivascular fibrosis.
ER stress in tissue sections was visualized by immunostaining with anti-KDEL antibody that binds both GRP78 and GRP94 (Figure 6C). In normal heart only weak perinuclear staining was visible. In contrast, cardiomyocytes and nonmyocyte cells surrounding scars and perivascular fibrosis after 4 wks ALDOST were intensely stained. ZnSO4 cotreatment attenuated GRP78 expression detected by immunostaining.
DISCUSSION
HHD accompanies the arterial hypertension of chronic ALDOST (49). HHD is manifest at the organ level as concentric LV hypertrophy with relative increase in LV wall thickness and reduction in EDD that normalizes systolic force per cross-sectional area of myocardium, or systolic wall stress (49). The hypertrophy of cardiomyocytes and rise in cellular protein synthesis is invoked by hemodynamic burden, or pressure overload, imposed by arterial hypertension. At the tissue level, however, there are marked alterations in architecture due to extracellular matrix remodeling and expressed as fibrosis. Myocardial fibrosis is inevitably the root cause of heterogeneity in tissue structure and cell size (12).
Our study led to several major findings. First, smaller cardiomyocytes neighboring on the normal valvular apparatus, including its chordae tendineae, or scar tissue and remodeled vessels with perivascular fibrosis are encircled by fibrillar collagen. These encircled cardiomyocytes have reduced workload leading to a fall in myocyte area (<1000 μm2). This disuse atrophy did not have a preferential distribution within the subendo- or subepicardium. At 4 wks ALDOST, myocyte atrophy is comparable to the proportional decrease in cell length and cross-sectional area or even more extensive than that seen with the nonworking perfused heart after heterotopic cardiac transplantation, where fibrosis is absent (50). Atrophy of myocytes that favors the endomyocardium is seen with the scarring that follows isoproterenol-induced cardiomyocyte necrosis (51). We found considerable heterogeneity in cardiomyocyte size in HHD associated with ALDOST, exceeding regional variations that are present in the normal rat heart (50). In fact, the concordant hypertrophic and atrophic remodeling of myocardium found in rats with 4 wks ALDOST strikingly resembles that seen in human aldosteronism and essential hypertension (6–8, 10, 40). A reversal of such maladaptive cardiomyocyte remodeling is a suggested therapeutic objective for the failing heart (52).
Our second major finding indicates that atrophic remodeling of cardiomyocytes to be simultaneous and associated with the pathophysiologic events subsequent to appearance of oxidative stress, activation of UPS ligases and re-expression of β-myosin heavy chain at sites of scarring, a footprint of necrosis. Cardiomyocyte necrosis is an outcome to events that begin with augmented enteral and urinary excretory losses of Ca2+, Mg2+, and Zn2+ that accompanies ALDOST. This leads to ionized hypocalcemia and hypomagnesemia, and hypozincemia (44, 45, 53). Secondary hyperparathyroidism follows with parathyroid hormone (PTH)-mediated intracellular Ca2+ overloading and induction of oxidative stress leading to opening of the inner membrane permeability pore of subsarcolemmal mitochondria (45, 53–55). We consider this pathophysiologic scenario to represent a mitochondriocentric signal-transducer-effector pathway to nonischemic cardiomyocyte necrosis (54–56). In peripheral blood mononuclear cells (PBMC; monocytes and lymphocytes) these pathophysiologic events contribute to their activation with the ensuing immunostimulatory state leading to invasion of coronary and systemic arterioles where consequent vasculopathy accounts for the appearance of hypertension (57–60).
In addition to the disuse atrophy hypothesis, paracrine signaling involving cardiomyocytes and myofibroblasts residing at microdomains of fibrosis, with or without heterocellular coupling, may also be operative (61–63). Fibrous tissue is metabolically active through a population of persisting, phenotypically transformed fibroblast-like cells termed myofibroblasts (myoFb) that contain α-smooth muscle actin microfilaments. The myoFb secretome has all essential components for de novo generation of angiotensin (Ang) peptides (64, 65) and reactive oxygen species (39) whose paracrine properties may induce intramyocytic Ca2+ overloading with oxidative stress that activates the UPS for proteolytic breakdown and/or autophagy and atrophy of these neighboring myocytes. Using gene chip array, one of us (ICG, personal communication) detected upregulation of ubiquitin-specific peptidase (USP)2 in the rat myocardium at 4 wks ALDOST, together with downregulated uncoupling protein (UCP)3. Upregulated USP2 and downregulated UCP3 occur with heterotopic transplantation of the rat heart and after LV circulatory assist in man (23, 66). Matrix remodeling and myocyte atrophy, therefore, appear to be concurrent and intertwined.
