

Abstract
We describe a highly efficient method for exact gene replacement in budding yeast. Induction of rapid and efficient recombination in an entire cell population results in at least 50% of the recombinants undergoing a specific switch of the endogenous copy to a specific mutated allele, with no remaining markers or remnant of foreign DNA, without selection. To accomplish this, a partial copy of the replacement allele, followed by an HO cut site, is installed adjacent to the wild-type locus, in a GAL-HO MATa-inc background. HO induction results in near-quantitative site cleavage and recombination/gene conversion, resulting in either regeneration of wild-type, or switch of the endogenous allele to the mutant, with accompanying deletion of intervening marker sequences, yielding an exact replacement. Eliminating the need for selection (over days) of rare recombinants removes concerns about second-site suppressor mutations, and also allows direct phenotypic analysis even of lethal gene replacements without the need of a method to make the lethality conditional, or to employ regulated promoters of unknown strength compared to the endogenous promoter. To test this method, we tried two known lethal gene replacements, substituting the non-essential CDH1 gene with a dominantly lethal version mutated for its Cdk phosphorylation sites, and substituting the essential CDC28 gene with two recessively lethal versions, one containing an early stop codon and another one inactivating Cdc28 kinase activity. We also tested a gene replacement of unknown phenotypic consequences: replacing the non-essential CLB3 B-type cyclin with a version lacking its destruction box.
Introduction
Exact gene replacement is an extremely useful method for establishing physiological relevance of a protein or a protein modification. In yeast, the conventional method involves a two-step integration-excision reaction, with initial duplicative insertion with URA3 or another counterselectable marker between duplicated regions (one of which contains the desired mutation) (Boeke et al., 1987; Scherer and Davis, 1979). Selection against the intervening marker selects homologous recombinants between the flanking duplications; depending on the location of recombination, wild-type or mutant can be generated. This method therefore relies on selection of rare recombinants, the frequencies of which depend on spontaneous recombination and therefore cannot be predicted in advance. If a given replacement results in inviability or poor growth, this could go undetected since the only positives that can be obtained might have acquired second-site suppressors during isolation, and the argument for complete lethality of the replacement proceeds solely from negative evidence. For potentially recessive lethal replacements, the recombination can be performed in diploids, followed by recovery and analysis of meiotic segregants carrying the replacement. This method will not work for dominant gain-of-function lethal replacements, or for replacements that block meiosis. Remedying the latter problem requires development of a means to make lethality conditional. For example, the lethal removal of the destruction box of the main mitotic cyclin CLB2 could be recovered upon conditional overexpression of the Sic1 cyclin kinase inhibitor (Wäsch and Cross, 2002). In another example, lethal removal of the destruction box of the PDS1 securin could be accomplished by modest conditional overexpression of the target of securin inhibition, the separase ESP1(Lu and Cross, 2009). Introduction of unphosphorylatable Cdh1 (causing lethal uncontrolled proteolysis of many targets) could be accomplished by overexpression of the stoichiometric Acm1 inhibitor of Cdh1, or by using hypomorphic alleles of components of the anaphase-promoting complex target of Cdh1 (Martinez et al., 2006; Robbins and Cross). Such cases require some guesswork as well as considerable knowledge of the system under consideration.
Null phenotypes for essential genes can be determined by the conventional ‘runout’ assay, where an essential gene is placed under control of a regulated promoter, which is then shut off, and the fates of cells ultimately depleted for the product are examined. This procedure can suffer from the requirement for mis- or over-expression of the gene before shutoff, as well as from ambiguities resulting from the inevitable expression of at least some of the regulated gene even after shutoff.
The method presented here bypasses all of these difficulties. A partial copy of the ultimate replacement allele is followed by an HO cut site and URA3, and integrated adjacent to the native gene. In such transformants, no phenotype is expected since the replacement allele is truncated and the native gene is expressed normally. Induction of GAL-HO (in a MATa-inc background so that mating-type locus cutting is prevented (Mascioli and Haber, 1980)) results in rapid, near-quantitative cleavage of the HO site, promoting gene conversion that eliminates the intervening URA3 and resulting (depending on ‘crossover’ point) in wild-type or mutant gene replacements. (In the case of multiply mutated replacements, ‘crossover’ can separate individual mutations; this can be an advantage, or can be bypassed in some cases by specific setup in the initial strain). This method produces a high yield (up to ~80% in some cases) of mutant gene replacement, with at least 90% viability of induced cells throughout, so the ‘extragenic suppressor’ problem is eliminated since the native gene replacement phenotype can be determined immediately, in multiple isolates, and in direct comparison to exactly isogenic wild-type restorations. In contrast to promoter shutoff procedures, wild-type gene expression is normal before gene replacement, and after replacement, the native gene is gone, so background (‘promoter off’) expression is irrelevant.
