

Abstract
Proteomics strategies have been widely used in the field of male reproduction, both in basic and clinical research. Bioinformatics methods are indispensable in proteomics-based studies and are used for data presentation, database construction and functional annotation. In the present review, we focus on the functional annotation of gene lists obtained through qualitative or quantitative methods, summarizing the common and male reproduction specialized proteomics databases. We introduce several integrated tools used to find the hidden biological significance from the data obtained. We further describe in detail the information on male reproduction derived from Gene Ontology analyses, pathway analyses and biomedical analyses. We provide an overview of bioinformatics annotations in spermatogenesis, from gene function to biological function and from biological function to clinical application. On the basis of recently published proteomics studies and associated data, we show that bioinformatics methods help us to discover drug targets for sperm motility and to scan for cancer-testis genes. In addition, we summarize the online resources relevant to male reproduction research for the exploration of the regulation of spermatogenesis.
Keywords: bioinformatics, database, male reproduction, proteomics, spermatogenesis
Introduction
Spermatogenesis is a complex process involving successive mitotic, meiotic and post-meiotic phases important for male reproduction.1 Approximately 60% of infertility is due—either directly or indirectly—to male factors.2 Various methods have been applied to identify biomarkers of male infertility at the level of the genome, transcriptome and proteome.3 Genomics and transcriptomics are mainly focused on the regulation of gene expression, whereas proteomics provides accurate information about protein composition, quantification and post-translational modifications on a larger scale and with high sensitivity and specificity. Thus, mass spectrometry-based proteomics has become a useful tool for the study of cellular biology and discovery of clinical biomarkers.4,5
In the field of male reproduction, proteomics has been serving as a tool for biological research of spermatogenesis and the clinical research of male infertility.6,7 There are generally two aspects of the proteomics analysis: protein identification and protein quantification. Protein identification concerns the presence of proteins in a given type of cell or tissue. Because of the discrepancy between the expression of mRNA and corresponding proteins in mammalian cells, which was identified in previous research,8 it is important to establish specific qualitative protein profiles for the biological study of spermatogenesis. The human sperm and testis proteomes have been established using gel-based proteomics methods.9,10 Guo et al.10 also identified 52 phosphorylated proteins heterogeneously expressed in the human testis. Quantitative proteomics is widely used for relative quantification of proteins in samples of different physiological or pathological states and yields lists of differentially expressed genes to elucidate biological processes. In the field of reproductive medicine, Zhao et al.11 identified proteins involved in the regulation of sperm motility by comparing protein expression profiles in asthenozoospermic patients with normozoospermic donors. In addition, Liao et al.12 identified differentially expressed proteins between normal and round-headed spermatozoa.
Bioinformatics tools were developed during the work on Human Genome Project,13 where they were successfully used in the database construction and gene annotation of the human genome. Now, bioinformatics is an indispensable toolkit in current biological research. Proteomics studies generally generate lists of proteins, and bioinformatics acts as a bridge that connects those raw lists to biological significance. Bioinformatics approaches have proven useful in the discovery and understanding of the reproduction-related biomarkers.14
In the present review, we summarize general strategies of bioinformatics analysis, integrated tools and online resources. We show how the study of male reproduction benefits from the use of bioinformatics. We provide an overview on how to derive biological significance from a raw list of proteins and show how bioinformatics aids in discovering clinical biomarkers. The current limitations and future prospects in this field are also discussed.
General strategy for data mining of proteomics-based studies
The initial aim of a proteomics analysis is to identify or quantify proteins. Technical methods for processing of proteomic data have been previously reviewed.15,16 In this review, we focus on data mining methods for revealing biological or medical significance associated with male reproduction. Bioinformatics methods can be used for data presentation, database construction and functional annotation. Many bioinformatics analyses produce good visualisation graphs for data presentation. For example, VennPlex draws a Venn diagram for the comparison of different gene lists.17 The construction of proteomics databases also helps to share the data for further analysis. Starting with a list of proteins, various tools and databases can be applied for biological and medical data mining. For example, in the bioinformatics analysis of proteins involved in mouse spermatogenesis,18 the KEGG pathway and Gene Ontology analyses were performed using DAVID to obtain a functional overview. For visualisation, the Pathway Studio program was used to draw a complex network of vesicle events and the associated genes. In addition, the ciliary proteome database19 and Mouse Genome Informatics (MGI) database20 that were used to identify the genes that might affect sperm flagella and influence male reproduction also helped to identify candidate biomarkers for male contraception and male infertility.
The meta-analysis of published datasets can also provide new insights. For example, Liu et al.21 provided insights and the foundation for the development of diagnostic markers and therapeutic targets for infertility and male contraception using integrated bioinformatics analysis of mouse testis protein profiles. A combination of multiple ‘omic' datasets may also help to decipher the intercellular molecular networks that drive sperm cell biology.22 The post-genome projects, including FANTOM and ENCODE, have provided resources for the functional and regulatory annotations of the mouse and human genes.23,24 In the Human Metabolome Database, detailed information has been collected about small molecule metabolites from the studies of metabolic processes.25 In addition, the RNA-Seq Atlas provides improved gene expression data using next-generation sequencing technology on multiple human tissues, including the testis.26 Although these data were not directly associated with the study of male reproduction, further integrated analyses can be performed to mine the data for spermatogenesis-related genes. Thus, with the help of bioinformatics, proteomics and proteomics-based integrated ‘omics', there will be more opportunities for the analysis of male reproduction.
