

Abstract
Increasing evidence shows a relationship between epigenetic regulation and male infertility. The GTF2A1L gene promoter contains the DNA methylation site of a tissue-specific differentially methylated region (TDMR). Eighty-six patients with non-obstructive azoospermia were assessed for the DNA methylation state of CpG islands in the GTF2A1L promoter using testicular genomic DNA. Based on histological criteria, 26 of the 86 patients had normal spermatogenesis (controls), 17 had hypospermatogenesis and 26 had a Sertoli cell-only phenotype or tubular sclerosis. GTF2A1L TDMR methylation was significantly lower in testes DNA from control samples than from hypospermatogenic samples (P=0.029). Patients with hypospermatogenesis were divided into two subgroups: high DNA methylation (HM, n=5) and low DNA methylation (LM, n=12). The GTF2A1L TDMR methylation rate differed significantly between the HM and LM groups (P=0.0019), and GTF2A1L expression was significantly higher among the LM than in the HM patients (P=0.023). High TDMR methylation was correlated with low GTF2A1L gene expression levels. Both groups demonstrated relatively good outcomes with respect to sperm retrieval, fertilisation, pregnancy and childbirth rates. We observed that aberrant GTF2A1L gene expression was not correlated with fertilisation rates. The testicular sperm extraction (TESE) technique may be used to overcome male infertility due to aberrant TDMR methylation.
Keywords: ALF, azoospermia, CpG island, hypospermatogenesis, MALDI-TOF MS, testicular sperm extraction (TESE)
Introduction
Male factors of total infertility may account for up to 40%–50% of infertile couples,1 and many cases of male infertility are unreported. Testicular sperm extraction (TESE) and intracytoplasmic sperm injection (ICSI) are commonly used techniques to overcome male infertility, and sperm retrieval is successful in 30%–70% of patients with non-obstructive azoospermia.2 In hypoplasia, 97.7% of patients had late spermatids or sperm recovered using the assisted reproductive technique (ART).3 In healthy males, infertility may result from a number of unknown factors including genetic or epigenetic disorders.
Epigenetic modifications, in particular, result in stable or semi-stable changes in gene expression without affecting the DNA nucleotide sequence.4,5 DNA methylation plays a role in gene regulation during development and has also been involved in genomic imprinting and X-inactivation.
CpG dinucleotides, also known as CpG islands, are located in the promoter regions of many genes and have variable DNA methylation states. CpG islands are generally not methylated during normal cell development, with the exception of imprinted genes. These islands are preferentially located at the start of genes and on the differentially methylated regions of imprinted genes.6
However, tissue-specific differentially methylated regions (TDMRs) are in a somewhat different category. These regions can be identified by DNA methylation array methods and are demethylated in some tissues but fully methylated in others.7,8 Therefore, the DNA methylation of TDMRs in gene loci causes transcriptional repression,9,10,11 is interspersed throughout the entire genome and ensures that the DNA methylation status corresponds to specific tissue types.
Methylation analysis performed on testicular DNA has revealed numerous differentially methylated loci in the male germline.12,13 Therefore, some genes with demethylated TDMRs are expressed in a tissue-specific manner.14,15 Numerous CpG islands that contain TDMRs are potential methylation sites in normal cells and tissues. The expression of genes associated with identified TDMRs has been shown to correlate with methylation status; thus, aberrant methylation of TDMRs may adversely affect clinical manifestations.
GTF2A1L (NM_006872) encodes the Homo Sapiens general transcription factor IIA, 1-like factor formerly known as ALF. The synthesis of eukaryotic mRNA requires the assembly and stability of RNA polymerase II and various general transcription factors. The GTF2A1L gene encodes a germ cell-specific counterpart of the large (alpha/beta) subunit of the general transcription factor TFIIA, which is able to stabilize the binding of TATA-binding protein to DNA and may be uniquely important to testis biology.16,17,18 The GTF2A1L gene is selectively transcribed in reproductive tissues and is coexpressed in late pachytene spermatocytes and in haploid round spermatids with the TATA-binding protein-related factor 2, and these proteins form stable complexes in testis extracts.19
Aberrant DNA methylation at TDMRs has raised concerns about the status of male fertility. In this study, we have confirmed the TDMR of the GTF2A1L promoter by applying matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS) to human testicular tissue. We also describe the TDMR methylation patterns of GTF2A1L CpG islands, characterize the methylation status of TDMR in some cases of male infertility and discuss the associated ART outcomes.