The physiological role of the HO endonuclease is to cut the mating type locus at a site in MATa or MATalpha specific sequences (Haber, 1992). The cut locus finds the silent copy of the other mating type locus elsewhere on the chromosome, and gene conversion results in replacement of the MATa or MATalpha information at the mating-type locus with the other mating type. This mechanism requires common homologous sequences to the left and right of MATa and MATalpha at expressed and silent chromosomal positions.
If homology is missing on one side, then gene conversion without any deletion is no longer possible by homologous recombination. Instead, with the right orientation of material, the HO cut is repaired by a combination of gene conversion and deletion of intervening material, as was shown more than twenty years ago using tandem copies of URA3 and ura3-52 (Rudin and Haber, 1988). Here we characterize the use of the method to construct and analyze potentially lethal gene replacements. The key advantage of this method compared to conventional gene replacement methods is its very high efficiency, such that almost all cells carry out recombination in a matter of hours with negligible loss of viability. We describe the application of this method in three different cases, to illustrate the generality and utility of the procedure.
Materials and Methods
Yeast strains and plasmids. All strains were W303-congenic. Strain DY3023 (MATa-inc LYS2::GAL-HO) was a gift from J. Nickoloff. MATa-inc and LYS2-GAL-HO were crossed from DY3023 into the W303 background by 6 backcrosses with tetrad analysis. GAL-HO could be followed in MATalpha segregants by poor viability on galactose medium, and could be followed in either mating type by ability to switch from Ura+ to Ura- upon growth in galactose medium in segregants containing an HO-targeted URA3-containing cassette. MATa-inc could be followed by vigorous growth on galactose in segregants deduced to contain GAL-HO. A 117-bp fragment containing the MATa HO cut site (Rudin and Haber, 1988) was cloned in a series of plasmids based on RS406 (integrating URA3-containing plasmid (Sikorski and Hieter, 1989) containing truncated or full-length mutant genes. Where necessary, a unique restriction site was placed by mutagenesis in the promoter region of the genomic insert, to target integration 5′ of the target gene. Plasmid maps are shown in Fig. 1. Sequence files are available upon request.
Figure 1.
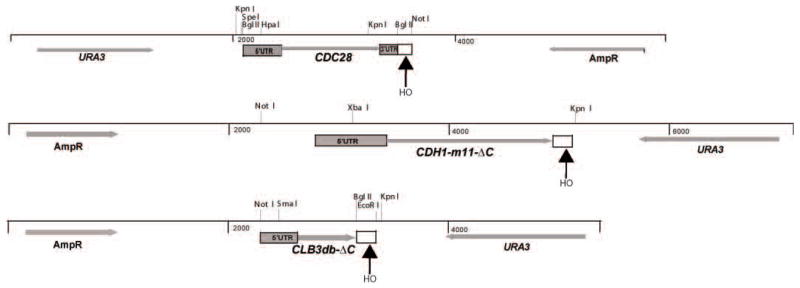
Plasmids used in this study. The backbone was RS406 in all cases. A 113-bp HO-site-containing fragment (open box) was inserted at the indicated locations, either downstream of 3′UTR (CDC28 plasmid) or truncating open reading frame to inactivate the (potentially) dominant-lethal coding sequence. The HpaI site and the SmaI site in the 5′ UTR of CDC28 and CLB3 were introduced by mutagenesis, and these sites, and the natural XbaI site in the CDH1 5′UTR, were used for targeting integration.
Gene replacement. For synchronous gene replacement, log phase cultures were pregrown in raffinose medium lacking uracil. Raffinose was used to allow rapid induction of GAL-HO by galactose addition; uracil dropout medium was employed because otherwise a significant population of Ura- popouts accumulated in some experiments even before GAL-HO addition, complicating results. Galactose was added to 3% final concentration, and at intervals cells were plated on solid YEP-glucose medium (stopping GAL-HO induction) and separated to known positions on the plate with a microdissector; at the same timepoints, approximately 10^8 cells were harvested to make yeast genomic DNA for PCR analysis. In other experiments where synchrony was less important, a fresh overnight plate stock on glucose-uracil dropout medium was transferred to YEP-galactose solid medium. After ~4 hrs, budded cells were separated from the cell population with a microdissector, and after another ~3 hours, cells that had divided and rebudded were identified and separated to fresh positions on the plate, and allowed to form colonies. This procedure was carried out to be sure that all isolated colonies had been through at least one complete division on galactose medium, which lowered the likelihood of sectored colony fates due to late or asynchronous GAL-HO induction.