Construction of the proteomics databases
There are several common repository databases for the proteomics-based data. The World-2DPAGE Portal is a dynamic portal for simultaneous query of the worldwide gel-based proteomic databases.27 PRIDE28 and Tranche29 are standard compliant databases for the storage and analysis of mass spectrometry (MS)-based proteomic data. Such databases aim to collect and share raw files and present the results in a standard format. Currently, only a few datasets are available from the field of male reproduction that have been deposited in PRIDE or Tranche. Over the past few years, our laboratory has uploaded the data from the mouse proteome studies involving spermatogenesis18 and male meiosis I30 to PRIDE (accession numbers 9754 and 10118, respectively). However, because the raw data from proteomics experiments are usually stored in very large files, it is inefficient to transfer such data to users via a third-party repository centre. In addition, the methods and objectives of each proteomic-based study are different, making it difficult to unify the results. Thus, it is necessary for the data holder to construct relevant databases.
In 2006, Pilch and Mann31 developed a proteome database of human seminal plasma that provided browsing and search functions (http://proteome.biochem.mpg.de/seminal/). In 2008, our laboratory established the REPRODUCTION-2DPAGE (http://reprod.njmu.edu.cn/2d), which provides gel maps associated with reproduction using the Make2D-DB II package.32 It includes maps of human testis proteins, including phosphorylated proteins,33 and the mouse testis proteins at different ages.34 REPRODUCTION-2DPAGE has created cross-references with the Universal Protein Resource, SWISS-2DPAGE and World-2DPAGE. Recently, with the help of high-resolution MS, we identified 4675 sperm proteins and 7346 testis proteins.35,36 As a result, we released two MS-based proteomic databases: the Human Sperm Proteome Database (HSPD at http://reprod.njmu.edu.cn/hspd/) and the Human Testis Proteome Database (HTPD at http://reprod.njmu.edu.cn/htpd/) using locally developed websites. These websites were designed to be user-friendly, where the proteome could be easily downloaded. We provided detailed information for the identified proteins, peptides and corresponding MS/MS spectra. Moreover, for each identified protein, the associated reproductive phenotypes for human and mouse were annotated based on the data from human reproductive diseases37 and the MGI database (Figure 1).
Figure 1.
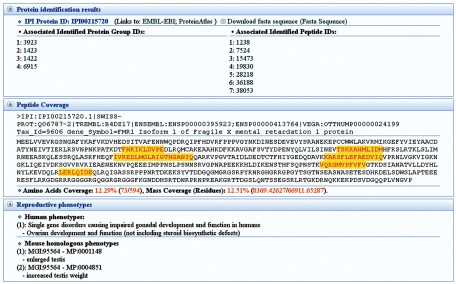
A representative snapshot of the human testis proteome database. The identification results for the protein ‘IPI00215720' in HTPD are shown. For each identified protein, HTPD provides detailed information about the identification results, protein coverage and reproductive associated phenotypes. HTPD, Human Testis Proteome Database.
Gene list-based annotations
Given a list of interesting proteins differentially expressed or qualitatively identified, the main task is to uncover the hidden biological significance. Protein entries are usually converted to gene names for convenient functional annotation. Many integrated bioinformatics tools have been developed that take a gene list as an input and make data interpretation easier and more efficient. A list of the representative tools is shown in Table 1.38,39,40,41,42,43 DAVID (http://david.abcc.ncifcrf.gov/), for example, collects various functional data for its knowledgebase, including the data related to Gene Ontology, protein domains, protein interactions, pathways and diseases. Gene lists can be uploaded and analysed in the sub-class of each field (e.g., by Gene Ontology (GO)-Biological Processes or by the KEGG pathway). For each term, DAVID can perform an enrichment statistical analysis. Identified enriched terms may shed light on the biological significance of an uploaded gene list. Other tools have similar functionality. The new version of Babelomics (version 4, http://babelomics.bioinfo.cipf.es/) now supports quantitative data. In addition to GO, PANTHER (http://www.pantherdb.org/) annotates protein classes and pathways through the evolutionary modelling. WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt/), ToppFun (http://toppgene.cchmc.org/) and Ingenuity Pathway Analysis (IPA at www.ingenuity.com) can be helpful for the discovery of biomarkers and drug targets. In addition, Pathway Studio is used for the navigation and analysis of biological pathways, gene regulation networks and protein interaction maps. It provides text mining-based databases and assists in the construction of complex networks for visualisation. In general, GO annotation and pathway annotation analyses are commonly performed on gene list-based annotations. Biomedical analysis is important for translating such basic research into clinical applications. We will focus on these three methods to show how the analysis of male reproduction benefits from the use of bioinformatics.