Materials and methods
Patients and samples
This study was approved by the Ethics Committee of the Kanazawa University Graduate School of Medical Science. All participants granted their informed consent for this study. Patients (n=86) with azoospermia in their ejaculate were recruited between May 2006 and April 2011, and patients with abnormal karyotypes or Y-chromosome microdeletions were excluded from the study, as described previously.20 Testicular tissue specimens were obtained from patients who underwent either a diagnostic testicular biopsy or a sperm retrieval procedure from testicular tissues to evaluate histology while attempting a testicular biopsy or microdissection TESE. Histological examinations of at least 50 seminiferous tubules were performed as previously described.21 The most advanced spermatogenetic cell that was identified in each analysis determined the histological classification of the sample. Men with azoospermia who were found to have normal sperm upon histological examination of testis samples were classified as having normal spermatogenesis.
Patient categorisation
Patients were classified into five groups using histological diagnosis as demonstrating normal spermatogenesis (NS, n=26), hypospermatogenesis (HS, n=17), maturation arrest (MA, n=17), Sertoli cell only phenotype (SCO, n=21) and tubular sclerosis (TS, n=5). Sperm were present in the tissues of 26 patients who were thought to have non-obstructive azoospermia due to obstructive azoospermia. We defined these patients as the control group. The mean ages (mean±s.d.) of NS (control), HS, MA, SCO and TS patients were 36.0±5.9, 38.2±3.9, 34.8±4.1, 35.2±3.7 and 37.2±5.0 years, respectively.
Isolation of genomic DNA and total RNA
Human genomic DNA from several normal tissues (kidney, muscle, heart, brain and colon tissue) were kindly provided by Professor Hiroki Nagase, Department of Advanced Medical Science, Division of Cancer Genetics, Nihon University School of Medicine. These samples were obtained by organ donations from autopsy cases at the Pathology Division of the Nihon University School of Medicine in Tokyo, Japan. When the tissues were received through organ donation, the bereaved families or relatives provided their informed, written consent.22 Premium Total RNA (Clontech, Mountain View, CA, USA) from various normal human tissues was used as a control for the gene expression studies. Total RNA and testicular genomic DNA were isolated from each of the testicular tissue specimens. TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) was used for RNA and DNA extraction from the same specimens. Total RNA was treated with the TURBO DNA-free kit (Ambion, Austin, TX, USA) to remove any residual genomic DNA. Single-stranded cDNA was synthesized using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). The additional primer pair sequences used for GTF2A1L amplification was 5′-CTGCCTCAACCCGGTGCCTAAAC-3′ and 5′-GCTGAACCACTGAGCACTGACTCCAC-3′ (product size, 798 bp). The primer pair sequences used for GAPDH amplification was 5′-GACCACAGTCCATGCCATCA-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ (product size, 453 bp).
Bisulphite treatment and quantitative DNA methylation analysis
Genomic DNA (1 µg) isolated from testicular specimens was treated with sodium bisulphite using the EZ DNA methylation kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. Quantitative DNA methylation analysis at single CpG dinucleotides was performed using the MassARRAY Compact system (Sequenom, San Diego, CA, USA) to quantify the methylation status of CpG islands in the GTF2A1L promoter, as previously described.13 MALDI-TOF MS was used for the high-throughput quantitative DNA methylation assay, and to analyse the base-specific cleaved amplification products. Each methylation evaluation was conducted using EpiTyper software v1.0 (Sequenom), which was able to generate quantitative results for each cleavage fragment (known as a CpG unit), and includes individual CpG dinucleotides or aggregates of multiple CpG sites.