Results
CDC28
CDC28 encodes an essential cell-cycle-regulatory cyclin dependent kinase. This kinase functions at multiple points in the cell cycle. Confusingly, most temperature-sensitive alleles are reported to arrest predominantly in G1 before Start (Lorincz and Reed, 1986; Reed, 1980), while an analog-sensitive allele arrests after Start at low analog doses, and at multiple points in the cell cycle at high analog doses (Bishop et al., 2000). A single temperature-sensitive allele, cdc28-1N, arrests late in the cell cycle (Piggott et al., 1982).
We constructed several alleles of CDC28 followed by a 113-bp HO cut site from MATa (Rudin and Haber 1988). This construct was integrated to the left of wild-type CDC28 with intervening URA3 by conventional duplicative integration (Fig. 2) in a GAL-HO MATa-inc strain (MATa-inc is not cleavable by HO, so that the only cleavable HO site in this strain is the one 3′ to CDC28).
Figure 2.

Synchronous gene conversion of CDC28(mutant)-HO-URA3-CDC28 to CDC28 or CDC28(mutant). MATa-inc GAL-HO strains with the integrated structure shown at bottom were pregrown in synthetic raffinose medium lacking uracil, GAL-HO was induced, and cultures processed as described in Methods. Top left: colonies from single viable cells isolated at the indicated timepoints were grown on YEPD, then assayed for a Ura+ phenotype. Top right: microscopic analysis of microcolonies derived from single cells micromanipulated on YEPD from a CDC28(mutant)-HO-URA3-CDC28 induced for GAL-HO for the indicated time, exhibiting a wild-type morphology, a cdc28-STOP-arrested morphology (few large round cells; see Fig. 3), and a sectored morphology (few large round cells on the edge of a microcolony of wild-type cells). Bottom: PCR analysis of two strains, one with CDC28(STOP) (introduced stop codon marked by a BglII site), and one with CDC28-rsc1 (rsc1 mutation marked with an SfoI site). Middle: PCR with oligos priming in the indicated locations. The right-hand oligonucleotide primes in the HO-cut-site fragment to the left of the HO cut site. Disappearance of product is indicative of processing/loss of DSB-adjacent sequences after HO cleavage. Bottom: the left-hand oligonucleotide primes in two places in the starting construct, in the duplicated CDC28 5′ UTR sequence. The right-hand oligonucleotide primes in sequences in the CDC28 3′ UTR that are not duplicated. Appearance of digestion products (arrows) with BglII or SfoI are indicative of exact gene replacement with the mutant CDC28.
One cdc28 allele contained two tandem stop codons introduced at codons 57-58, marked with a BglII site (‘cdc28-STOP’). Another allele was the cdc28-rsc1 mutant (R159G) characterized previously (marked here with an SfoI site). R159 is a highly conserved residue in the protein kinase superfamily. Surprisingly, though, Cdc28-rsc1 apparently had biological activity in combination with the G1 cyclin Cln2 but not with the mitotic cyclin Clb2 (Levine et al., 1998). Despite functioning genetically with Cln2, Cdc28-rsc1 exhibited essentially no in vitro kinase activity in association with either Cln2 or Clb2, and cells containing cdc28-rsc1 in place of wild-type CDC28 were completely inviable (Levine et al., 1998). The R159G mutation was marked with an SfoI site. Another construct contained the cdc28-1N temperature-sensitive allele (Piggott et al., 1982) (P250L), and a control construct contained wild-type CDC28.
Incubation of these strains on galactose medium to induce GAL-HO, followed by selection of single cells by microdissection after ~6 hrs incubation, gave the following results when plates were examined at ~24 hours. The wild-type CDC28 transformants all exhibited normal colony formation. The cdc28-1N transformants (incubated at 23 degrees) exhibited about 50% normal colonies and 50% colonies with elongated cell morphology; the latter colonies all proved temperature-sensitive and therefore are presumed to be cdc28-1N replacements. The rsc1 and STOP alleles both gave approximately equal proportions of apparently wild-type colonies (presumably wild-type recombinants) that grew to full size, and inviable microcolonies, consistent with the essentiality of CDC28 and the complete inactivation of its mitotic function by the rsc1 mutation. The similar recovery of wild-type and mutant colonies suggests that recombination occurs with similar frequencies to the right and left of the mutation sites; this roughly corresponds to physical distance. We did not attempt to accumulate sufficient data to determine how exact this match was.