Table 1. Representative tools for gene list-based functional annotation.
| Name | Description | Version | License |
|---|---|---|---|
| DAVID | Database for annotation, visualisation and integrated discovery | Web-based | Academic |
| Babelomics | Integrative platform for the analysis of transcriptomics, proteomics and genomic data | Web-based | Academic |
| PANTHER | Protein analysis through evolutionary relationships | Web-based | Academic |
| WebGestalt | Web-based gene set analysis toolkit | Web-based | Academic |
| ToppFun | One-stop portal for gene list enrichment analysis | Web-based | Academic |
| IPA | Ingenuity pathway analysis | Web-based | Commercial |
| Pathway studio | Pathway analysis software | Desktop | Commercial |
The column ‘Version' indicates whether the tool is online or desktop-based. The column ‘License' indicates whether the tool is free to academia or requires payment for usage.
GO analysis: from gene localisation to gene function
Gene Ontology (GO)44 classifies gene product functions through the use of structured and controlled vocabularies. It is available for a broad range of species. The ontology consists of three parts: the cellular component, biological process and molecular function. ‘Cellular component' refers to the parts of the cell or its extracellular environment, underscoring the importance of the localisation of a gene product as it reflects its function. A sperm cell is highly differentiated and is different from germ cells as well as normal body cells. Therefore, one should be cautious when analysing the cellular components of the sperm in GO, even though GO contains terms such as ‘sperm flagellum' and ‘acrosomal vesicle' that are highly specific for the sperm. The term ‘Biological process' represents a collection of molecular events with defined beginning and end. GO uses terms related to each step of male reproduction: ‘male sex differentiation', ‘male meiosis', ‘spermatogenesis', ‘sperm axoneme assembly', ‘sperm motility', ‘sperm capacitation', ‘sperm ejaculation', ‘sperm competition', ‘sperm–egg recognition' and sperm–oocyte fusion'. ‘Molecular function' represents the fundamental molecular activities of a gene product and provides basic information on the regulation of spermatogenesis at the molecular level.
Previous research has found that metabolic regulation and mitochondria are important for spermatogenesis.45 Mitochondria of the sperm are different from somatic cells in their morphology and biochemistry.46 Thus, mitochondria and their involvement in energy metabolism play key role in sperm motility. Among the GO terms, the energy metabolism-associated activities such as ‘coenzyme binding' and ‘NAD' or ‘NADH binding' are important for sperm motility. We recently performed a GO analysis using the latest human sperm proteome.36 As shown in Table 2, about 715 gene products were located in the mitochondrion. Biological processes associated with the energy metabolism, such as glycolysis and the tricarboxylic acid cycle, were also enriched. Consistent with the biological processes, enriched molecular functions included catalytic activity and NAD or NADH binding. These results provide an overview of the energy metabolism processes related to sperm motility.
Table 2. Representative enriched GO terms for the human sperm proteome.
| Class | Accession | Description | Count | Enrichment | FDR |
|---|---|---|---|---|---|
| CC | GO:0043226 | Organelle | 4037 | 1.2 | 1.2E-155 |
| CC | GO:0005739 | Mitochondrion | 715 | 1.8 | 4.9E-91 |
| CC | GO:0043234 | Protein complex | 1357 | 1.5 | 1.7E-74 |
| CC | GO:0042470 | Melanosome | 69 | 2.2 | 1.3E-11 |
| CC | GO:0030117 | Membrane coat | 50 | 2.2 | 2.3E-08 |
| CC | GO:0070469 | Respiratory chain | 56 | 2.1 | 7.8E-08 |
| BP | GO:0055114 | Oxidation reduction | 367 | 1.5 | 7.4E-22 |
| BP | GO:0006119 | Oxidative phosphorylation | 71 | 1.9 | 2.0E-08 |
| BP | GO:0006732 | Coenzyme metabolic process | 98 | 1.7 | 1.6E-07 |
| BP | GO:0006007 | Glucose catabolic process | 47 | 2.2 | 2.0E-07 |
| BP | GO:0006096 | Glycolysis | 37 | 2.1 | 1.2E-04 |
| BP | GO:0006099 | Tricarboxylic acid cycle | 22 | 2.5 | 2.4E-04 |
| MF | GO:0003824 | Catalytic activity | 2624 | 1.3 | 7.8E-122 |
| MF | GO:0000166 | Nucleotide binding | 1263 | 1.5 | 2.4E-82 |
| MF | GO:0016491 | Oxidoreductase activity | 392 | 1.5 | 6.4E-23 |
| MF | GO:0048037 | Cofactor binding | 164 | 1.8 | 2.2E-16 |
| MF | GO:0050662 | Coenzyme binding | 127 | 1.9 | 1.5E-15 |
| MF | GO:0051287 | NAD or NADH binding | 36 | 2.0 | 4.7E-04 |
Abbreviations: BP, biological process; CC, cellular component; GO, Gene Ontology; MF, molecular function.
The enrichment analysis was performed with Fisher's exact test using DAVID; the human genome was set as the background. The FDR represents false discovery rate, which was set to FDR<0.05 to control for significant enrichment.
