

Abstract
There has been extensive research on the use of nanovectors for cancer therapy. Targeted delivery of nanotherapeutics necessitates two important characteristics; the ability to accumulate at the disease locus after overcoming sequential biological barriers and the ability to carry a substantial therapeutic payload. Successful combination of the above two features is challenging, especially in solid porous materials where chemical conjugation of targeting entities on the particle surface will generally prevent successful loading of the therapeutic substance. In this study, we propose a novel strategy for decorating the surface of mesoporous silicon particles with targeting entities (bacteriophage) and gold nanoparticles (AuNP) while maintaining their payload carrying potential. The resulting Bacteriophage Associated Silicon Particles (BASP) demonstrates efficient encapsulation of macromolecules and therapeutic nanoparticles into the porous structures. In vitro targeting data show enhanced targeting efficiency with about four orders of magnitude lower concentration of bacteriophage. In vivo targeting data suggest that BASP maintain their integrity following intravenous administration in mice and display up to three fold higher accumulation in the tumor.
1. INTRODUCTION
Nanovectors are being extensively studied to improve outcome of cancer therapy through the targeted delivery of therapeutics to the disease site.1 To achieve this goal, a nanovector should have the following two characteristics: (1) the ability to accumulate at the disease locus after overcoming sequential biological barriers; and (2) the ability to carry a substantial therapeutic payload. The main challenge, however, lies in the successful combination of the above two features. It was previously shown that site specific accumulation is governed by the nanovector design, namely size,2 shape,3–5 surface charge6–8 and deformability,9 and is affected by surface decoration of the nanovectors with targeting moieties towards elements in the disease microenvironment.10, 11
The main difficulty in enhancing localization of the therapeutics in the disease loci is the presence of successive and heterogeneous biological barriers, which cannot be overcome by a single agent or particle. Thus, there is an emerging requirement for a multimodular nanoassembly, in which different components with individual and distinct functions act in a synergistic manner to circumvent these barriers. To address this necessity, multistage drug delivery vectors (MSV) were developed.12 MSV comprise several nanocomponents, or stages. In this system, Stage 1 Mesoporous silicon Particles (S1MP) are rationally designed with a non-spherical geometry to enable superior vascular margination and cell surface interactions.13, 14 Mesoporous silicon is a promising material for biomedical applications due to its biocompatibility15–17 and ability to degrade under physiological conditions.15, 18 Electrochemically etched mesoporous silicon offers a tunable structure for drug loading and high surface area for conjugation of multivalent targeting ligands.19 S1MP is fabricated by photolithographic patterning enabling the tailoring of various geometries of particles20 which affects their biodistribution and tumor accumulation.14, 21 Following arrival at the disease site, S1MP can deliver payload-carrying second stage nanoparticles (S2NP) encapsulated in their pores. Various S2NP have been encapsulated into the S1MP including quantum dots (QD),12 liposomes,22 gadolinium nanotubes,23 super-paramagnetic iron oxide nanoparticles (SPION)24 and hollow gold nanospheres (HAuNS).25 The major task of S1MP is to protect and simultaneously transport S2NP from the administration site to the disease lesion. S1MP can be chemically modified with negatively/positively charged groups, polyethylene glycol (PEG) and other polymers,15 fluorescent probes,16 contrast agents, and biologically active tumor-microenvironment targeting moieties including antibodies,26 peptides27 and thioaptamers.28
Studies involving active targeting of the S1MP to elements in the tumor microenvironment have shown efficient accumulation in tumor sites. For example, E-Selectin targeted thioaptamer (ESTA) conjugation to the S1MP surface resulted in up to 20% of injected dose (ID) accumulation in the bone marrow vasculature.28 In another study using a melanoma mouse model, arginine-glycine-aspartic acid (RGD) peptide conjugation to the S1MP surface enabled localization of up to 10% of ID in the tumor vasculature.27 While efficient therapeutic/contrast agent loading or targeting have been shown using S1MP, effective coupling of both drug loading and targeting encounters several challenges. Specifically, loading of S1MP with S2NP before the chemical conjugation of targeting ligands renders the surface of the S1MP unsuitable for targeting moiety conjugation. On the contrary, prior conjugation of the targeting moieties to the S1MP surface was shown to significantly reduce the surface area of pores available for loading, thus lowering the encapsulation efficiency of the S1MP by 75%, as in the case of ESTA conjugation.28 In addition, there are several technical challenges associated with the covalent chemical conjugation of peptides, antibodies and thioaptamers on the S1MP surface to confer specificity, including the complexity of their synthesis, and the use of organic solvents during the conjugation process. These could potentially result in the loss of receptor-ligand binding capability29 and may damage delicate S2NP.
Genetically engineered bacteriophage expressing specific peptides on their pIII minor capsid proteins are complex units with good targeting capabilities,30 considerable stability and long shelf-life, and can be synthesized with high-yield, reproducibly and at low-cost. Bacteriophage, well characterized for mapping antigen-antibody epitopes and protein binding sites using phage display technology31 has been recently studied for its ability to deliver cargo to tumors.29 Souza et al. demonstrated that gold nanoparticles (AuNP) and bacteriophage organize spontaneously into dense networks with enhanced absorption in the near-infrared spectrum that can be used as signal reporters for dark-field microscopy and Surface Enhanced Raman Scattering (NIR-SERS) spectroscopy.30 The cell-targeting attributes of the bacteriophage and biocompatibility of the AuNP are preserved in these networks.
In the present study, we propose to utilize electrostatic interactions for the attachment of targeting moieties to S1MP surface while still preserving their cargo loading and carrying potentials. As a targeting moiety we are using the networks of bacteriophage and gold nanoparticles (AuNP). Electrostatic interactions between S1MP, bacteriophage and AuNP result in the formation of Bacteriophage Associated Silicon Particles (BASP, Figure 1). We demonstrate that the encapsulation of S2NP such as SPION into the porous structures of the BASP as well as the degradation of the S1MP is not compromised by the attachment of AuNP-bacteriophage networks on the S1MP surface. In vitro targeting studies using BASP demonstrate the ability to use four orders of magnitude lower concentration of bacteriophage for efficient targeting. In vivo data suggests that BASP maintain their integrity following intravenous administration to mice as well as demonstrate three fold higher accumulation of targeted BASP in orthotopic mammary tumors as compared to non-targeted S1MP.
Figure 1. Schematic presentation of BASP.

showing the interaction of the three components: S1MP (grey), AuNP (yellow) and targeting peptide (red) displaying filamentous bacteriophage (orange). The inset shows the pores of S1MP loaded with S2NP (blue).