Interestingly, the morphology of these microcolonies was strikingly different between the two mutants, with the rsc1 mutants exhibiting strong bud hyperpolarization (as would be expected for effective interaction with the G1 cyclin Cln2 but not with mitotic cyclins, (Levine et al., 1998) while the STOP mutants formed small clusters of very large and round cells (Fig. 3). The hyperpolarized cdc28-rsc1 microcolonies were highly clumped and could not be pulled apart with a microneedle, but gave a clear impression of containing more cells than the cdc28-STOP microcolonies; the cdc28-STOP microcolonies could be readily pulled apart, and turned out to be a mix of budded and unbudded cells (in one experiment, 60/85 cells (71%) were large and unbudded; 25 were large cells with buds of varying morphology.)
Figure 3.

Morphology of CDC28 gene replacements. A: ~18 hrs after galactose induction. cdc28-STOP mutant microcolonies contain ~4–10 large round cells; cdc28-rsc1 cells contain an apparently larger number of extremely hyper-polarized cells. B. 3 days incubation. Both cdc28 mutant replacements are completely inviable: compare microcolony size of a wild-type replacement (top) to the mutants (below). Limited proliferation and/or hyperpolarized bud growth continues for longer in the cdc28-rsc1 microcolonies than the cdc28-STOP microcolonies. C: cdc28-STOP microcolonies, in a wild-type or cdh1 mutant background, were microdissected away from each other and compared to wild-type cells. cdc28-STOP cells are extremely large and round; a minority of cdc28-STOP CDH1 cells are budded, and this budded proportion decreases in a cdh1 background. D: clb2 cdc28-1N synthetic lethality.
Thus, this simple depletion assay suggests biological activity of Cdc28-rsc1 with G1 cyclins but not with mitotic cyclins, as was concluded previously based on more complicated genetic assays (Levine et al., 1998). The arrest phenotype of the STOP mutant largely resembles that of G1-arresting cdc28 temperature-sensitive alleles (although we lack a convenient way to measure their DNA content directly), and suggests the simple hypothesis that overall lowering of Cdc28 protein or overall activity might first reduce G1 cyclin-Cdc28 activity below a critical threshold, so that cells that have executed Start can divide with residual mitotic cyclin-Cdc28 activity, but then arrest before the succeeding Start.
Switches to cdc28-STOP allowed only a few cell divisions before all switched cells arrested irreversibly (Fig. 3). We found previously that there is about a five-fold excess of Cdc28 over cyclins in growing cells (Cross et al., 2002), suggesting that the critical level may be approximately the point when there is not enough free Cdc28 to bind to most or all available cyclin (Cdc28 degradation is unlikely to play a major role since it is a stable protein).
Bishop et al. (2000), using an analog-sensitive cdc28-as1 allele, reported that low analog concentrations resulted in G2/M arrest with long, hyperpolarized buds, while high analog concentrations resulted in arrest as large round unbudded cells with G1 DNA content. They drew the inference from this result that the cell cycle step with the highest requirement for Cdc28 activity was mitosis, driven by mitotic cyclin-Cdc28 complexes, while the budding requirement for Cdc28, driven by G1 Cln-Cdc28 complexes, required less Cdc28 activity and was therefore harder to inhibit. This result contrasts in an interesting way with the cdc28-STOP experiments reported here, in which most cells, budding fails before mitosis upon depletion of wild-type Cdc28.