Pathway analysis: from gene function to biological function
A single gene may play many roles in a cell. However, a specific group of genes is involved in a specific biological function. Pathway analysis focuses on the regulated relationship of a group of genes in a defined biological process. This process is different from GO analysis, as GO simply describes how many genes are involved, whereas pathway analysis illustrates their relationships in a complex network. A pathway can be drawn as a map for better visualisation and understanding. Current well-known pathway databases are KEGG,47 REACTOME48 and WikiPathways.49 KEGG, for example, provides a collection of pathways involved in metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, human diseases and drug development. Although there are no spermatogenesis-specific pathways annotated in KEGG or other current pathway databases, pathway analysis can still provide indirect insight for the analysis of male reproduction. For example, pathway analysis output, such as ‘oxidative phosphorylation' or ‘tight junction', could lead to further analysis of the sperm energy metabolism or the blood–testis barrier.
We performed a KEGG pathway analysis using the latest human testis proteome.36 Pathways associated with the energy metabolism, such as ‘glycolysis/gluconeogenesis' and ‘citrate cycle (TCA cycle)', were enriched. As an example, the KEGG map of the glycolysis pathway is shown in Figure 2 with the human testis genes highlighted. The human testis genes were enriched in the ‘coenzyme binding' and ‘catalytic activity' categories according to the GO analysis; hence, their precise functional roles and complex relationships are shown on the map. We also showed that the pathway analysis was able to verify the GO analysis. In addition, the visualisation of the pathway map compensated for the deficiencies in the GO analysis that only provided the classification results.
Figure 2.

The KEGG pathway of glycolysis with human testis genes highlighted. Generated by the KEGG online, this graph shows the genes and their role in glycolysis. The human testis genes are highlighted in red. The genes with purple or green background represent disease genes and drug targets, respectively.
Biomedical analysis: from biological function to medical application
Translating basic research into medical research will prove beneficial in understanding the biological function and development of medical applications. We have previously mentioned that the ‘omics' research can help to discover potential biomarkers for spermatogenesis and male infertility.33,50,51 It is also necessary to search for known male reproduction-associated genes or phenotypes in the obtained list of interesting genes. The Online Mendelian Inheritance in Man database provides detailed information about the human genes and associated phenotypes.52 The mouse is a commonly used model organism for human diseases. The MGI20 database provides a full annotation of the phenotypes and human disease associations for the mouse models (by their genotypes) using the terms from the Mammalian Phenotype Ontology53 and disease names from the Online Mendelian Inheritance in Man52 resources. The MGI database includes phenotypes such as ‘abnormal spermatid morphology', ‘abnormal testis morphology', ‘abnormal spermatogenesis', ‘asthenozoospermia', ‘oligozoospermia', ‘azoospermia', ‘teratozoospermia', ‘testis tumour' and ‘male infertility. A phenotype-based analysis can provide hypotheses for the basic research in spermatogenesis and its clinical applications, such as contraceptive targets or infertility markers. Databases such as DrugBank and the Therapeutic Target Database provide information about the drugs, target genes, and corresponding diseases and pathways, which helps in the search for candidate drugs and target genes for clinical use.54,55
In the latest study of the human sperm proteome performed in our laboratory,35 we explored candidate targets for the development of male contraceptive drugs. First, we annotated sperm proteins using the DrugBank data. Next, using GO and the ciliary proteome database, we found that among the 500 drug-targeted proteins, 154 proteins were located in the mitochondria and 162 proteins were associated with the cilia.19 We successfully verified four drugs that affect sperm motility. Thus, we showed that bioinformatics could be helpful in the mining of proteomic data for biomedical significance.
Another example of combining biological and medical research toward better understanding of male reproduction is the research on CT (cancer-testis) antigens. CT antigens are normally only expressed in the human testis, but can also be expressed in various types of tumours.56 The ‘gametic recapitulation' theory explains the common features of spermatogenesis and tumourigenesis57 and states that the activation of normally silent germline-specific genes in tumour cells is the cause of tumourigenesis. The CT genes research could, therefore, stimulate the research in spermatogenesis and tumourigenesis. There are currently 251 known CT genes according to the CT database.58 The CT genes, such as melanoma antigen and New York oesophageal squamous cell carcinoma 1, are expressed in spermatogonia and associated with meiosis. Figure 3 shows a representative network of the CT genes involved in spermatogenesis and tumourigenesis. For male reproduction, CT genes are not only evolved in mitosis, meiosis and spermatogenesis but also in sperm motility, fertilisation and even embryonic development. However, many CT genes have only been studied with respect to their expression and not their functionality. Thus, CT genes could be a resource for further analysis in the regulation of spermatogenesis. The immunogenic proteins encoded by CT genes are also actively pursued as potential cancer diagnostic, prognostic and therapeutic biomarkers. For example, PRAME can be used as a diagnostic or prognostic biomarker for breast cancer and serous ovarian adenocarcinomas.59,60 In addition, the melanoma antigen A3 vaccine is under phase III trials for the immunotherapy of non-small-cell lung cancer and melanoma.61
Figure 3.

A representative network of the CT genes in spermatogenesis and tumourigenesis. Drawn by Pathway Studio, this graph shows the relationship of 11 representative biological terms (including four disease terms and seven cell processes) and 64 CT genes. Red, yellow and purple labels represent the CT genes, cell processes and disease terms, respectively. Grey, blue and green lines represent the relationships with the regulation, expression and promoter binding, respectively.