Cotreatment with non-vasoactive ZnSO4 did not attenuate the elevation in blood pressure or hemodynamic stimulus to hypertrophy. Its antioxidant properties would be operative in both PTH-driven oxidative stress in cardiomyocytes and redox signaling in myoFb. ZnSO4 attenuated necrosis and fibrosis, and simultaneously reduced the number of atrophic cells. In so doing, it markedly diminished tissue heterogeneity while optimizing the hypertrophic response that allowed a further increase in myocyte cell area and width and improved systolic function.
Our third major finding is the unequivocal evidence of oxidative and ER stress responses in the atrophic signaling associated with HHD during ALDOST. Oxidative stress has been demonstrated at subcellular levels involving subsarcolemmal mitochondria in cardiomyocytes as well as in PBMC, plasma and urine in ALDOST, and with aortocaval fistula where renal ischemia and secondary aldosteronism are expected (45, 67, 68). In the myocardium, biomarker evidence of oxidative stress with immunohistochemical localization of 3-nitrotyrosine and redox-sensitive nuclear factor (NF)-κB activation have been identified at sites of scarring and the coronary vasculopathy at 4 wks ALDOST (39). Moreover, these pathophysiologic responses were attenuated by cotreatment with N-acetylcysteine, an antioxidant, or a Zn2+ ionophore, pyrrolidine dithiocarbamate (PDTC). Herein, ZnSO4 was shown to attenuate lipid peroxidation with reduced tissue 8-isoprostane, reduce cardiomyocyte necrosis and scarring, and reduce the number of atrophic cells and mean cell area.
ER stress is an inherent protective strategy of cells, including cardiomyocytes to sustain intracellular homeostasis in response to cell injury from degenerating proteins. At 4 wks ALDOST, we found upregulation of ER stress biomarkers, including GRP78, XBP-1, HERPUD1 and PDI, which were localized to cardiomyocytes and nonmyocyte cells surrounding scars at sites of injury and perivascular fibrosis. Cotreatment with ZnSO4 attenuated these responses without reducing blood pressure suggesting injury is not related to this hemodynamic factor. We therefore propose the Zn2+-dependent cardioprotective response is related to increased cytosolic [Zn2+]i-mediated induction of MTF-1 and upregulation of MT1, an antioxidant gene it regulates (43). MT1 expression was shown to be increased nearly 3-fold. At sites of healing MT1 binds and confines Zn2+, where it contributes to transcription and cell replication (44).
We readily admit our study has some obvious limitations. For example, we did not use an antihypertensive agent to address more specifically the role of elevated blood pressure. The choice of agent is inherently problematic for the following reasons: a) spironolactone, an aldosterone receptor antagonist, attenuates the heightened excretory losses of Ca2+ and Mg2+ seen during ALDOST, and thereby prevents SHPT (45); b) an angiotensin converting enzyme inhibitor or receptor antagonist would impact the myoFb-based fibrous tissue response (64); c) a Ca2+ channel blocker would prevent intracellular Ca2+ overloading with oxidative stress and necrosis (67); and d) hydralazine, a nonspecific vasodilator, is sympathomimetic. Furthermore, we did not monitor the hypertrophic signaling pathway, involving calcineurin (69), and optimized by ZnSO4.
In summary, HHD that accompanies ALDOST involves LV hypertrophy, appearance of fibrous tissue with concordant cardiomyocyte atrophy, induction of oxidative and ER stress and mRNA upregulation of relevant genes. The ER stress response is consistent with the activation of atrophic signaling from protein degradation. It seems highly likely that the quantity of myocardium is not a credible risk factor for adverse cardiovascular events in HHD. Instead, myocardial quality appears to be far more relevant. Furthermore, cotreatment with a nonvasoactive antioxidant, ZnSO4, attenuated atrophy and optimized hypertrophy thus enhancing systolic function. It is conceivable that in targeting microdomains of fibrosis with the objective of rescuing and rightsizing atrophied cardiomyocytes that have been exiled from the mainstream contractile mass, a novel strategy to rebuild myocardium with these autologous cells may be at hand. Such an approach would be complementary to current interest in regenerating myocardium using stem cells and/or progenitor cells.