Putative GTF2A1L promoter regions were chosen for this analysis. Arbitrarily chosen genomic regions, approximately 1-kb upstream of the GTF2A1L start site, were analysed to determine whether there were promoter-specific changes in DNA methylation. Bisulphite-treated DNA was amplified using polymerase chain reaction (PCR) with a reverse primer (5′-CAGTAATACGACTCACTATAGGGAGAAGGCTTTAAAACAAACCATAACAACACC-3′) tagged with a T7 promoter sequence and a forward primer (5′-AGGAAGAGAGGGATTGAGGAA ATAATTTGTGAA-3′). The amplification products were transcribed in vitro, base-specifically cleaved by RNaseA, and subjected to MALDI-TOF MS for the quantitative DNA methylation assay. The graphic data obtained from methylation analysis using MassARRAY were expressed as an epigram. DNA methylation standards (0%, 20%, 40%, 60%, 80% and 100% methylated genomic DNA) and correction algorithms based on the R statistical computing environment were used for data correction and normalisation.13
LightCycler amplification
GTF2A1L mRNA was quantified using the LightCycler TaqMan Master (Roche Applied Science, Basel, Switzerland). The Universal probe No. 62 (Roche) with forward primer 5′-TCCTGGTTATCCCATTCATGT-3′ and reverse GTF2A1L primer 5′-CTGTCACCATAATTGGTACATTGAC-3′ were used for amplification. As an internal reference standard, GAPDH expression was also measured using universal probe No. 60 (Roche) with forward primer 5′-AGCCACATCGCTCAGACA-3′ and reverse primer 5′-TCAGGAAATTTGACTTTCCATTC-3′. Each universal probe was designed according to the Universal Probe Assay Design Centre (https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp). All PCR reactions were performed in a total volume of 20 µl comprising 4 µl of 5 × LightCycler TaqMan Master (Roche Diagnostics, Mannheim, Germany), 0.3 µl of 10 µmol l−1 TaqMan probe, 1 µl of 10 µmol l−1 each primer, 2 µl of sample cDNA and 11.6 µl of DEPC-treated water.
Amplification of GTF2A1L and of GAPDH was performed in triplicate for each sample. The thermal cycling conditions used were: 10 min at 95 °C, followed by 50 cycles at 95 °C for 10 s and 60 °C for 20 s for both GTF2A1L and GAPDH. The number of GTF2A1L and GAPDH transcripts in each sample was calculated with the LightCycler software using the experimentally generated standard curves.
Statistical analysis
The data were analysed using the GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego, CA, USA; http://www.graphpad.com). Non-parametric statistical analyses were used because only a small number of subjects were available, and most variables demonstrated a skewed data distribution. The Mann–Whitney U test was used to compare the mean methylation rates for non-parametric data. The Student's t-test was used to examine the correlation between GTF2A1L expression and hormone levels, and differences were considered to be statistically significant at P<0.05.
Results
Confirmation of TDMR in the GTF2A1L of normal tissues
The expression of GTF2A1L in several different human tissue types (testis, kidney, muscle, heart, brain and colon) was evaluated using reverse transcription-PCR. The GTF2A1L transcript was detected in testis tissue only (Figure 1).
Figure 1.
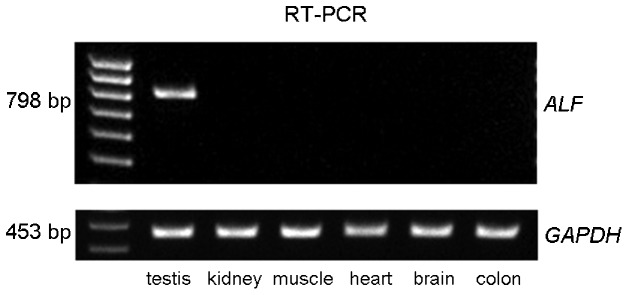
Tissue-specific expression of GTF2A1L in normal human tissues. cDNAs were prepared from total RNA isolated from testis, kidney, muscle, heart, brain and colon tissues. GAPDH was used as the internal standard for comparison.
The DNA methylation status of the GTF2A1L regulatory region in several different human tissue types was investigated using testes genomic DNA. We assayed DNA methylation in testis, kidney, muscle, heart, brain and colon, and focused on the regions upstream of the GTF2A1L gene. These amplicons were screened using the MALDI-TOF MS system (MassARRAY) to quantify methylation at CpG islands. Methylation at CpG islands in the GTF2A1L promoter was lower in the testis (methylation rate: 21.6%±2.83%) than in the five other tissue types: kidney (85.3%±6.21%), muscle (83.1%±9.31%), heart (84.8%±5.66%), brain (88.9%±5.85%) and colon (84.8%±8.16%). These differences indicate the presence of a TDMR in the 5′-upstream GTF2A1L promoter region somewhere between −258 bp and +41 bp of the transcription start site. This region, a 299 bp-amplicon, contains a promoter CpG island with 22 CpG dinucleotides (Figure 2). Therefore, the TDMR of GTF2A1L was classified as having low methylation at CpG islands in the normal testes.
Figure 2.

Chromosomal location of GTF2A1L. This map is based on the March 2006 human reference sequence (NCBI Build 36.1). The GTF2A1L gene is shown as an arrow, with an arrowhead indicating the direction of transcription. CpG islands are shown with the number of CpGs. The amplicon was generated from the −258 bp to +41 bp (299 bp) CpG region near the transcription start site.