In terms of the transcripts, massively parallel signature sequencing has been used to identify candidate CT genes.62 Recently, our laboratory performed a proteomic-based study to scan candidate CT genes.36 By combining the information on the proteins predominantly expressed in the testis and the genome-wide association study data from several cancers, we identified six novel cancer/testis genes. These genes could be candidates for cancer diagnostic biomarkers, as well as a valuable resource for the analysis of spermatogenesis.
Prediction analysis: digging into the dark matter
The general annotation strategy depends on the previously defined information. For example, GO annotates genes based on four levels of evidence: experimental evidence, evidence through computational analysis, author statement evidence and curator statement evidence. Proteins not annotated by GO or other databases are functionally ‘dark matter'. Prediction methods help to sift through this dark matter. The Centre for Biological Sequence Analysis (CBS at http://www.cbs.dtu.dk/services/) provides various sequence-based prediction tools involving subcellular location, post-translational modifications, protein function and protein structure. These sequence-based prediction tools are useful for annotating the results of new genomic and proteomic analyses. Blast2GO is another sequence-based functional annotation tool.63 Blast2GO interprets uncharacterized proteins by GO, KEGG or other annotation databases using BLAST to find homologous sequences. The homology annotation strategy could be helpful in the analysis of male reproduction in humans. Genes involved in male fertility have been identified and studied using mouse models. However, the corresponding research in humans is limited. Thus, by homology annotation of human sperm genes using the reproductive phenotype data of MGI we can find candidate drug targets for sperm motility.35
Prediction tools can be used for indirect data mining in spermatogenesis. For example, because the microRNA biogenesis is required for germ cell development and spermatogenesis,64 databases such as TargetScan65 can be used for the prediction of microRNA targets. Another tool, microDoR,66 predicts miRNA-mediated gene silencing based on the miRNA-target duplex features. During spermatogenesis, protein synthesis is temporally controlled, whereas during the sperm deformation the degradation of a large fraction of proteins is performed by the ubiquitination evolved for this purpose.67,68 Tools that can be used to study protein degradation include UbPred, a predictor of protein ubiquitination sites69 and SProtP that predicts the half-life of a protein.70
Online resources on male reproduction in mammals
The online resources on male reproduction help us to explore the regulation of spermatogenesis with gene lists. Table 3 summarizes specialized websites that contain resources on mammalian male reproduction (excluding the proteome databases). These websites cover a broad range of reproductive information, including gene function, gene expression and gene regulation. Mammalian Reproductive Genetics provides information on the genes and literature associated with the reproduction (currently focused on mouse and rat). SpermatogenesisOnline collects and annotates the genes involved in spermatogenesis for multiple species. ReCGiP provides the reproduction candidate genes in pig and is based on the bibliomics.71 Germ SAGE collects male germ cell transcriptomes from the Serial Analysis of Gene Expression for the mouse type A spermatogonia, pachytene spermatocytes and round spermatids.72 K-SPMM provides detailed information on the promoter regions in Sertoli cells, spermatogonia, spermatocytes and spermatids.73 GermOnline is an integrated online tool based on the microarray expression databases74 that focuses on germline development, meiosis, gametogenesis and the mitotic cell cycle. It combines information from the high-throughput expression data, sample annotations, protein-DNA binding data, protein–protein interaction data, genome annotations and orthologs.
Table 3. Representative online resources on male reproduction in mammals.
| Name | Description | Website |
|---|---|---|
| Mammalian Reproductive Genetics | Information regarding genes and corresponding literature related to mammalian reproduction | http://mrg.genetics.washington.edu/ |
| SpermatogenesisOnline | Annotation of genes or proteins involved in spermatogenesis | http://mcg.ustc.edu.cn/sdap1/spermgenes/ |
| ReCGiP | Database of reproduction candidate genes in pig based on the bibliomics | http://klab.sjtu.edu.cn/ReCGiP/ |
| GermSAGE | A collection of the male germ cell transcriptome information derived from the Serial Analysis of Gene Expression | http://germsage.nichd.nih.gov/germsage/ |
| K-SPMM | A database of murine Spermatogenic Promoters Modules and Motifs | http://compbio.med.wayne.edu/software/kspmm/ |
| GermOnline | A cross-species microarray expression database that focuses on the germline development, meiosis and gametogenesis as well as the mitotic cell cycle | http://www.germonline.org/ |
Except for these male reproduction-associated databases, other common online resources could also be used for exploring the expression patterns of genes to provide additional clues for the biology of spermatogenesis. On the protein level, the Universal Protein Resource database75 provides access to many other proteomic databases. The Human Protein Atlas76 database provides the antibody-based protein expression and localisation profiles for 48 normal human tissues and 20 different cancers. Each annotated protein is presented with high-resolution immunohistochemistry and immunofluorescence images. STRING is another web tool for visualizing the known and predicted protein–protein interactions.77 On the transcript level, the Genomics Institute of the Novartis Research Foundation expression data stored in BioGPS,78 which provides an overview of the gene expression in multiple tissues and cells. The Tissue-Specific Genes Database79 provides comprehensive information on the tissue-specific genes derived from various gene expression profiles from human and mouse tissues. Tissue-Specific Genes Database defines testis-specific genes, including the genes in testis germ cells, testis interstitium, testis Leydig cells and the seminiferous tubules.