Acknowledgments
This work was supported, in part, by NIH grants R01-HL73043 and R01 HL90867 (KTW). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Authors have no conflicts of interest to disclose.
References
- 1.Weber KT. Enhanced cardiovascular risk in hypertensive heart disease. Cardiovasc Risk Factors. 1995;5:87–92. [Google Scholar]
- 2.Diez J, Lopez B, Gonzalez A, Querejeta R. Clinical aspects of hypertensive myocardial fibrosis. Curr Opin Cardiol. 2001;16:328–335. doi: 10.1097/00001573-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Sugihara N, Genda A, Shimizu M, Suematsu T, Kita Y, Minamoto M, Kawagoshi H, Umeda K, Chin S, Takeda R. Diastolic dysfunction and its relation to myocardial fibrosis in essential hypertension. J Cardiol. 1988;18:353–361. [PubMed] [Google Scholar]
- 4.Sugihara N, Genda A, Shimizu M, Suematu T, Kita Y, Horita Y, Takeda R. Quantitation of myocardial fibrosis and its relation to function in essential hypertension and hypertrophic cardiomyopathy. Clin Cardiol. 1988;11:771–778. doi: 10.1002/clc.4960111109. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro LM, McKenna WJ. Left ventricular hypertrophy: relation of structure to diastolic function in hypertension. Br Heart J. 1984;51:637–642. doi: 10.1136/hrt.51.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotran RS, Kumar V, Robbins SL. The heart. In: Cotran RS, Kumar V, Robbins SL, editors. Robbins Pathologic Basis of Disease. 4th ed W B Saunders; Philadelphia: 1989. pp. 597–656. [Google Scholar]
- 7.Karsner HT. Human Pathology. JB Lippincott; Philadelphia: 1955. [Google Scholar]
- 8.Pearlman ES, Weber KT, Janicki JS, Pietra GG, Fishman AP. Muscle fiber orientation and connective tissue content in the hypertrophied human heart. Lab Invest. 1982;46:158–164. [PubMed] [Google Scholar]
- 9.Huysman JAN, Vliegen HW, Van der Laarse A, Eulderink F. Changes in nonmyocyte tissue composition associated with pressure overload of hypertrophic human hearts. Pathol Res Pract. 1989;184:577–581. doi: 10.1016/S0344-0338(89)80162-1. [DOI] [PubMed] [Google Scholar]
- 10.Rossi MA. Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J Hypertens. 1998;16:1031–1041. doi: 10.1097/00004872-199816070-00018. [DOI] [PubMed] [Google Scholar]
- 11.Díez J, Laviades C, Mayor G, Gil MJ, Monreal I. Increased serum concentrations of procollagen peptides in essential hypertension. Relation to cardiac alterations. Circulation. 1995;91:1450–1456. doi: 10.1161/01.cir.91.5.1450. [DOI] [PubMed] [Google Scholar]
- 12.Swynghedauw B. Molecular mechanisms of myocardial remodelling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz K, Boheler KR, de la Bastie D, Lompre AM, Mercadier JJ. Switches in cardiac muscle gene expression as a result of pressure and volume overload. Am J Physiol. 1992;262:R364–369. doi: 10.1152/ajpregu.1992.262.3.R364. [DOI] [PubMed] [Google Scholar]
- 14.Pandya K, Kim HS, Smithies O. Fibrosis, not cell size, delineates β-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci U S A. 2006;103:16864–16869. doi: 10.1073/pnas.0607700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.López JE, Myagmar BE, Swigart PM, Montgomery MD, Haynam S, Bigos M, Rodrigo MC, Simpson PC. β-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res. 2011;109:629–638. doi: 10.1161/CIRCRESAHA.111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa T, Kasajima T, Naruse M, Naruse K, Demura H, Hiroe M, Nakazawa M, Nakajima Y, Nagata M. Immunohistochemical study on human atrial natriuretic polypeptide in the ventricle of hearts with endocardial fibroelastosis. Am J Cardiovasc Pathol. 1990;3:247–251. [PubMed] [Google Scholar]