Quantification of the GTF2A1L methylation state in the testicular genome
We analysed DNA methylation for the GTF2A1L gene promoter in genomic DNA samples from testes using MassARRAY. Testes DNA in all samples from the control group (n=26) had low methylation at the GTF2A1L promoter, and the mean methylation rate was 24.4%±10.6% (95% CI: 20.1%–28.7%). Testis DNA from all samples from the HS group (n=17) also demonstrated low DNA methylation, and the mean methylation rate was 35.1%±12.0% (95% CI: 29.0%–41.3%). The mean GTF2A1L DNA methylation rates in patients with HS were significantly higher compared with the controls (P=0.029). However, the mean methylation rates in patients with MA (n=17), SCO (n=21) and TS (n=5) were 66.5%±0.07% (95% CI: 62.9%–70.1%), 76.5%±0.08% (95% CI: 73.1%–79.9%) and 79.4%±0.07% (95% CI: 70.2%–88.7%), respectively. Testes DNA from all samples from the MA, SCO or TS groups demonstrated high DNA methylation (Figure 3). Patients with SCO and TS were extreme cases, because the genomic DNA extracted from those testes biopsies may not present with any germ cells. Although all samples revealed high GTF2A1L DNA methylation except NS (control) and some HS samples, differences in methylation levels between control and HS groups were statistically significant in this study.
Figure 3.

Rate of methylation status in patients characterized histological diagnosis. Normal spermatogenesis controls (NS, closed circles, n=26), hypospermatogenesis (HS, closed squares, n=17), maturation arrest phenotype (MA, closed triangles, n=17), Sertoli cell only phenotype (SCO, inverted closed triangles, n=21) and tubular sclerosis (TS, closed diamonds, n=5). The asterisk indicates a statistically significant difference between values (P<0.05).
Characterisation of patients with hypospermatogenesis
To further characterize the patients with HS, this group was divided into two subgroups according to methylation level: low methylation (LM) and high methylation (HM). The mean CpG methylation rates were 28.6%±6.9% (95% CI: 24.1%–33.0%, n=12) in the LM group versus 50.9%±2.3% (95% CI: 48.0%–53.7%, n=5) in the HM group, and these means were significantly different (P=0.0019, Figure 4a). These results indicate that some subjects with HS exhibit relatively high rates of high methylation in the GTF2A1L promoter region.
Figure 4.
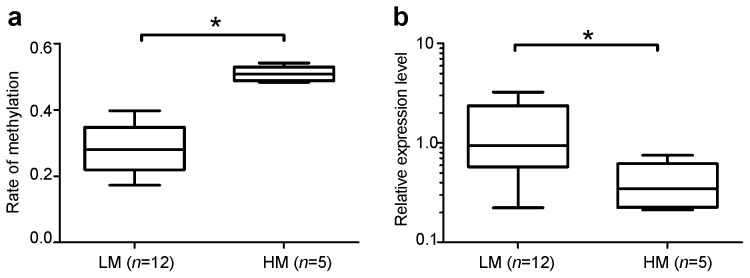
Classification of high- and low-methylation TDMR groups in patients with hypospermatogenesis. (a) Box–Whiskers plot (min. to max.) of patients with hypospermatogenesis subdivided according to the GTF2A1L methylation rate: low methylation (LM, n=12) and high methylation (HM, n=5). (b) The relative expression level of GTF2A1L in patients with hypospermatogenesis and low methylation (LM, n=12) or high methylation (HM, n=5). The asterisk indicates a statistically significant difference between values (P<0.05). TDMR, tissue-specific differentially methylated region.
GTF2A1L expression in the HM and LM groups
The GTF2A1L mRNA was measured using real-time quantitative reverse transcription-PCR with GAPDH mRNA as an internal reference standard. The GTF2A1L expression rate in the control group was 0.91±0.42 (95% CI: 0.74–1.08), whereas the rates in the MA, SCO and TS groups were 0.18±0.10 (95% CI: 0.1–0.23), 0.095±0.039 (95% CI: 0.08–0.11) and 0.03±0.02 (95% CI: 0.001–0.060), respectively. Both patients with SCO and TS also exhibited high DNA methylation, and the GTF2A1L expression was very low, as expected.