2. EXPERIMENTAL SECTION
2.1. Particle synthesis and fabrication
S1MP were fabricated by integration of photolithography and electrochemical etching of p-type silicon wafers in the Microelectronics Research Centre at The University of Texas at Austin, as previously described.12, 21 Briefly, the dimensions of S1MP were defined by photolithography while the porous structure was formed by electrochemical etching of silicon in a mixture of hydrofluoric acid (HF) and ethanol. Particles with different geometries and porosities were evaluated in this study including quasi-hemispherical and discoidal particles with mean pore sizes of 20–30nm or 50–60nm. Photolithography followed by the electrochemical etching was used to make quasi-hemispherical S1MP.20 For discoidal S1MP, the electrochemical etching was performed before photolithography.32 The current density was varied to obtain different pore sizes. The unmodified S1MP were retained on the substrate for washing steps, and then released by sonication in isopropyl alcohol (IPA). The particles were dried, and oxidized in a piranha solution (3:1 solution of sulfuric acid and 30% hydrogen peroxide) at 100–110ºC for 2h to yield S1MP with negative charge [S1MP(−)]. To produce S1MP with positive zeta potential [S1MP(+)], S1MP(−) were reacted with 2% 3-aminopropyltriethoxysilane (APTES, Sigma-Aldrich Co. LLC, USA) for 2h at 35°C. The modified particles were re-suspended and stored in isopropyl alcohol. Immediately before use, the particles were washed thrice and re-suspended in distilled water. The concentration (counts/mL) of the S1MP was obtained using a MultisizerTM 4 Coulter Counter (Beckman Coulter, USA). The zeta potential was measured with ZetaPALS (Brookhaven Instruments Corporation, USA). For identification during confocal microscopy studies, S1MP(+) were loaded with fluorescein isothiocyanate conjugated bovine serum albumin (BSA-FITC, Sigma-Aldrich Co. LLC, USA). For this purpose 107 S1MP(+) were desiccated to remove any residual IPA. To the dry S1MP(+) 200μL of 5mg/mL aqueous solution of BSA-FITC was added and the system was incubated for 2h at 4ºC under rotation. The loaded particles were washed thrice in distilled water and used immediately.
Gold nanoparticles (AuNP) were prepared using the common Turkevich method33 with a ratio of 1:0.8 of Gold(III) chloride and trisodium citrate (Sigma-Aldrich Co. LLC, USA) to yield ~45nm particles. The resulting AuNP colloid solution was concentrated by sequential centrifugation steps at 2600xg in Sorval Legend X1R Centrifuge (USA) for 10, 15 and 20min at 4°C to achieve an absorbance of approximately 5A.U. at 530nm. The hydrodynamic radius and the zeta potential measurements of the AuNP were performed using ZetaPALS.
Bacteriophage particles were obtained as previously described 34. Briefly, filamentous bacteriophage, CDCRGDCFC (i.e. RGD-4C) peptide displaying (Integrin targeted) and YRCTLNSPFFWEDMTHECHA peptide-displaying (Integrin mimicking), were grown by infection of K91 Escherichia coli (E.coli) bacteria overnight at 37°C and were subsequently isolated by PEG precipitation.29, 35. The final pellet was resuspended in phosphate-buffered saline (PBS). Bacteriophage titers or transducing units (TU) were determined by infecting E.coli and screening for tetracycline resistant colonies. Fluorescently labeled bacteriophage was used for identification by confocal microscopy when needed. The bacteriophage particles were conjugated with a 1000 fold excess of Dylight 594 (Thermo Fisher Scientific Inc., USA) for 1.5h and precipitated using PEG.
2.2. Assembly of Bacteriophage Associated Silicon Particles (BASP)
AuNP-bacteriophage networks were obtained based on the previously published method30 with a bacteriophage input of 2.5×107 TU/μL. Two-fold serial dilutions of bacteriophage were done in distilled water and equal volumes of AuNP were added to each dilution and left undisturbed overnight at room temperature. The formation of AuNP-bacteriophage networks was noted by a change in colour of the solution from red to purple.30 AuNP-bacteriophage networks were concentrated and incubated with S1MP for 1h with intermittent vortexing to form BASP.
To optimize the design of BASP, we evaluated the formation of systems containing various concentrations of the three constituents, various geometries, porosities and surface chemistries of S1MP.
2.3. Characterization of BASP
The morphology of the BASP was examined using various microscopic techniques. Fluorescently labeled BASP containing BSA-FITC loaded S1MP(+) and Dylight 594 conjugated bacteriophage were visualized under a Leica DM6000 upright confocal microscope (Leica Microsystems, Germany) equipped with a 63X oil immersion objective. For transmission electron microscopy (TEM), samples were placed on 100 mesh coated copper grids treated with poly-l-lysine for 1h. Excess samples were blotted with filter paper and the BASP were stained with filtered aqueous 1% ammonium molybdate + 0.02% BSA, pH 7.0 for 1min. The samples were blotted, allowed to dry and imaged using the AMT Imaging System (Advanced Microscopy Techniques Corp., USA) JEM 1010 TEM (JEOL USA, Inc.) at 80kV fitted with an AMT Advantage digital CCD camera system. Scanning electron microscopy (SEM) images were acquired using FEI Quanta 400 FEG ESEM (FEITM, USA) equipped with an ETD (SE) detector. Samples were prepared by drying BASP solutions on SEM stub overnight in a desiccator and sputter-coated with gold or platinum (5nm).
2.4. Evaluation of contact angle and spectral characteristics of the systems
The hydrophilicity of the systems was determined by measuring the contact angles made by the sample solution resting on a flat horizontal solid surface using captive bubble contact angle goniometry.
Light spectroscopy was used to study the absorption characteristics of the BASP during their formation. Shifts in the absorbance spectra of AuNP towards the NIR wavelengths were evaluated using SpectraMax M2 spectrophotometer (Molecular Devices, USA) or Biotek Synergy H4 hybrid multimode microplate reader (BioTek® Instruments, Inc., USA).
The enhanced absorbance in the NIR region prompted the use of a NIR laser to test the heating potential of the BASP. 200μL of the sample solution was exposed to Delta 810nm diode laser (AngioDynamics UK Ltd.) with an output of 10J/cm2 for 250msec at an interval of 100msec for a total of 10min using a 2mm fixed focus spot hand-piece. The resulting change in temperature, and for 10min after the cessation of laser irradiation, was measured using an OxyLiteTM probe (Oxford optotronix Ltd., USA). For SERS, BASP were excited by a fiber-optic probe delivering 785nm laser light for 10s. The resulting Raman scattering data (average of 3 results) were gathered on a PerkinElmer Raman Station 400F (PerkinElmer Inc., USA) after background subtraction with water. The spectra were analyzed with Spectrum software (PerkinElmer Inc., USA).