- 17.Saito Y, Nakao K, Arai H, Nishimura K, Okumura K, Obata K, Takemura G, Fujiwara H, Sugawara A, Yamada T, Itoh H, Mukoyama M, Hosoda K, Kawai C, Ban T, Yasue H, Imura H. Augmented expression of atrial natriuretic polypeptide gene in ventricle of human failing heart. J Clin Invest. 1989;83:298–305. doi: 10.1172/JCI113872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takemura G, Fujiwara H, Horike K, Mukoyama M, Saito Y, Nakao K, Matsuda M, Kawamura A, Ishida M, Kida M, Ueguito T, Tanaka M, Matsumori A, Fujiwara Y, Fujiwara T, Imura H, Kawai C. Ventricular expression of atrial natriuretic polypeptide and its relations with hemodynamics and histology in dilated human hearts. Circulation. 1989;80:1137–1147. doi: 10.1161/01.cir.80.5.1137. [DOI] [PubMed] [Google Scholar]
- 19.Takemura G, Fujiwara H, Yoshida H, Mukoyama M, Saito Y, Nakao K, Fujiwara T, Uegaito T, Imura H, Kawai C. Identification and distribution of atrial natriuretic polypeptide in ventricular myocardium of humans with myocardial infarction. J Pathol. 1990;161:285–292. doi: 10.1002/path.1711610404. [DOI] [PubMed] [Google Scholar]
- 20.Takemura G, Fujiwara H, Mukoyama M, Saito Y, Nakao K, Kawamura A, Ishida M, Kida M, Uegaito T, Tanaka M, Matsumori A, Fujiwara T, Imura H, Kawai C. Expression and distribution of atrial natriuretic peptide in human hypertrophic ventricle of hypertensive hearts and hearts with hypertrophic cardiomyopathy. Circulation. 1991;83:181–190. doi: 10.1161/01.cir.83.1.181. [DOI] [PubMed] [Google Scholar]
- 21.Di Nardo P, Minieri M, Carbone A, Maggiano N, Micheletti R, Peruzzi G, Tallarida G. Myocardial expression of atrial natriuretic factor gene in early stages of hamster cardiomyopathy. Mol Cell Biochem. 1993;125:179–192. doi: 10.1007/BF00936447. [DOI] [PubMed] [Google Scholar]
- 22.Vikstrom KL, Bohlmeyer T, Factor SM, Leinwand LA. Hypertrophy, pathology, and molecular markers of cardiac pathogenesis. Circ Res. 1998;82:773–778. doi: 10.1161/01.res.82.7.773. [DOI] [PubMed] [Google Scholar]
- 23.Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, Taegtmeyer H. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–364. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- 24.Wohlschlaeger J, Sixt SU, Stoeppler T, Schmitz KJ, Levkau B, Tsagakis K, Vahlhaus C, Schmid C, Peters J, Schmid KW, Milting H, Baba HA. Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J Heart Lung Transplant. 2010;29:125–132. doi: 10.1016/j.healun.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 25.Hardziyenka M, Campian ME, Reesink HJ, Surie S, Bouma BJ, Groenink M, Klemens CA, Beekman L, Remme CA, Bresser P, Tan HL. Right ventricular failure following chronic pressure overload is associated with reduction in left ventricular mass evidence for atrophic remodeling. J Am Coll Cardiol. 2011;57:921–928. doi: 10.1016/j.jacc.2010.08.648. [DOI] [PubMed] [Google Scholar]
- 26.Gruber C, Nink N, Nikam S, Magdowski G, Kripp G, Voswinckel R, Mühlfeld C. Myocardial remodelling in left ventricular atrophy induced by caloric restriction. J Anat. 2012;220:179–185. doi: 10.1111/j.1469-7580.2011.01453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pansani MC, Azevedo PS, Rafacho BP, Minicucci MF, Chiuso-Minicucci F, Zorzella-Pezavento SG, Marchini JS, Padovan GJ, Fernandes AA, Matsubara BB, Matsubara LS, Zornoff LA, Paiva SA. Atrophic cardiac remodeling induced by taurine deficiency in Wistar rats. PLoS One. 2012;7:e41439. doi: 10.1371/journal.pone.0041439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE, Rodriguez JE, Glass DJ, Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaglia T, Milan G, Franzoso M, Bertaggia E, Pianca N, Piasentini E, Voltarelli VA, Chiavegato D, Brum PC, Glass DJ, Schiaffino S, Sandri M, Mongillo M. Cardiac sympathetic neurons provide trophic signal to the heart via β2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc Res. 2013;97:240–250. doi: 10.1093/cvr/cvs320. [DOI] [PubMed] [Google Scholar]