The relative GTF2A1L expression level in the HS group is shown in Figure 4b. Expression of GTF2A1L mRNA was significantly higher in the LM group (median=0.9370, 25% percentile=0.5745, 75% percentile=7.425) than in the HM group (median=0.3460, 25% percentile=0.2251, 75% percentile=0.6211) (P=0.019). These results demonstrated that highly methylated TDMRs correlate with low levels of gene expression. Therefore, the alteration of methylation within the GTF2A1L promoter may greatly affect its expression and result in spermatogenesis failure.
Clinical characteristics of patients with LM and HM
The general characteristics and clinical parameters of patients with hypospermatogenesis are summarized in Tables 1 and 2. These patients were classified into two groups based on the GTF2A1L methylation state as HM (Table 1) and LM (Table 2); the mean patients' age in each group was 33.8±2.63 years and 37.7±2.1 years, respectively. Levels of follicle stimulating hormone (FSH), luteinizing hormone (LH), and testosterone (T) in the LM group were 3.2±0.42 mU ml−1, 2.7±0.24 mU ml−1 and 5.7±0.99 ng ml−1, respectively. In contrast, the mean FSH, LH and T levels in the HM group were 5.7±0.77 mU ml−1, 2.7±0.44 mU ml−1 and 4.4±0.30 ng ml−1, respectively. No significant difference was found in mean levels of FSH (P=0.08), LH (P=0.96) or T (P=0.12) between the LM and HM groups (Tables 1 and 2). Following various ARTs, including TESE–ICSI, both groups had relatively good outcomes with respect to sperm retrieval and fertilisation as well as pregnancy and childbirth rates. We observed that aberrant GTF2A1L promoter methylation was not correlated with reproductive outcome following TESE–ICSI procedures. Moreover, the sperm positive control group (n=26) revealed normal endocrinology profile and retrieval, and fertilisation rate are 100%.
Table 1. Clinical features of HS patients with high methylation.
| Age (year) | FSH (mU ml−1) | LH (mU ml−1) | Testosterone (ng ml−1) | TESE | Sperm retrieval | ICSI | Fertilisation | Pregnancy | Childbirth |
|---|---|---|---|---|---|---|---|---|---|
| 34 | 2.25 | 2.42 | 4.6 | Yes | Yes | Yes | Yes | Yes | Yes |
| 41 | 3.4 | 3.29 | 8.2 | Yes | Yes | Yes | Yes | Yes | Yes |
| 42 | N/P | N/P | N/P | Yes | Yes | Yes | Yes | Yes | Yes |
| 31 | 4.23 | 2.7 | 3.71 | Yes | Yes | Yes | Yes | Yes | Yes |
| 29 | 2.8 | 2.2 | 6.2 | Yes | Yes | Yes | Yes | Yes | Yes |
Abbreviations: HS, hypospermatogenesis; ICSI, intracytospermic sperm injection; N/P, not performed; TESE, testicular sperm extraction including microdissection.
Table 2. Clinical features of HS patients with low methylation.
| Age (year) | FSH (mU ml−1) | LH (mU ml−1) | Testosterone (ng ml−1) | TESE | Sperm retrieval | ICSI | Fertilisation | Pregnancy | Childbirth |
|---|---|---|---|---|---|---|---|---|---|
| 31 | 2.8 | 1 | 4.1 | Yes | Yes | Yes | Yes | No | No |
| 31 | 3.4 | 2.2 | 3.8 | Yes | Yes | Yes | Yes | Yes | Yes |
| 32 | 7.2 | 5.4 | 6.9 | Yes | Yes | Yes | Yes | Yes | Yes |
| 45 | 5.4 | 2.5 | 4.5 | Yes | Yes | Yes | Yes | No | No |
| 35 | 3 | 3.1 | 5.1 | Yes | Yes | Yes | Yes | Yes | Yes |
| 38 | 4.5 | 1.5 | 3.7 | Yes | Yes | Yes | Yes | No | No |
| 54 | 5.4 | 0.8 | 4.4 | Yes | Yes | Yes | Yes | Yes | Yes |
| 36 | 6.7 | 4 | 4.1 | Yes | Yes | Yes | Yes | Yes | Yes |
| 58 | N/P | N/P | N/P | Yes | Yes | Yes | Yes | Yes | Yes |
| 38 | 4.93 | 2.06 | 4.7 | Yes | Yes | Yes | Yes | Yes | Yes |
| 34 | 11.81 | 2.42 | 2.9 | Yes | Yes | N/P | N/P | N/P | N/P |
| 41 | 7.21 | 4.64 | 4.2 | Yes | Yes | Yes | Yes | Yes | Yes |
Abbreviations: FSH, follicle-stimulating hormone; HS, hypospermatogenesis; ICSI, intracytospermic sperm injection; LH, luteinizing hormone; N/P, not performed; TESE, testicular sperm extraction including microdissection.