2.5. Degradation of BASP
BASP or S1MP (5×107, 1.6μm quasi-hemispherical) were suspended in 1mL of 100% fetal bovine serum (FBS, Gibco, USA) and incubated at 37°C with constant mixing in a rotary shaker for the predetermined times. Aliquots (100μL) were taken at specified time intervals namely 0, 0.5, 1, 4, 8, 24 and 48h. For quantitative analysis of elemental silicon in the degradation medium, 90μL of the aliquots were filtered through a microcentrifuge filter to separate the non-degraded S1MP from the degradation products, diluted with water and analyzed using Varian 720-ES Inductively Coupled Plasma – Optical Emission Spectrometer (ICP-OES, Varian Inc., USA) set at 1kW, with plasma flow set to 15L/min, auxiliary flow of 1.5L/min and a nebulizer flow of 0.75L/min. Silicon contents in the degradation medium was detected at 250.69nm, 251.43nm, 251.61nm and 288.158nm using 1ppm of Yttrium as an internal control. The remaining 10μL were washed thrice with deionized water to remove salts and proteins and processed for SEM analysis as described above.
2.6. Loading of S2NP
S1MP(+) (1.5×108, 1×0.4μm discoidal, 20–30nm pore size) and E-Selectin thioaptamer conjugated S1MP (ESTA-S1MP) were dried in a desiccator. For ESTA-S1MP, the S1MP(+) were conjugated with a 107 fold excess of thioaptamers using PolyLink Protein Coupling kit for 1h as described earlier.28 Amine modified SPION (15nm) suspended in PEG was centrifuged at 2000rpm for 5min at room temperature to remove aggregated particles. The supernatant was used as the stock for loading experiments. SPION solution (25μL) was added to the S1MP and incubated for 30min with intermittent sonication. The SPION loaded S1MP were washed twice with water and then incubated with 15μL AuNP-bacteriophage networks for 1h to allow the assembly of BASP. The BASP were washed an additional time to remove any unbound SPION. NMR relaxation profiles of the SPION loaded particles were measured using a Bruker Minispec mq-60 bench-top relaxometer (Bruker Optics Inc., USA) operating at 60MHz and 37°C. The solutions were then digested for evaluation of elemental iron by heating them with nitric acid at 120°C thrice until the acid evaporated. The sediments were then resuspended in water. The iron content was detected at 238.204nm and 259.940nm using ICP-OES as previously described.
S1MP(+) (1.5×108, 1×0.4μm discoidal, 20–30nm pore size) were loaded with 20μl of 2μM carboxylated quantum dots (QD) in a process similar to the SPION loading. Following BASP formation, the particles were visualized by fluorescence microscopy to see if the QD are confined to the S1MP in the BASP or are released and entrapped in the AuNP-bacteriophage networks during BASP formation. The samples were also digested using nitric acid and analysed for Cadmium content using ICP-OES.
2.7. In vitro targeting
Kaposi sarcoma cells were purchased from American Tissue Culture Collection (ATCC), grown and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (GIBCO, Life Technologies™, USA) supplemented with 10% fetal bovine serum (FBS) (GIBCO, Life Technologies™, USA) and 1% penicillin (100units/mL) and streptomycin (100μg/mL). The cells were seeded at 80% confluency in 16 chamber glass slides and allowed to grow overnight at 37°C. Cells were blocked with 30% FBS for 1h and then incubated with integrin targeted bacteriophage or BASP (containing 1.6μm quasi-hemispherical S1MP) at concentrations of 104 TU/μL for 6h. The cells were subsequently washed and fixed with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences, USA), permeabilized with 0.2% triton X-100 (Sigma-Aldrich Cp. LLC, USA) and blocked with 1% BSA (Sigma-Aldrich Cp. LLC, USA) for 1h. The cells were then incubated with anti-phage antibody (Sigma-Aldrich Cp. LLC, USA) followed by Dylight 594 conjugated goat anti-rabbit secondary antibody (Thermo Fisher Scientific Inc., USA). The nuclei were counterstained with DAPI (Sigma-Aldrich Cp. LLC, USA), fixed and imaged using a Nikon upright fluorescence microscope.
2.8. In vivo integrity and targeting of BASP
Breast cancer xenografts were grown in immunocompetent Balb/c mice by injecting 5×104 EF43.fgf4 cells orthotopically in the mammary fat pad. After 10–14 days, the mice were administered intravenously with 5×108 S1MP or integrin mimicking BASP (1.6μm quasi-hemispherical). The bacteriophage used in these experiments were either labeled with Dylight 594 or unlabeled. Four hours following the administration of the systems, the animals were sacrificed and the major organs including liver, lung, kidney and tumor were collected for histological analysis (conventional and hyperspectral microscopy) and silicon content analysis. Paraffin embedded tissue sections were stained for bacteriophage by incubating with anti-bacteriophage antibody (Sigma Aldrich Co. LLC., USA) at 1:750 dilution for 30min. Frozen tissue sections from mice injected with Dylight 594 labeled bacteriophage containing BASP were counterstained for nucleus with Draq5 (Biostatus Limited, UK). Non-stained paraffin embedded tissue sections were fixed with 4% paraformaldehyde, mounted with Prolong gold mounting media (Invitrogen, USA) and imaged using a darkfield condenser from CytoViva attached to an Olympus BX41 microscope and DAGE camera/software (CytoViva, Auburn, AL 36830) hyper-spectral imaging system that can identify material at resolutions of less than 100nm based on the material’s unique spectral signatures. The biodistribution of the injected particles was determined by analysing the silicon content in the tissues by ICP-OES. The tissues were homogenized in 0.5N sodium hydroxide solution with 20% ethanol and centrifuged after 48h. The clear supernatant was then diluted in distilled water and analysed as previously described.
3. RESULTS AND DISCUSSION
In previously reported works on silicon and silica particles, the addition of molecular targeting moieties to the drug carriers was performed through a covalent surface conjugation of antibodies, peptides or other entities.15, 16, 21, 27, 28, 36 Such chemical reactions may not only impair the targeting ability of the biological entities29 but in porous particles it may also drastically reduce the volume of pores available for loading of active substances in molecular or nanoparticle-based form.28 Here, we report for the first time the possibility of immobilizing molecularly targeted filamentous bacteriophage on porous silicon drug carriers without losing the drug carrying potential of the porous silicon-based S1MP. We hypothesized that a simple co-incubation of the three components namely S1MP, bacteriophage and AuNP could yield closely associated BASP. We have shown that AuNP-bacteriophage networks spontaneously assemble onto S1MP by electrostatic physical adsorption to form BASP. This enabled us to load the S1MP with S2NP and instantaneously cover with the AuNP-bacteriophage networks, thus avoiding loss of the loaded S2NP as schematically represented in Figure 1.