Summary and outlook
Proteomic strategies have been widely used in the field of male reproduction for both basic and clinical research. Bioinformatics methods are indispensable for proteomic-based studies because they help us understand the biology of spermatogenesis and aid in the discovery of potential biomarkers for the diagnosis and therapy of male infertility. In this review, we focused on the methods of annotation of gene lists, obtained either qualitatively or quantitatively and summarized general strategies for the bioinformatics analysis, including integrated annotation tools. These methods operate on gene lists and could also be applied to other high-throughput studies, such as transcriptomics. The construction of specialized proteomic databases is helpful for data presentation, sharing, evaluation and further analysis. The integrated analysis tools such as DAVID make it easier to mine for hidden biological significance through various databases. We also provided an overview of bioinformatics annotation techniques applied to spermatogenesis, from gene function to biological function and from biological function to clinical application. Through recently published proteomic data and studies, we showed that bioinformatics methods help to discover drug targets for sperm motility and scan for cancer-testis genes. In addition, the online resources associated with male reproduction help to explore the regulation of spermatogenesis. In the field of male reproduction, bioinformatics provides us with powerful connections between a list of genes and the hidden biological significance. It also provides a bridge between the basic research and clinical applications.
There are limitations in the use of annotations. Because annotations of gene lists are based mainly on known or predicted knowledge databases, undefined genes or biological functions are also not annotated. For example, the core database for the KEGG pathways lacks pathways directly associated with spermatogenesis. As for annotations with DAVID, genes that do not match the DAVID database might be lost. These “unknown” genes can be functionally annotated based on their sequences using the prediction tools such as Blast2GO. We showed that combining proteomic data with genome-wide association study data could help to identify novel cancer-testis genes. As the sequencing technology and other post-genome projects develop, bioinformatics will help us to interpret the integration of large datasets from genomics, transcriptomics, proteomics and metabolomics studies.
Author contributions
XJG conceived of the project. TZ and ZMZ performed analysis. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by grants from the National 973 Program (Nos. 2011CB944304 and 2013CB911400), the National Natural Science Foundation of China (No. 81222006) and the Qing Lan Project.
The authors declare no competing financial interests.
References
- de Kretser DM, Loveland KL, Meinhardt A, Simorangkir D, Wreford N. Spermatogenesis. Hum Reprod. 1998;13 Suppl 1:1–8. doi: 10.1093/humrep/13.suppl_1.1. [DOI] [PubMed] [Google Scholar]
- Esteves SC, Miyaoka R, Agarwal A. An update on the clinical assessment of the infertile male [corrected]. Clinics (Sao Paulo) 2011;66:691–700. doi: 10.1590/S1807-59322011000400026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac JR, Pastuszak AW, Lamb DJ. The use of genomics, proteomics, and metabolomics in identifying biomarkers of male infertility. Fertil Steril. 2013;99:998–1007. doi: 10.1016/j.fertnstert.2013.01.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann DJ, Jr, McGuigan MD, Patel AR, Tomov S, Ross S, et al. Clinical proteomics and biomarker discovery. Ann NY Acad Sci. 2004;1022:295–305. doi: 10.1196/annals.1318.045. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Huang XY, Sha JH. Proteomics of spermatogenesis: from protein lists to understanding the regulation of male fertility and infertility. Asian J Androl. 2011;13:18–23. doi: 10.1038/aja.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MA. The ‘omics' revolution and our understanding of sperm cell biology. Asian J Androl. 2011;13:6–10. doi: 10.1038/aja.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960–9. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Johnston DS, Wooters J, Kopf GS, Qiu Y, Roberts KP. Analysis of the human sperm proteome. Ann NY Acad Sci. 2005;1061:190–202. doi: 10.1196/annals.1336.021. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang P, Huo R, Zhou Z, Sha J. Analysis of the human testis proteome by mass spectrometry and bioinformatics. Proteomics Clin Appl. 2008;2:1651–7. doi: 10.1002/prca.200780120. [DOI] [PubMed] [Google Scholar]
- Zhao C, Huo R, Wang FQ, Lin M, Zhou ZM, et al. Identification of several proteins involved in regulation of sperm motility by proteomic analysis. Fertil Steril. 2007;87:436–8. doi: 10.1016/j.fertnstert.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Liao TT, Xiang Z, Zhu WB, Fan LQ. Proteome analysis of round-headed and normal spermatozoa by 2-D fluorescence difference gel electrophoresis and mass spectrometry. Asian J Androl. 2009;11:683–93. doi: 10.1038/aja.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Patrinos A, Jordan E, Chakravarti A, Gesteland R, et al. New goals for the U.S. Human Genome Project: 1998–2003. Science. 1998;282:682–9. doi: 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos AK, Tsiliki G, Spyrou G, Tsangaris GT. Bioinformatics approaches in the discovery and understanding of reproduction-related biomarkers. Expert Rev Proteomics. 2011;8:187–95. doi: 10.1586/epr.11.12. [DOI] [PubMed] [Google Scholar]