Discussion
Numerous CpG islands that contain TDMR are potential methylation sites in normal tissues. In this study, DNA methylation at the GTF2A1L CpG island was apparently lower in testes samples with normal spermatogenesis; however, aberrant methylation at the GTF2A1L CpG island was associated with hypospermatogenesis in some azoospermic patients. We revealed that the aberrant methylation of TDMRs was associated with human spermatogenesis. However, only five patients exhibited relatively high methylation at the GTF2A1L CpG island, but these low numbers are likely not sufficient to generate statistical significance. Other subjects, including patients with MA, SCO and TS, exhibited high GTF2A1L promoter methylation, but low GTF2A1L promoter methylation is considered to be the normal status for TDMR. Although larger numbers of infertile male subjects may be necessary to detect the influence of aberrant methylation on hypospermatogenesis incidence, the relatively higher GTF2A1L gene methylation observed in the five patients may still yield useful data.
Previously, CpG islands were considered to be almost entirely unmethylated except within imprinted regions and on the inactive X chromosome.23 However, accumulating evidence suggests that methylation of TDMRs is associated with modulated gene expression24,25,26 and impaired spermatogenesis.13,22,27
Unlike imprinted genes, many TDMRs may play a role in defining cellular identity and tissue-specific regulation of genome function. Moreover, the TDMRs may be broadly distributed in intragenic and intergenic regions that have CpG islands. Approximately 4% of more than 5000 autosomal genes with unmethylated CpG island promoters were methylated in normal peripheral blood.28 Aberrant TDMR methylation may increase the prevalence of male infertility and might be involved in idiopathic male infertility.
TESE is a powerful technique used to retrieve sperm from patients with azoospermia. ARTs are regularly used to facilitate fertilisation and pregnancy. ART may be associated with epigenetic changes in imprinted genes that can lead to human disease. Various artificial fertilisation procedures may be implicated in the susceptibility to epigenetic defects such as Beckwith–Wiedemann syndrome and Angelman syndrome after ART.29 However, the absolute incidence of imprinting disorders is very low.30,31,32
Epigenetic regulation of the germline has recently become a clear candidate process that results in male infertility. Tissue-specific CpG island methylation was identified at developmental gene loci, which became demethylated during the differentiation into adult tissues.33,34 Moreover, high CpG dense regions were mostly unmethylated, and low CpG dense regions were demonstrated as the preferential targets for de novo methylation in a 5-methyl cytosine antibody study.35
Additionally, testicular spermatozoa from men with abnormal spermatogenesis often have methylation defects, which further supports an association between ARTs and the occurrence of imprinting errors.30 Studies of the methylation levels in paternally and maternally imprinted genes and aberrantly methylated imprints revealed a significant association with abnormal semen parameters, but did not appear to influence ART outcomes.36
In the present study, sperm retrieval, fertilisation, pregnancy and child birth rates were relatively high. An atypical GTF2A1L expression was due to an aberrant methylation of GTF2A1L TDMR in testicular DNA; however, these aberrations did not cause serious damage in the testicular spermatozoa recovered via TESE procedures. One possible explanation for this observation was that aberrant TDMR methylation was considered to result in weak CpG island promoters, which are the preferential targets for de novo methylation.
The GTF2A1L protein is coexpressed in late pachytene spermatocytes and in haploid round spermatids19 and may form part of a transcriptional network that is vital for the completion of meiosis and for the preparation of post-meiotic differentiation.37
Hypospermatogenesis is characterized histologically by a reduced number of spermatozoa in any seminiferous tubule .21 Although some post-meiotic failure is believed to result from aberrant methylation in promoters of some testis-specific genes, the genetic regulation of post-meiotic spermiogenesis remains unclear (e.g., germ cell-specific epigenetic processes, including histone to protamine exchange in haploid spermatids or genomic imprinting in the parent). Once germ cells complete meiosis, male infertility caused by aberrant methylation can be overcome using TESE.