3.1. Assembly of BASP
BASP are comprised of three components: S1MP, AuNP and bacteriophage. Optimization processes in the assembly of BASP (using 1μm silica beads) involved the incubation of various concentrations of the components in a systematic manner. It was seen that low concentrations of AuNP (OD < 4) and high concentrations of silica beads (>106/μL) prevented the formation of BASP (Supplemental figure 1). Also, it was found that formation of BASP was most efficient when assembled in a two-step process involving: (1) the formation of dense AuNP-bacteriophage networks; (2) the coating of S1MP by these networks. As shown in the Supplemental figure 2, when the bacteriophage were added to the silica particles and AuNp, no adsorption of the phage on the particle surface is seen, pointing towards inefficient formation of the BASP. A possible explanation for that is in the fact that the AuNP-bacteriophage networks are required to efficiently attach to the surface of the silica/silicon particles. The density of the AuNP-bacteriophage networds can be indirectly monitored through the shift in the spectral properties of the system, since the gold particles that are packed closer (denser) in the network casue a stronger quantum effect. The addition of AuNP to bacteriophage causes instantaneous agglomeration of AuNP noted by the color change from red to purple 30 and the condensation of the system into fibrous structures as seen in Figure 2A, similar to what was previously reported by Souza et al.30 Addition of these condensed AuNP-bacteriophage networks to S1MP resulted in spontaneous association between the components forming BASP. To verify the formation of BASP we used confocal microscopy and TEM. Fluorescently labeled bacteriophage used in the assembly of BASP clearly showed co-localization of the bacteriophage (red) and the BSA-FITC loaded S1MP (green) (Figure 2B). Further, TEM micrographs showed the close association of the fibrous bacteriophage (white lines) and the AuNP (black dots) to the S1MP (a large porous structure) (Figure 2C). Physical adsorption of bacteriophage on various surfaces for use as antibacterial agents/surfaces has been studied previously.37 While most of these reports relate to chemical or genetic modifications of the bacteriophage, the technique described here is based on an efficient and spontaneous assembly through the electrostatic interactions between the various components of BASP.
Figure 2. Visualization of the BASP formation.

(A) Macroscopic view on network/gel formation corresponding to the association of AuNP-bacteriophage networks with S1MP to form BASP at serial dilutions of the bacteriophage starting from the concentration of 3.87×105 TU/μL; (B) Confocal microscopic image of BASP with BSA-FITC loaded S1MP(+) (Green) and Dylight 594 labeled phage (Red) showing the specific co-localization of the two; (C) TEM image of BASP showing the S1MP surrounded by AuNP (arrow heads) and bacteriophage (arrows).
Among the variables to design BASP with desirable spectral characteristics and targeting abilities, we evaluated geometry, porosity and zeta potential of S1MP. Recent research has recognized the importance of physical characteristics such as geometry of drug carriers in their interaction with biological systems, both at the cellular and vascular level.38–40 At the cellular level, phagocytosis of drug carriers showed dependence on particle geometry. While the effect of the size of particles on cellular uptake was not immediately perceived, the shape of the particles affected their frequency of uptake. It was observed that non-spherical (ellipsoidal) particles had a differential uptake in macrophages, depending on which surface of the particle came in contact with the cell first when compared to a spherical particle that had uniform uptake due to its symmetry.41 Also, it has been mathematically and experimentally derived that size, shape and density of particles determine their vascular interaction potential.39, 42 Discoidal particles show higher adhesion to endothelial cells under shear flow compared to spherical particles. The interaction of particles with endothelial cells in a vessel is of utmost importance for efficiently delivering the cargo at the intended site. Thus, here we compared non-porous spherical silica beads to quasi-hemispherical and discoidal mesoporous silicon particles with different pore sizes in their ability to form BASP. Although the shape of the S1MP used did not have much effect on the self-assembly of BASP (Supplemental figure 3), the association of the AuNP to porous particles was evidently better than their association to non-porous silica beads (Figure 3A). Increased surface area in porous materials provides more sites for interactions of biological entities with the particles. Our observation that porosity of the S1MP enhances the adhesion of the AuNP-bacteriophage networks is in agreement with previous studies on the effect of particle porosity on the efficiency of surface adsorption.43
Figure 3. Effect of porosity and surface zeta potential of S1MP on BASP formation.

(A) SEM micrographs demonstrating the importance of porosity in the association of the AuNP-bacteriophage networks with S1MP having (i) 20–30nm pores, (ii) 40–60nm pores, (iii) nonporous surface; (B) Zeta potential of BASP and their components (inset) SEM image of BASP with S1MP(-) and S1MP(+) showing better adhesion of AuNP-bacteriophage networks to the S1MP(+) in comparison to S1MP(-); (C) Contact angle measurements of BASP showing an increase in hydrophilicity in comparison to AuNP or AuNP-bacteriophage.
The surface potential of drug delivery vehicles is another important feature to be considered in their design. The effect of zeta potential of S1MP on the formation of BASP was evaluated by comparing BASP containing 1.6μm quasi-hemispherical S1MP(+) and S1MP(−). The initial and final zeta potential values of the systems are presented in Figure 3B. For S1MP(+) and S1MP(−), the initial zeta potential values were 8.9 ± 1.9mV and −25.7 ± 1.3mV respectively. The AuNP-bacteriophage networks have a net negative charge (Figure 1). After the formation of BASP, both systems yielded similar negative zeta potentials meaning that the significant shift in zeta potential values occurred only during the formation of BASP containing S1MP(+). Morphological examination of the resulting systems by SEM confirmed this behavior. In agreement with the zeta potential data, the SEM micrographs show more efficient association of the AuNP-bacteriophage networks with the S1MP(+) particles (Figure 3B inset). Positively charged particles have the greatest efficiency in cell-membrane penetration and internalization, however, they also interact more prominently with components of the immune system, such as macrophages and are prompt to produce non-specific interactions with various cells 44. Particles with negative zeta potential, on the other hand, have a lesser propensity to be sequestered by the immune system. In our studies, we saw that positively charged S1MP formed stronger associations with negatively charged AuNP-bacteriophage networks to form BASP with a final negative zeta potential. Hydrophilicity measurements on the BASP showed that both types of BASP, containing either S1MP(+) or S1MP(−) (Figure 3C) are more hydrophilic than AuNP solution alone, which can be important for avoiding non-specific uptake by cells of the reticuloendothelial system, as shown for nanovectors modified on their surface with hydrophilic polymers, such as PEG.45 We also observed that there is a reduction in the uptake of the BASP by macrophages when compared to S1MP (Supplemental figure 4).