- 30.Novaes RD, Penitente AR, Gonçalves RV, Talvani A, Peluzio MC, Neves CA, Natali AJ, Maldonado IR. Trypanosoma cruzi infection induces morphological reorganization of the myocardium parenchyma and stroma, and modifies the mechanical properties of atrial and ventricular cardiomyocytes in rats. Cardiovasc Pathol. 2013 doi: 10.1016/j.carpath.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Hunter RB, Mitchell-Felton H, Essig DA, Kandarian SC. Expression of endoplasmic reticulum stress proteins during skeletal muscle disuse atrophy. Am J Physiol Cell Physiol. 2001;281:C1285–1290. doi: 10.1152/ajpcell.2001.281.4.C1285. [DOI] [PubMed] [Google Scholar]
- 32.Kim HJ, Jamart C, Deldicque L, An GL, Lee YH, Kim CK, Raymackers JM, Francaux M. Endoplasmic reticulum stress markers and ubiquitin-proteasome pathway activity in response to a 200-km run. Med Sci Sports Exerc. 2011;43:18–25. doi: 10.1249/MSS.0b013e3181e4c5d1. [DOI] [PubMed] [Google Scholar]
- 33.Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC, Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. J Physiol. 2011;589:2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122:339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 36.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. J Cardiovasc Transl Res. 2010;3:355–364. doi: 10.1007/s12265-010-9200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schips TG, Wietelmann A, Höhn K, Schimanski S, Walther P, Braun T, Wirth T, Maier HJ. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res. 2011;91:587–597. doi: 10.1093/cvr/cvr144. [DOI] [PubMed] [Google Scholar]
- 38.Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricle in experimental hypertension. Circ Res. 1990;67:1355–1364. doi: 10.1161/01.res.67.6.1355. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart. Role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell SE, Diaz-Arias AA, Weber KT. Fibrosis of the human heart and systemic organs in adrenal adenoma. Blood Press. 1992;1:149–156. doi: 10.3109/08037059209077510. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Song Y, Elsherif L, Song Z, Zhou G, Prabhu SD, Saari JT, Cai L. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation. 2006;113:544–554. doi: 10.1161/CIRCULATIONAHA.105.537894. [DOI] [PubMed] [Google Scholar]
- 42.Gandhi MS, Deshmukh PA, Kamalov G, Zhao T, Zhao W, Whaley JT, Tichy JR, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT. Causes and consequences of zinc dyshomeostasis in rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2008;52:245–252. doi: 10.1097/FJC.0b013e3181833eb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamalov G, Ahokas RA, Zhao W, Zhao T, Shahbaz AU, Johnson PL, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol. 2010;55:248–254. doi: 10.1097/FJC.0b013e3181cf0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas M, Vidal A, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT. Zinc dyshomeostasis in rats with aldosteronism. Response to spironolactone. Am J Physiol Heart Circ Physiol. 2007;293:H2361–H2366. doi: 10.1152/ajpheart.00200.2007. [DOI] [PubMed] [Google Scholar]
- 45.Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation. 2005;111:871–878. doi: 10.1161/01.CIR.0000155621.10213.06. [DOI] [PubMed] [Google Scholar]
- 46.Kamalov G, Deshmukh PA, Baburyan NY, Gandhi MS, Johnson PL, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Coupled calcium and zinc dyshomeostasis and oxidative stress in cardiac myocytes and mitochondria of rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2009;53:414–423. doi: 10.1097/FJC.0b013e3181a15e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 48.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 49.Weber KT, Janicki JS. Instantaneous force-velocity-length relations in isolated dog heart. Am J Physiol. 1977;232:H241–H249. doi: 10.1152/ajpheart.1977.232.3.H241. [DOI] [PubMed] [Google Scholar]
- 50.Campbell SE, Korecky B, Rakusan K. Remodeling of myocyte dimensions in hypertrophic and atrophic rat hearts. Circ Res. 1991;68:984–996. doi: 10.1161/01.res.68.4.984. [DOI] [PubMed] [Google Scholar]