In summary, we demonstrated that aberrant TDMR methylation at the GTF2A1L promoter may have caused a decrease in GTF2A1L expression that was associated with hypospermatogenesis. We suggest that TESE is a valuable procedure for retrieving sperm from patients with hypospermatogenesis. However, this issue must be further examined in a larger patient population to provide adequate data for statistically significant results and to confirm our conclusion.
Author contributions
KS and EK contributed to the study design, performed experiments, analyzed data and wrote the manuscript. MI, MT and YM contributed to the collection of written informed consent from all patients and controls and the purification of the DNAs from all participants. KS and EK analysed the data and assisted with writing the paper. MN designed the study, obtained the funding and revised the paper.
Acknowledgments
The authors would like to thank Professor Hiroki Nagase, Department of Advanced Medical Science, Division of Cancer Genetics, Nihon University School of Medicine, who kindly provided human genomic DNA from several normal tissues. This study supported in part by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Science, Sports, and Culture (No. 24791638).
All authors declare that there are no competing financial interests.
References
- Cram DS, O'Bryan MK, de Kretser DM. Male infertility genetics—the future. J Androl. 2001;22:738–46. [PubMed] [Google Scholar]
- Ezeh UI, Taub NA, Moore HD, Cooke ID. Establishment of predictive variables associated with testicular sperm retrieval in men with non-obstructive azoospermia. Hum Reprod. 1999;14:1005–12. doi: 10.1093/humrep/14.4.1005. [DOI] [PubMed] [Google Scholar]
- Sousa M, Cremades N, Silva J, Oliveira C, Ferraz L, et al. Predictive value of testicular histology in secretory azoospermic subgroups and clinical outcome after microinjection of fresh and frozen–thawed sperm and spermatids. Hum Reprod. 2002;17:1800–10. doi: 10.1093/humrep/17.7.1800. [DOI] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- Kobayashi H, Suda C, Abe T, Kohara Y, Ikemura T, et al. Bisulphite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet Genome Res. 2006;113:130–7. doi: 10.1159/000090824. [DOI] [PubMed] [Google Scholar]
- Matsuyama T, Kimura MT, Koike K, Abe T, Nakano T, et al. Global methylation screening in the Arabidopsis thaliana and Mus musculus genome: applications of virtual image restriction landmark genomic scanning (Vi-RLGS) Nucleic Acids Res. 2003;31:4490–6. doi: 10.1093/nar/gkg488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T, et al. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci USA. 2005;102:3336–41. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Hattori N, Tanaka S, Shiota K. DNA methylation-mediated control of Sry gene expression in mouse gonadal development. J Biol Chem. 2004;279:22306–13. doi: 10.1074/jbc.M309513200. [DOI] [PubMed] [Google Scholar]
- Ko YG, Nishino K, Hattori N, Arai Y, Tanaka S, et al. Stage-by-stage change in DNA methylation status of Dnmt1 locus during mouse early development. J Biol Chem. 2005;280:9627–34. doi: 10.1074/jbc.M413822200. [DOI] [PubMed] [Google Scholar]
- Tomikawa J, Fukatsu K, Tanaka S, Shiota K. DNA methylation-dependent epigenetic regulation of dimethylarginine dimethylaminohydrolase 2 gene in trophoblast cell lineage. J Biol Chem. 2006;281:12163–9. doi: 10.1074/jbc.M513782200. [DOI] [PubMed] [Google Scholar]
- Shiota K. DNA methylation profiles of CpG islands for cellular differentiation and development in mammals. Cytogenet Genome Res. 2004;105:325–34. doi: 10.1159/000078205. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Koh E, Sin HS, Maeda Y, Narimoto K, et al. Tissue-specific differentially methylated regions of the human VASA gene are potentially associated with maturation arrest phenotype in the testis. J Hum Genet. 2009;54:450–6. doi: 10.1038/jhg.2009.59. [DOI] [PubMed] [Google Scholar]
- Cho JH, Kimura H, Minami T, Ohgane J, Hattori N, et al. DNA methylation regulates placental lactogen I gene expression. Endocrinology. 2001;142:3389–96. doi: 10.1210/endo.142.8.8347. [DOI] [PubMed] [Google Scholar]