Spectral characterization of BASP
As mentioned above, the formation of the AuNP-bacteriophage networks can be monitored in real-time using UV-Vis spectroscopy. Instantaneous interaction between the components of AuNP-bacteriophage networks in successive serial dilutions of the bacteriophage solution (5×106 TU/μL to 5×105 TU/μL) is indicated by the broadening and reduction of UV-Vis absorbance at 530nm and simultaneous increase in the absorbance at NIR wavelengths between 650nm and 800nm.30 This observed 3–4 fold increase in NIR absorbance occurs within 20sec of the addition of AuNP to bacteriophage. This change in absorbance from the visible to NIR wavelengths has been previously shown and can be explained by the dipole-dipole interactions between the AuNP when they come within distances lesser than their diameters.46 However, this red shift disappears with further changes in the bacteriophage concentration. At higher concentrations of bacteriophage, the sites available for the AuNP to bind are plenty, leading to the placement of the AuNP at distances large enough to avoid the dipole-dipole interactions. In contrast, below 5×105 TU/μL the concentration of the bacteriophage particles is so dilute that the majority of the AuNP exists in colloidal form and hence do not interact with each other. Thus, it is important to achieve a delicate balance in the relative concentrations of the phage and AuNP during formation of the networks. The AuNP-bacteriophage with enhanced NIR absorbance are then pooled together and incubated with S1MP to form BASP. The increase of the absorbance in the NIR region was maintained following the formation of BASP (Figure 4A).
Figure 4. Spectral characterization of BASP.
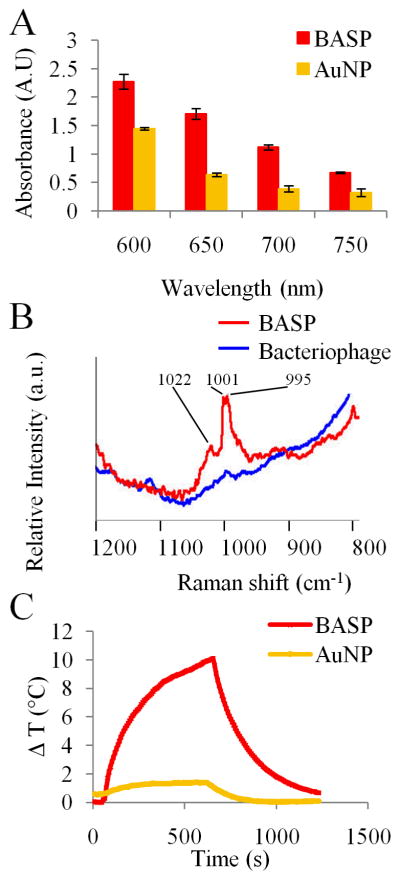
(A) UV-Vis spectra of BASP show 2–3 fold increase in the NIR absorbance of BASP in comparison with the same concentration of AuNP; (B) Detection of BASP using NIR-SERS signal of bacteriophage in close association with AuNP and S1MP; (C) Temperature as a function of NIR laser exposure time of AuNP and BASP.
The high absorbance in the NIR region enables the use of spectral properties of the systems for diagnostic and therapeutic purposes. As an example, the use of SERS as a diagnostic tool was investigated and the NIR-SERS spectra of the BASP were recorded. As shown above, AuNP are closely associated with bacteriophage and S1MP in BASP. The close association of AuNP to bacteriophage facilitated an increase (~2000 fold) in the signature Raman peaks of the bacteriophage major coat proteins (such as phenylalanine and alanine). The data presented in Figure 4B illustrates the enhancement of unique peaks, namely wavenumbers 1022cm−1, 1001cm−1 and 995cm−1,47–49 in the NIR-SERS spectra of bacteriophage associated with the BASP. The observed peaks are “fingerprints” of BASP, since they appear only when bacteriophage and S1MP are in close association with AuNP. Thus, they can be used as a more significant reporter of the formation and presence of BASP than UV-Vis spectroscopy. NIR-SERS is an optical imaging technique that allows noninvasive longitudinal analysis based on SERS reporters to provide real-time, in vivo access to critical information at the molecular scale.
Deep tissue imaging exploits the NIR window (650–900nm), where hemoglobin is transparent,50 to discern function, localization, affinity and fate of the nanovectors through their innate scattering properties.51 These photonic properties of BASP originating from the quantum confinement of the AuNP in the BASP are tunable based on the design of the system and offer a direct ability to assess their interaction within biological systems, providing diagnostic capability.
We also investigated the NIR photothermal conversion property of the BASP in deionized water. When irradiated by a NIR laser the temperature in BASP increased by 10°C from room temperature within 10min while the AuNP did not heat up significantly (Figure 4C). The samples were irradiated with the NIR laser for 10min, after which the laser source was turned off and the sample was left to recover at room temperature. The AuNP did not undergo any visible agglomerations after the irradiation. The proximity of the AuNP seen in BASP caused a more efficient absorption of NIR photons from lasers resulting in temperature increase. This heat generation triggered by NIR laser irradiation could potentially be used for thermal ablation.
3.2. Biodegradation of BASP
Degradation of the drug delivery system in biological fluids is an important characteristic that determines drug release kinetics as well as the biocompatibility of the vector. We evaluated the degradation kinetics of BASP in simulated in vivo conditions such as 100% FBS at 37°C under constant motion. SEM micrographs (Figure 5A) and ICP-OES analysis (Figure 5B) show that the presence of AuNP-bacteriophage networks on the S1MP in BASP does not affect the degradation kinetics of the S1MP. The quantification of the percentage silicon release by ICP-OES showed complete degradation of both S1MP and BASP within 24h, consistent with previously published work.15 Although the degradation profile of the BASP was not different from naked S1MP, the adhesion of serum proteins on particles was significantly decreased in the presence of the AuNP-bacteriophage networks as seen by the open pores in BASP in contrast to the covered appearance of the naked S1MP (Figure 5A). This could potentially translate into the reduction of opsonization and hence non-specific uptake of particles by cells under in vivo conditions (data not shown).
Figure 5. Time dependent degradation of S1MP and BASP in 100% FBS.

(A) SEM micrographs showing the pattern of degradation of the porous silicon particles over time (Scale bar = 500nm); (B) ICP-OES quantification of the degradation profile expressed as a percentage of total silicon content released from S1MP over time.