- Marengo E, Robotti E, Bobba M. 2D-PAGE maps analysis. Methods Mol Biol. 2008;428:291–325. doi: 10.1007/978-1-59745-117-8_16. [DOI] [PubMed] [Google Scholar]
- Matthiesen R, Jensen ON. Analysis of mass spectrometry data in proteomics. Methods Mol Biol. 2008;453:105–22. doi: 10.1007/978-1-60327-429-6_4. [DOI] [PubMed] [Google Scholar]
- Cai H, Chen H, Yi T, Daimon CM, Boyle JP, et al. VennPlex—a novel Venn diagram program for comparing and visualizing datasets with differentially regulated datapoints. PLoS ONE. 2013;8:e53388. doi: 10.1371/journal.pone.0053388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Shen J, Xia Z, Zhang R, Zhang P, et al. Proteomic analysis of proteins involved in spermiogenesis in mouse. J Proteome Res. 2010;9:1246–56. doi: 10.1021/pr900735k. [DOI] [PubMed] [Google Scholar]
- Gherman A, Davis EE, Katsanis N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet. 2006;38:961–2. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE. The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012;40 Database issue:D881–6. doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wang H, Li J. An integrated bioinformatics analysis of mouse testis protein profiles with new understanding. BMB Rep. 2011;44:347–51. doi: 10.5483/BMBRep.2011.44.5.347. [DOI] [PubMed] [Google Scholar]
- Calvel P, Rolland AD, Jegou B, Pineau C. Testicular postgenomics: targeting the regulation of spermatogenesis. Philos Trans R Soc Lond B Biol Sci. 2010;365:1481–500. doi: 10.1098/rstb.2009.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–52. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41 Database issue:D801–7. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp M, Marquardt JU, Sahin U, Galle PR, Castle J, et al. RNA-Seq Atlas—a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics. 2012;28:1184–5. doi: 10.1093/bioinformatics/bts084. [DOI] [PubMed] [Google Scholar]
- Hoogland C, Mostaguir K, Appel RD, Lisacek F. The World-2DPAGE Constellation to promote and publish gel-based proteomics data through the ExPASy server. J Proteomics. 2008;71:245–8. doi: 10.1016/j.jprot.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Cote R, Reisinger F, Barsnes H, Foster JM, et al. The Proteomics Identifications Database: 2010 update. Nucleic Acids Res. 2010;38 Database issue:D736–42. doi: 10.1093/nar/gkp964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BE, Hill JA, Gjukich MA, Andrews PC. Tranche distributed repository and ProteomeCommons.org. Methods Mol Biol. 2011;696:123–45. doi: 10.1007/978-1-60761-987-1_8. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhang P, Qi Y, Chen W, Chen X, et al. Proteomic analysis of male 4C germ cell proteins involved in mouse meiosis. Proteomics. 2011;11:298–308. doi: 10.1002/pmic.200900726. [DOI] [PubMed] [Google Scholar]
- Pilch B, Mann M. Large-scale and high-confidence proteomic analysis of human seminal plasma. Genome Biol. 2006;7:R40. doi: 10.1186/gb-2006-7-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaguir K, Hoogland C, Binz PA, Appel RD. The Make 2D-DB II package: conversion of federated two-dimensional gel electrophoresis databases into a relational format and interconnection of distributed databases. Proteomics. 2003;3:1441–4. doi: 10.1002/pmic.200300483. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhao C, Wang F, Zhu Y, Cui Y, et al. Investigation of human testis protein heterogeneity using 2-dimensional electrophoresis. J Androl. 2010;31:419–29. doi: 10.2164/jandrol.109.007534. [DOI] [PubMed] [Google Scholar]
- Huang XY, Guo XJ, Shen J, Wang YF, Chen L, et al. Construction of a proteome profile and functional analysis of the proteins involved in the initiation of mouse spermatogenesis. J Proteome Res. 2008;7:3435–46. doi: 10.1021/pr800179h. [DOI] [PubMed] [Google Scholar]
- Wang G, Guo Y, Zhou T, Shi X, Yu J, et al. In-depth proteomic analysis of the human sperm reveals complex protein compositions. J Proteomics. 2012;79C:114–22. doi: 10.1016/j.jprot.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Liu M, Hu Z, Qi L, Wang J, Zhou T, et al. Scanning of novel cancer/testis proteins by human testis proteomic analysis. Proteomics. 2013;13:1200–10. doi: 10.1002/pmic.201200489. [DOI] [PubMed] [Google Scholar]
- Achermann JC, Ozisik G, Meeks JJ, Jameson JL. Genetic causes of human reproductive disease. J Clin Endocrinol Metab. 2002;87:2447–54. doi: 10.1210/jcem.87.6.8622. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37 Web Server issue:W305–11. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Kir J, Liu D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35 Web Server issue:W169–75. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38 Web Server issue:W210–3. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38 Database issue:D204–10. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33 Web Server issue:W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio—the analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–7. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium Gene Ontology annotations and resources. Nucleic Acids Res. 2012;41 Database issue:D530–5. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rato L, Alves MG, Socorro S, Duarte AI, Cavaco JE, et al. Metabolic regulation is important for spermatogenesis. Nat Rev Urol. 2012;9:330–8. doi: 10.1038/nrurol.2012.77. [DOI] [PubMed] [Google Scholar]