To determine the role of mitotic cyclins in the phenotype of cdc28-STOP arrest, we carried out cdc28-STOP replacement in a cdh1 background, which allows mitotic cyclin accumulation in G1 cells (Schwab et al., 1997; Visintin et al., 1997). cdh1 deletion increased the proportion of large round unbudded cells in the cdc28-STOP microcolonies to 91% (47/50) (Fig. 3). Mitotic cyclins present in G1 may compete effectively for the low amount of Cdc28 present, blocking Start.
cdc28-1N is synthetically lethal even at low temperatures with deletion of the major mitotic cyclin CLB2, and reciprocally, CLB2 overexpression rescues cdc28-1N temperature-sensitivity (Surana et al., 1991). We examined the result of replacement of CDC28 with cdc28-1N in a clb2 background, using the same assay as above. Strikingly, while CDC28-wt controls with or without CLB2 yielded healthy microcolonies with high efficiency, about 50% of colonies derived from cdc28-1N-HO-CDC28 clb2 strains formed only a few cells with a remarkably hyperpolarized bud phenotype (Fig. 3). These are presumably the cdc28-1N clb2 double mutants, since none of the viable colonies obtained from the clb2 background were temperature-sensitive, while ~50% of colonies derived from cdc28-1N-HO-CDC28 CLB2 controls were temperature-sensitive.
We characterized genomic DNA from cells containing the cdc28-STOP and cdc28-rsc1 mutations (marked with BglII and SfoI sites respectively) followed by the HO cut site, during a time-course of GAL-HO induction (note that since switching CDC28 alleles lacks phenotypic effect for at leas 6 hours due to phenotypic lag (presumably the need to dilute out wild-type protein by growth and division), DNA of wild-type and mutant cells should be equally represented in this experiment). Using an oligonucleotide priming in the CDC28 promoter with another priming to the left of the HO cut site, we observed gradual loss of product over 4–6 hours of galactose induction, suggesting time required for processing and recombination (Fig. 2). Simple cutting with HO, which occurs within a few hours (see below), would not be sufficient to eliminate this product. We scored recombination using oligonucleotides that amplified the entire CDC28 gene (with the right-hand oligo outside of the duplicated DNA in the tandem integration), and digesting PCR products with BglII or SfoI to detect recombination between site (marking the STOP mutation) or SfoI site (to detect the rsc1 mutation). Cut product was detectable beginning after 2–4 hrs of galactose induction (Fig. 2). PCR products derived from DNA to the right of the HO cut site, in non-homologous plasmid sequences fated to be lost during recombination, disappeared with approximately similar kinetics (data not shown).
These results overall indicate that after initial cutting with HO, processing and recombination occurs over at least a 4–6 hour period.
Serial dilutions of the cultures were performed onto rich glucose medium (turning off continued HO expression) at the same timepoints. After colonies were grown, the proportion that were Ura+ was determined. From platings at 2 hrs about 50% of the colonies were Ura-, and almost all were Ura- from platings at 6 hrs (Fig. 2). Thus ‘commitment’ to a Ura- fate preceded actual recombination by some hours, and probably reflects the time of HO cutting itself. A small residual population of cells does not become Ura- even after long galactose incubation; we have not analyzed these events further.
Isolation of single cells from the cultures at various timepoints, followed by microscopic observation of their fates after an overnight incubation, showed that by 6 hrs 50% or more of cells were committed to formation of small inviable microcolonies with cell morphologies indicating switch to the lethal cdc28 allele (Fig. 2); in contrast, digestion of PCR product with diagnostic enzymes showed that no more than 1/3 of the product was recombinant by 6 hrs. At early times, a small population of sectored colonies were observed, consisting of a minority of large arrested cells and a majority of small wild-type cycling cells, presumably reflecting independent switches occurring after cell isolation. Results are shown for cdc28-STOP; similar results were obtained for cdc28-rsc1.
Quantitatively similar findings were obtained with analysis of genomic DNA from the other two cases analyzed below (CDH1-11m and CLB3-db; see below), indicating that these conclusions are likely general: the HO cut site is rapidly cleaved, within a few hours of galactose addition. Cells are then ‘committed’ to recombination, which occurs roughly concomitant with loss of non-homologous sequences over the succeeding ~4–6 hrs, and probably longer in some cells.
Thus the reaction is somewhat slow, but ultimately highly efficient, since when all recombinants are potentially viable (as with a wild-type CDC28 control, or CLB3-db; see below), individual cells form viable colonies with at least 80% efficiency. Therefore, observation of a high frequency of a lethal phenotype in these cells can be attributed reliably to the replacement allele.
CDH1-11m
Recent work (Robbins and Cross 2010) demonstrates that exact gene replacement of CDH1 with CDH1-11m, a version with all Cdk sites mutated (Zachariae et al., 1998), is lethal due to unrestrained APC-Cdh1 activity. CDH1-11m yields completely penetrant dominant lethality in heterozygous diploids even as an exact gene replacement (Robbins and Cross, 2010), which makes it impossible to handle by standard genetic methods.