3.3. Loading efficiency in BASP
Payload carrying capacity is another important consideration when designing nanovectors. In multi-stage vectors,12, 52 S1MP chemically modified on their surface have a significantly lower loading capacity of S2NP than unmodified S1MP, due to the unavailability of sufficiently open pores.28 Here we evaluated the encapsulation efficiency of BASP by incorporating either amine functionalized SPION (15nm) or carboxylated QD (15nm). The amount of SPION loading was determined by measuring the iron content in the pellet. Post-assembly of AuNP-bacteriophage networks on the S1MP to form BASP retains the loaded SPION (1.17±0.34ng of iron/μg of silicon, i.e. ~100%) when compared to naked S1MP (Figure 6A). In contrast, ESTA-S1MP showed 70% lower loading (0.33ng/μg of silicon). This demonstrates the ability of BASP to maintain the loading efficiency of S1MP while coupling targeted bacteriophage to their surface. This simple technique of coupling the targeting moiety to the vector enables both targeting as well as efficient payload delivery. NMR relaxometry data also corresponded to the loading efficiency, with SPION loaded S1MP exhibiting the shortest transverse relaxation time as shown in previous work.24 Correspondingly, the r2 relaxivity (normalized to iron oxide concentration) of the SPION loaded BASP (162.09mM−1s−1) was half that of the SPION loaded S1MP (258.75mM−1s−1) while that of the SPION loaded ESTA-S1MP was significantly lower (98.63mM−1s−1) (Figure 6B). Conversely, the T2 relaxation times were short for the SPION loaded S1MP and BASP (Figure 6C). The relaxation time of BASP (68.31msec) was almost twice as long as that of the S1MP (39msec) but about 5 times shorter than that of ESTA-S1MP (354.4msec). These data show that BASP loaded with SPION maintain their ability of exhibiting transverse relaxation and hence may be used for magnetic imaging applications.
Figure 6. Loading of S2NP into BASP.

(A) Iron content analysis in S1MP, BASP and ESTA-S1MP following loading of SPION using the wet incipient method as quantified by ICP-OES; (B) Comparison of transverse relaxivity, r2 and (C) relaxation times, T2, of SPION loaded particles; (D) Fluorescence microscopic images of QD (green) loaded S1MP and BASP (red) showing S1MP colocalization with QD (yellow). In a control experiment, free QD nonspecifically bound to AuNP-bacteriophage networks (arrow heads) enable the visualization of the bacteriophage fibers (green) while the AuNP appear as red dots.
Next we considered if loaded S2NP were closely associated with S1MP pores or were released from S1MP during BASP formation. QD loading into S1MP and BASP was comparable with 4.5±0.45ng of cadmium loaded in 1.5×108 silicon particles in both naked S1MP and BASP suggesting that QD were not lost during BASP formation. To determine if the QD escaped out of the S1MP and were trapped in the AuNP-bacteriophage networks, we visualized the systems using fluorescence microscopy. We would expect unassociated QD (Figure 6D, green) to bind non-specifically to the AuNP-bacteriophage. However, we observed specific localization of the QD (yellow due to co-localization) with S1MP (Figure 6D, red) and an absence of any non-specific staining outside the S1MP. AuNP-bacteriophage networks incubated with QD served as controls. We observed non-specific labeling of bacteriophage fibers (green) interspersed with AuNP (red dots).
3.4. In vivo integrity of BASP
Since the BASP is not a chemically conjugated assembly and is based on electrostatic interactions, which can be impaired in complex biological fluids (e.g. blood), it was important to verify the integrity of the systems following in vivo intravenous administration. We administered integrin mimicking BASP through the tail vein to orthotopic breast tumor bearing mice and analyzed tumor and liver tissue sections for the integrity of the systems in vivo. We used bacteriophage displaying a peptide sequence (YRCTLNSPFFWEDMTHECHA)53 targeted to CRKL to enhance accumulation of BASP in the tumor vessels. CRKL, an intracellular adaptor protein present in many cell types, is over-expressed and secreted into the tumor microenvironment, where it then interacts with the extracellular domain of β1-integrin leading to further downstream signaling events.53, 54 Tumor tissue from mice injected with BASP containing fluorescently labeled bacteriophage show co-localization of the bacteriophage (red) and S1MP (Figure 7A). Similarly, immunohistochemistry staining for bacteriophage (Figure 7B, brown) in tumor tissue shows specific association with S1MP. Thus, we can infer that there is a close association of the bacteriophage and the S1MP after intravenous administration. Hyperspectral imaging provides unique reflectance spectral analysis of materials. Combined with dark field microscopy (Figure 7C(i)), hyperspectral imaging was used to identify the location of BASP. Once the particles were identified, spectra from different pixels on the particles were collected and grouped into libraries (S1MP and AuNP) (Figure 7C(ii)) according to the shape of the spectra. Mapping these libraries to the image of liver tissues shows co-localization of the silicon spectra and AuNP spectra (Figure 7C(iii)). The spectra collected for the AuNP show a peak at approximately 650nm, which correlates with the red shift in the UV-Vis absorbance of BASP. These spectra based mapping confirm proximity of AuNP and S1MP and thus, the integrity of BASP.
Figure 7. In vivo integrity of BASP.

Mice bearing EF43.fgf4 mammary xenografts were injected intravenously with BASP. After a 4h circulation time, the animals were sacrificed and the tumor and liver were harvested and processed for histological analysis. (A) Fluorescence microscopy and (B) Immunohistochemistry analyses for bacteriophage (brown) show that the (i) bacteriophage remains associated with S1MP and the assembly is not impaired in the circulation (ii) Negative staining control; (C) Hyperspectral images of the liver show superimposed spectral mapping of silicon and AuNP also confirming the close association of the AuNP and S1MP in the BASP.
3.5. In vitro targeting
We hypothesized that the presence of S1MP in BASP would enhance the targeting efficiency of the bacteriophage. As a proof of concept, we tested targeting efficiency of integrin targeted BASP in Kaposi sarcoma cells. The BASP showed enhanced internalization of the targeted bacteriophage in Kaposi sarcoma cells in comparison to cells incubated with the bacteriophage alone at concentrations of 104 TU/μL. Although RGD-4C is a good targeting peptide, the amount of bacteriophage needed to exhibit internalization is very high; in the order of 108TU/μL (data not shown). One of the major disadvantages faced in bacteriophage based nanosystem targeting is the concentrations needed. Here it was observed that when incorporated into BASP, the concentration bacteriophage required for targeting in vitro is reduced by about 4 orders of magnitude (Figure 8). This could serve as an innovative way of overcoming the scalability of bacteriophage for in vivo applications.