- Hattori N, Abe T, Hattori N, Suzuki M, Matsuyama T, et al. Preference of DNA methyltransferases for CpG islands in mouse embryonic stem cells. Genome Res. 2004;14:1733–40. doi: 10.1101/gr.2431504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya AB, Lee SH, DeJong J. Identification of a general transcription factor TFIIAalpha/beta homolog selectively expressed in testis. J Biol Chem. 1999;274:18040–8. doi: 10.1074/jbc.274.25.18040. [DOI] [PubMed] [Google Scholar]
- Upadhyaya AB, Khan M, Mou TC, Junker M, Gray DM, et al. The germ cell-specific transcription factor ALF. Structural properties and stabilization of the TATA-binding protein (TBP)–DNA complex. J Biol Chem. 2002;277:34208–16. doi: 10.1074/jbc.M204808200. [DOI] [PubMed] [Google Scholar]
- Kim M, Li D, Cui Y, Mueller K, Chears WC, et al. Regulatory factor interactions and somatic silencing of the germ cell-specific ALF gene. J Biol Chem. 2006;281:34288–98. doi: 10.1074/jbc.M607168200. [DOI] [PubMed] [Google Scholar]
- Catena R, Argentini M, Martianov I, Parello C, Brancorsini S, et al. Proteolytic cleavage of ALF into alpha- and beta-subunits that form homologous and heterologous complexes with somatic TFIIA and TRF2 in male germ cells. FEBS Lett. 2005;579:3401–10. doi: 10.1016/j.febslet.2005.04.083. [DOI] [PubMed] [Google Scholar]
- Fukushima M, Koh E, Choi J, Maeda Y, Namiki M, et al. Reevaluation of azoospermic factor c microdeletions using sequence-tagged site markers with confirmed physical positions from the GenBank database. Fertil Steril. 2006;85:965–71. doi: 10.1016/j.fertnstert.2005.09.060. [DOI] [PubMed] [Google Scholar]
- Cerilli LA, Kuang W, Rogers D. A practical approach to testicular biopsy interpretation for male infertility. Arch Pathol Lab Med. 2010;134:1197–204. doi: 10.5858/2009-0379-RA.1. [DOI] [PubMed] [Google Scholar]
- Kitamura E, Igarashi J, Morohashi A, Hida N, Oinuma T, et al. Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics. 2007;89:326–37. doi: 10.1016/j.ygeno.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) Genome Res. 2008;18:1518–29. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling E, Rehli M. Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics. 2007;90:314–23. doi: 10.1016/j.ygeno.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Hattori N, Imao Y, Nishino K, Hattori N, Ohgane J, et al. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes Cells. 2007;12:387–96. doi: 10.1111/j.1365-2443.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- Stoger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics. 2006;1:155–62. doi: 10.4161/epi.1.4.3400. [DOI] [PubMed] [Google Scholar]
- Dasoula A, Georgiou I, Kontogianni E, Sofikitis N, Syrrou M. Methylation status of the SNRPN and HUMARA genes in testicular biopsy samples. Fertil Steril. 2007;87:805–9. doi: 10.1016/j.fertnstert.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–36. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowdin S, Allen C, Kirby G, Brueton L, Afnan M, et al. A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod. 2007;22:3237–40. doi: 10.1093/humrep/dem268. [DOI] [PubMed] [Google Scholar]
- Marques CJ, Francisco T, Sousa S, Carvalho F, Barros A, et al. Methylation defects of imprinted genes in human testicular spermatozoa. Fertil Steril. 2010;94:585–94. doi: 10.1016/j.fertnstert.2009.02.051. [DOI] [PubMed] [Google Scholar]
- Tierling S, Souren NY, Gries J, Loporto C, Groth M, et al. Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J Med Genet. 2010;47:371–6. doi: 10.1136/jmg.2009.073189. [DOI] [PubMed] [Google Scholar]
- Odom LN, Segars J. Imprinting disorders and assisted reproductive technology. Curr Opin Endocrinol Diabetes Obes. 2010;17:517–22. doi: 10.1097/MED.0b013e32834040a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Mahmood S, Ghosh S, Liang P, Smiraglia DJ, et al. Tissue specific differentially methylated regions (TDMR): changes in DNA methylation during development. Genomics. 2009;93:130–9. doi: 10.1016/j.ygeno.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- El Hajj N, Zechner U, Schneider E, Tresch A, Gromoll J, et al. Methylation status of imprinted genes and repetitive elements in sperm DNA from infertile males. Sex Dev. 2011;5:60–9. doi: 10.1159/000323806. [DOI] [PubMed] [Google Scholar]
- Horvath GC, Kistler MK, Kistler WS. RFX2 is a candidate downstream amplifier of A-MYB regulation in mouse spermatogenesis. BMC Dev Biol. 2009;9:63. doi: 10.1186/1471-213X-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]