- Piomboni P, Focarelli R, Stendardi A, Ferramosca A, Zara V. The role of mitochondria in energy production for human sperm motility. Int J Androl. 2012;35:109–24. doi: 10.1111/j.1365-2605.2011.01218.x. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40 Database issue:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Eustachio P. Reactome knowledgebase of human biological pathways and processes. Methods Mol Biol. 2011;694:49–61. doi: 10.1007/978-1-60761-977-2_4. [DOI] [PubMed] [Google Scholar]
- Kelder T, van Iersel MP, Hanspers K, Kutmon M, Conklin BR, et al. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40 Database issue:D1301–7. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy Choudhury D, Small C, Wang Y, Mueller PR, Rebel VI, et al. Microarray-based analysis of cell-cycle gene expression during spermatogenesis in the mouse. Biol Reprod. 2010;83:663–75. doi: 10.1095/biolreprod.110.084889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette C, Platts AE, Johnson GD, Emery BR, Carrell DT, et al. Identification of human sperm transcripts as candidate markers of male fertility. J Mol Med (Berl) 2009;87:735–48. doi: 10.1007/s00109-009-0485-9. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33 Database issue:D514–7. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Goldsmith CA, Eppig JT. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6:R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, et al. DrugBank 3.0: a comprehensive resource for ‘omics' research on drugs. Nucleic Acids Res. 2011;39 Database issue:D1035–41. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Shi Z, Qin C, Tao L, Liu X, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012;40 Database issue:D1128–36. doi: 10.1093/nar/gkr797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci USA. 2008;105:20422–7. doi: 10.1073/pnas.0810777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- Almeida LG, Sakabe NJ, deOliveira AR, Silva MC, Mundstein AS, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37 Database issue:D816–9. doi: 10.1093/nar/gkn673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodison S, Urquidi V. The cancer testis antigen PRAME as a biomarker for solid tumor cancer management. Biomark Med. 2012;6:629–32. doi: 10.2217/bmm.12.65. [DOI] [PubMed] [Google Scholar]
- Partheen K, Levan K, Osterberg L, Claesson I, Fallenius G, et al. Four potential biomarkers as prognostic factors in stage III serous ovarian adenocarcinomas. Int J Cancer. 2008;123:2130–7. doi: 10.1002/ijc.23758. [DOI] [PubMed] [Google Scholar]
- Peled N, Oton AB, Hirsch FR, Bunn P. MAGE A3 antigen-specific cancer immunotherapeutic. Immunotherapy. 2009;1:19–25. doi: 10.2217/1750743X.1.1.19. [DOI] [PubMed] [Google Scholar]
- Chen YT, Scanlan MJ, Venditti CA, Chua R, Theiler G, et al. Identification of cancer/testis-antigen genes by massively parallel signature sequencing. Proc Natl Acad Sci USA. 2005;102:7940–5. doi: 10.1073/pnas.0502583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Chuva de Sousa Lopes SM, Kaneda M, Tang F, Hajkova P, et al. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS ONE. 2008;3:e1738. doi: 10.1371/journal.pone.0001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Cheng L, Zhou T, Guo X, Zhang X, et al. Predicting miRNA-mediated gene silencing mode based on miRNA-target duplex features. Comput Biol Med. 2012;42:1–7. doi: 10.1016/j.compbiomed.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Braun RE. Temporal control of protein synthesis during spermatogenesis. Int J Androl. 2000;23 Suppl 2:92–4. doi: 10.1046/j.1365-2605.2000.00027.x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Oughtred R, Wing SS. Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol Cell Biol. 2005;25:2819–31. doi: 10.1128/MCB.25.7.2819-2831.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–80. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhou T, Jia H, Guo X, Zhang X, et al. SProtP: a web server to recognize those short-lived proteins based on sequence-derived features in human cells. PLoS ONE. 2011;6:e27836. doi: 10.1371/journal.pone.0027836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhang X, Chen J, Wang Q, Wang L, et al. ReCGiP, a database of reproduction candidate genes in pigs based on bibliomics. Reprod Biol Endocrinol. 2010;8:96. doi: 10.1186/1477-7827-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TL, Cheung HH, Claus J, Sastry C, Singh S, et al. GermSAGE: a comprehensive SAGE database for transcript discovery on male germ cell development. Nucleic Acids Res. 2009;37 Database issue:D891–7. doi: 10.1093/nar/gkn644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Platts AE, Ostermeier GC, Krawetz SA. K-SPMM: a database of murine spermatogenic promoters modules & motifs. BMC Bioinformatics. 2006;7:238. doi: 10.1186/1471-2105-7-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardenois A, Gattiker A, Collin O, Chalmel F, Primig M. GermOnline 4.0 is a genomics gateway for germline development, meiosis and the mitotic cell cycle. Database (Oxford) 2010;2010:baq030. doi: 10.1093/database/baq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–32. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37 Database issue:D412–6. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SJ, Zhang C, Zou Q, Ji ZL. TiSGeD: a database for tissue-specific genes. Bioinformatics. 2010;26:1273–5. doi: 10.1093/bioinformatics/btq109. [DOI] [PMC free article] [PubMed] [Google Scholar]