Figure 8. In vitro targeting in Kaposi sarcoma cells.
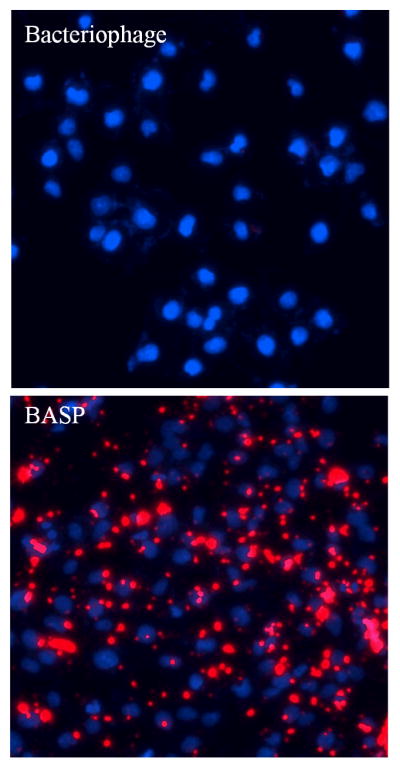
Kaposi sarcoma cells incubated with integrin targeted systems (104 TU/μL bacteriophage concentration) for 6h. Enhanced association of bacteriophage (red) with cells incubated with integrin targeted BASP can be seen when compared to bacteriophage only. Nuclei stained with DAPI (blue).
3.6. In vivo targeting
To evaluate the biodistribution of BASP comprised of quasihemispherical S1MP, integrin mimicking bacteriophage and AuNP, the systems were injected via the tail vein into mice bearing orthotopic EF43.fgf4 tumors. Figure 9 shows the biodistribution of S1MP and integrin mimicking BASP in the major organs (liver, lung, kidney) represented by the silicon content in each organ. The integrin mimicking BASP showed upto a 3 fold higher accumulation in the tumor tissue when compared to S1MP. This needs to be further evaluated and validated with other S1MP geometries.
Figure 9. Biodistribution of targeted BASP.

Silicon content in different organs as measured by ICP-OES. Balb/C mice bearing orthotopic EF43.fgf4 tumors were injected intravenously with either S1MP or integrin mimicking BASP. After a 4h circulation time, the animals were sacrificed and the organs were harvested and processed for silicon content analysis.
4. CONCLUSIONS
In this study we report a strategy for non-covalent attachment of targeting entities to porous silicon particle surfaces, which enables independent control over the loading of actives into the porous structure of the particles and their targeting to elements in the tumor microenvironment. The attachment of AuNP-bacteriophage networks on the S1MP surface does not impair the loading capacity of naked S1MP. The resulting BASP are biodegradable in the serum and maintain their integrity while injected intravenously. This shows that the electrostatic interactions of the components in the system are stable under constant motion as well as in vivo. Further, in vitro and in vivo studies show efficient targeting of BASP compared to bacteriophage alone and S1MP respectively. Collectively, based on the above observations, it is expected that the system described here might enhance the delivery of S1MP with significant therapeutic/imaging payload to the tumor microenvironment. Based on the targeting moieties, BASP can also be evaluated for use in inflammatory conditions and cardiovascular diseases.
Supplementary Material
Acknowledgments
The authors acknowledge a financial support from the following sources: NIH U54CA143837 (CTO, PSOC), NIH 1U54CA151668-01 (TCCN, CCNE), DODW81XWH-09-1-0212, DODW81XWH-07-2-0101 and ANH NASA/DOE Graduate Student Research Training Fellowship. We thank Kenneth Dunner, Jr. for assistance with transmission electron microscopy, Mathew Landry for designing the graphical presentations, Dr. Jeyarama S. Ananta Subramanian for help with NMR measurements and Dr. David Gorenstein for kindly providing the ESTA.
Notes and references
- 1.Moghimi SM, Peer D, Langer R. ACS Nano. 2011;5:8454–8458. doi: 10.1021/nn2038252. [DOI] [PubMed] [Google Scholar]
- 2.Gaumet M, Vargas A, Gurny R, Delie F. Eur J Pharm Biopharm. 2008;69:1–9. doi: 10.1016/j.ejpb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari M. Nat Nano. 2008;3:131–132. doi: 10.1038/nnano.2008.46. [DOI] [PubMed] [Google Scholar]
- 4.Kersey FR, Merkel TJ, Perry JL, Napier ME, DeSimone JM. Langmuir. 2012;28:8773–8781. doi: 10.1021/la301279v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoo JW, Chambers E, Mitragotri S. Curr Pharm Des. 2010;16:2298–2307. doi: 10.2174/138161210791920496. [DOI] [PubMed] [Google Scholar]
- 6.Lind K, Kresse M, Muller RH. Electrophoresis. 2001;22:3514–3521. doi: 10.1002/1522-2683(200109)22:16<3514::AID-ELPS3514>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Garbuzenko O, Zalipsky S, Qazen M, Barenholz Y. Langmuir. 2005;21:2560–2568. doi: 10.1021/la0479105. [DOI] [PubMed] [Google Scholar]
- 8.Christian DA, Garbuzenko OB, Minko T, Discher DE. Macromol Rapid Commun. 2010;31:135–141. doi: 10.1002/marc.200900589. [DOI] [PubMed] [Google Scholar]
- 9.Merkel TJ, Jones SW, Herlihy KP, Kersey FR, Shields AR, Napier M, Luft JC, Wu H, Zamboni WC, Wang AZ, Bear JE, DeSimone JM. Proc Natl Acad Sci U S A. 2011;108:586–591. doi: 10.1073/pnas.1010013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaspar R. Nat Nano. 2013;8:79–80. doi: 10.1038/nnano.2013.11. [DOI] [PubMed] [Google Scholar]
- 11.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 12.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F, Ferrari M. Nat Nanotechnol. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 13.Gentile F, Chiappini C, Fine D, Bhavane RC, Peluccio MS, Cheng MM, Liu X, Ferrari M, Decuzzi P. J Biomech. 2008;41:2312–2318. doi: 10.1016/j.jbiomech.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. J Control Release. 2010;141:320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Godin B, Gu J, Serda RE, Bhavane R, Tasciotti E, Chiappini C, Liu X, Tanaka T, Decuzzi P, Ferrari M. J Biomed Mater Res A. 2010;94:1236–1243. doi: 10.1002/jbm.a.32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tasciotti E, Godin B, Martinez JO, Chiappini C, Bhavane R, Liu X, Ferrari M. Mol Imaging. 2011;10:56–68. [PMC free article] [PubMed] [Google Scholar]
- 17.Bimbo LM, Sarparanta M, Santos HA, Airaksinen AJ, Mäkilä E, Laaksonen T, Peltonen L, Lehto VP, Hirvonen J, Salonen J. ACS Nano. 2010;4:3023–3032. doi: 10.1021/nn901657w. [DOI] [PubMed] [Google Scholar]