- 51.Jalil JE, Janicki JS, Pick R, Abrahams C, Weber KT. Fibrosis-induced reduction of endomyocardium in the rat after isoproterenol treatment. Circ Res. 1989;65:258–264. doi: 10.1161/01.res.65.2.258. [DOI] [PubMed] [Google Scholar]
- 52.Savinova OV, Gerdes AM. Myocyte changes in heart failure. Heart Fail Clin. 2012;8:1–6. doi: 10.1016/j.hfc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol. 2004;287:H2023–H2026. doi: 10.1152/ajpheart.00477.2004. [DOI] [PubMed] [Google Scholar]
- 54.Shahbaz AU, Kamalov G, Zhao W, Zhao T, Johnson PL, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Weber KT. Mitochondria-targeted cardioprotection in aldosteronism. J Cardiovasc Pharmacol. 2011;57:37–43. doi: 10.1097/FJC.0b013e3181fe1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheema Y, Sherrod JN, Shahbaz AU, Zhao W, Zhao T, Ahokas RA, Sun Y, Bhattacharya SK, Gerling IC, Weber KT. Mitochondriocentric pathway to cardiomyocyte necrosis in aldosteronism: cardioprotective responses to carvedilol and nebivolol. J Cardiovasc Pharmacol. 2011;58:80–86. doi: 10.1097/FJC.0b013e31821cd83c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khan MU, Cheema Y, Shahbaz AU, Ahokas RA, Sun Y, Gerling IC, Bhattacharya SK, Weber KT. Mitochondria play a central role in nonischemic cardiomyocyte necrosis: common to acute and chronic stressor states. Pflügers Arch. 2012;464:123–131. doi: 10.1007/s00424-012-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerling IC, Sun Y, Ahokas RA, Wodi LA, Bhattacharya SK, Warrington KJ, Postlethwaite AE, Weber KT. Aldosteronism: an immunostimulatory state precedes the proinflammatory/fibrogenic cardiac phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H813–H821. doi: 10.1152/ajpheart.00113.2003. [DOI] [PubMed] [Google Scholar]
- 58.Ahokas RA, Warrington KJ, Gerling IC, Sun Y, Wodi LA, Herring PA, Lu L, Bhattacharya SK, Postlethwaite AE, Weber KT. Aldosteronism and peripheral blood mononuclear cell activation. A neuroendocrine-immune interface. Circ Res. 2003;93:e124–e135. doi: 10.1161/01.RES.0000102404.81461.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Y, Zhang J, Zhang JQ, Ramires FJA. Local angiotensin II and transforming growth factor-β1 in renal fibrosis of rats. Hypertension. 2000;35:1078–1084. doi: 10.1161/01.hyp.35.5.1078. [DOI] [PubMed] [Google Scholar]
- 60.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6:848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 63.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun Y, Weber KT. Angiotensin II receptor binding following myocardial infarction in the rat. Cardiovasc Res. 1994;28:1623–1628. doi: 10.1093/cvr/28.11.1623. [DOI] [PubMed] [Google Scholar]
- 65.Katwa LC, Campbell SE, Tyagi SC, Lee SJ, Cicila GT, Weber KT. Cultured myofibroblasts generate angiotensin peptides de novo. J Mol Cell Cardiol. 1997;29:1375–1386. doi: 10.1006/jmcc.1997.0376. [DOI] [PubMed] [Google Scholar]
- 66.Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB, Taegtmeyer H. Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation. 2003;108:2536–2541. doi: 10.1161/01.CIR.0000096481.45105.13. [DOI] [PubMed] [Google Scholar]
- 67.Ahokas RA, Sun Y, Bhattacharya SK, Gerling IC, Weber KT. Aldosteronism and a proinflammatory vascular phenotype. Role of Mg2+, Ca2+ and H2O2 in peripheral blood mononuclear cells. Circulation. 2005;111:51–57. doi: 10.1161/01.CIR.0000151516.84238.37. [DOI] [PubMed] [Google Scholar]
- 68.Gladden JD, Zelickson BR, Wei CC, Ulasova E, Zheng J, Ahmed MI, Chen Y, Bamman M, Ballinger S, Darley-Usmar V, Dell'Italia LJ. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic Biol Med. 2011;51:1975–1984. doi: 10.1016/j.freeradbiomed.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berry JM, Le V, Rotter D, Battiprolu PK, Grinsfelder B, Tannous P, Burchfield JS, Czubryt M, Backs J, Olson EN, Rothermel BA, Hill JA. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ Res. 2011;109:407–417. doi: 10.1161/CIRCRESAHA.110.228452. [DOI] [PMC free article] [PubMed] [Google Scholar]