- 18.Low SP, Voelcker NH, Canham LT, Williams KA. Biomaterials. 2009;30:2873–2880. doi: 10.1016/j.biomaterials.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Nat Mater. 2009;8:331–336. doi: 10.1038/nmat2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiappini C, Tasciotti E, Fakhoury JR, Fine D, Pullan L, Wang YC, Fu L, Liu X, Ferrari M. Chemphyschem. 2010;11:1029–1035. doi: 10.1002/cphc.200900914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Advanced Functional Materials. 2012;22:4225–4235. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, et al. Cancer Res. 2010;70:3687–3696. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ananta JS, Godin B, Sethi R, Moriggi L, Liu X, Serda RE, Krishnamurthy R, Muthupillai R, Bolskar RD, Helm L, et al. Nat Nanotechnol. 2010;5:815–821. doi: 10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serda RE, Mack A, Pulikkathara M, Zaske AM, Chiappini C, Fakhoury JR, Webb D, Godin B, Conyers JL, Liu XW, Bankson JA, Ferrari M. Small. 2010;6:1329–1340. doi: 10.1002/smll.201000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He L, Malcolm AS, Dimitrijev M, Onaizi SA, Shen HH, Holt SA, Dexter AF, Thomas RK, Middelberg AP. Langmuir. 2009;25:4021–4026. doi: 10.1021/la802825c. [DOI] [PubMed] [Google Scholar]
- 26.Yokoi K, Godin B, Oborn CJ, Alexander JF, Liu X, Fidler IJ, Ferrari M. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Ven AL, Kim P, Haley O, Fakhoury JR, Adriani G, Schmulen J, Moloney P, Hussain F, Ferrari M, Liu X, Yun SH, Decuzzi P. J Control Release. 2012;158:148–155. doi: 10.1016/j.jconrel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mann AP, Tanaka T, Somasunderam A, Liu X, Gorenstein DG, Ferrari M. Advanced Materials. 2011;23:H278–H282. doi: 10.1002/adma.201101541. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Zurita AJ, Ardelt PU, Giordano RJ, Arap W, Pasqualini R. Chem Biol. 2004;11:1081–1091. doi: 10.1016/j.chembiol.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 30.Souza GR, Christianson DR, Staquicini FI, Ozawa MG, Snyder EY, Sidman RL, Miller JH, Arap W, Pasqualini R. Proc Natl Acad Sci U S A. 2006;103:1215–1220. doi: 10.1073/pnas.0509739103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoogenboom HR. Methods in molecular biology. Vol. 178. Clifton, N.J: 2002. pp. 1–37. [DOI] [PubMed] [Google Scholar]
- 32.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Adv Funct Mater. 2012;22:4225–4235. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Handley DA. In: Colloidal Gold: Principles, Methods and Applications. Hayat MA, editor. Vol. 1. Academic Press; 1989. pp. 23–27. [Google Scholar]
- 34.Christianson DR, Ozawa MG, Pasqualini R, Arap W. Methods in molecular biology (Clifton, NJ) 2007;357:385–406. doi: 10.1385/1-59745-214-9:385. [DOI] [PubMed] [Google Scholar]
- 35.Barbas CF, III, Burton DR, Scott JK, Silverman GJ. 2001 [Google Scholar]
- 36.Arap W, Pasqualini R, Montalti M, Petrizza L, Prodi L, Rampazzo E, Zaccheroni N, Marchio S. Curr Med Chem. 2013 doi: 10.2174/0929867311320170005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cademartiri R, Anany H, Gross I, Bhayani R, Griffiths M, Brook MA. Biomaterials. 2010;31:1904–1910. doi: 10.1016/j.biomaterials.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 38.Decuzzi P, Pasqualini R, Arap W, Ferrari M. Pharm Res. 2009;26:235–243. doi: 10.1007/s11095-008-9697-x. [DOI] [PubMed] [Google Scholar]
- 39.Lee SY, Ferrari M, Decuzzi P. J Biomech. 2009;42:1885–1890. doi: 10.1016/j.jbiomech.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 40.Ferrari M. Nat Nanotechnol. 2008;3:131–132. doi: 10.1038/nnano.2008.46. [DOI] [PubMed] [Google Scholar]
- 41.Champion JA, Katare YK, Mitragotri S. J Control Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adriani G, de Tullio MD, Ferrari M, Hussain F, Pascazio G, Liu X, Decuzzi P. Biomaterials. 2012;33:5504–5513. doi: 10.1016/j.biomaterials.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zong J, Zhu Y, Yang X, Li C. Materials Science and Engineering: C. In Press, Corrected Proof. [Google Scholar]
- 44.Verma A, Stellacci F. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 45.Harris JM, Martin NE, Modi M. Clin Pharmacokinet. 2001;40:539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 46.Souza GR, Levin CS, Hajitou A, Pasqualini R, Arap W, Miller JH. Anal Chem. 2006;78:6232–6237. doi: 10.1021/ac060483a. [DOI] [PubMed] [Google Scholar]
- 47.Garfinkel D, Edsall JT. Journal of the American Chemical Society. 1958;80:3823–3826. [Google Scholar]
- 48.Williams RW, Dunker AK, Peticolas WL. Biochim Biophys Acta. 1984;791:131–144. doi: 10.1016/0167-4838(84)90003-7. [DOI] [PubMed] [Google Scholar]
- 49.Aubrey KL, Thomas GJ., Jr Biophys J. 1991;60:1337–1349. doi: 10.1016/S0006-3495(91)82171-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weissleder R. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 51.Gobin AM, Lee MH, Halas NJ, James WD, Drezek RA, West JL. Nano Lett. 2007;7:1929–1934. doi: 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- 52.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Acc Chem Res. 2011 doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mintz PJ, Cardo-Vila M, Ozawa MG, Hajitou A, Rangel R, Guzman-Rojas L, Christianson DR, Arap MA, Giordano RJ, Souza GR, Easley J, Salameh A, Oliviero S, Brentani RR, Koivunen E, Arap W, Pasqualini R. Proc Natl Acad Sci U S A. 2009;106:2182–2187. doi: 10.1073/pnas.0807543105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ozawa MG, Cardo-Vila M, Mintz PJ, Arap W, Pasqualini R. Cell Cycle. 2010;9:8–9. doi: 10.4161/cc.9.1.10288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.