We cloned CDH1-11m, truncated near the C-terminus of the coding sequence (eliminating essential C-terminal sequences, so that this protein was non-functional) adjacent to an HO cut site, and integrated it to the left of wild-type CDH1 with intervening URA3 (Fig. 4). This construct yielded a normal phenotype since CDH1-11m was truncated and non-functional, and a full-length wild-type CDH1 gene was still present.
Figure 4.

Analysis as in Figure 2, with CDH1-m11-delC-HOcs-URA3-CDH1 strains. The extreme hyperpolarized lethal phenotype of CDH1-m11 cells (Robbins and Cross, 2010) is recovered with about 50% efficiency. PCR analysis: top: use of an oligonucleotide priming to the right of the HO cut site shows rapid and efficient cleavage. Bottom: use of an oligonucleotide that is allele-specific to the 9th phosphorylation site mutation (‘*’) in CDH10-m11 (Zachariae et al., 1998) combined with an oligonucleotide priming outside of the duplicated N-terminal coding sequence of CDH1-m11 demonstrates effective recombination starting at ~4 hrs of galactose induction.
Cultures of this strain were grown in raffinose medium, and induced with galactose for varying times. At intervals, genomic DNA was prepared, and in addition, serial dilutions of the culture made on YEPD (rich glucose) medium, to repress further HO expression, and incubated until colony formation. Finally, single cells from the culture at each time point were micromanipulated to solitary positions on a YEPD plate, and assayed microscopically at intervals.
The single cell analysis (Fig. 4) showed that ~50% of cells isolated from the culture at 4–6 hrs after galactose addition were inviable, showing the characteristic hyperpolarized bud growth phenotype of CDH1-11m (Fig. 4; some variability in microscopic phenotype was noted). Since this phenotype requires at least 5 Cdk site mutations starting from the N-terminus (Robbins and Cross, unpublished), this result suggests that ~50% of recombination junctions occurred beyond site 5 at aa 169, indicating a junction within the 1 kb distance from this site to the end of the insert; the remaining 50% of recombinants presumably recombined to the left of this position, in the remaining 0.5 kb of the insert. We assume, based on previous findings on HO-induced gene conversion, that a continuous conversion tract is the most likely outcome (Sweetser et al., 1994) (that is, it is unlikely that wild-type sites are interspersed between mutant sites). Since there is no homology to the right of the HO site in these constructs, all homologous recombination events are expected to result in deletion of the intervening plasmid and URA3 sequences. A small, variable proportion (~20%) of micromanipulated cells were inviable without the characteristic polarized cell morphology of CDH1-11m cells, suggesting a low but detectable rate of loss of viability through the procedure. Similar sporadic inviability was observed in the CDC28 experiments above.
PCR analysis of genomic DNA from these strains (Fig. 4) showed that efficient HO cutting occurred within ~2–4 hrs of galactose addition. Use of an allele-specific primer overlapping the S169A, T172A mutations in CDH1-11m allowed determination of the timecourse of recombination, which required longer than HO cutting, at least 4–6 hrs, as with the CDC28 experiments above. We could use such PCR products to map apparent recombination junctions, since some of the CDK site mutations were marked with restriction sites. A majority of PCR product obtained with the T172A-specific primer was digestible with SfoI, but only a minor proportion was digestible with ApaI (data not shown); these two sites mark the S227A and S436A mutations respectively. This result suggests that for recombination initiating to the right of T172A, the site of crossover was usually but not always between S227A and S436A.
These and other results show that multiple intervals across the duplicated CDH1 sequences are available for recombination, with no obvious preference for any scorable interval, consistent with the results with CDC28 above.
In conclusion, this method allows immediate direct determination of the lethal phenotype of a CDH1-11m exact gene replacement, without the need for interpretation of failure to recover a class of recombinants, or for developing a method for making lethality conditional. Dominant lethality of this replacement (Robbins and Cross, 2010) precludes construction of the replacement in a heterozygous diploid followed by sporulation. Previously, we found that either the cdc23-1 temperature-sensitive allele at permissive temperature, or overexpression of the Acm1 inhibitor of Cdh1, allowed recovery of CDH1-11m replacements; these methods required considerable knowledge of the system.
These results are complicated by recombination events that install only a subset of the multiple Cdk site mutations. This problem could be avoided by installing the recombination cassette adjacent to a disrupted cdh1 allele with all of the sequence encoding the Cdk site mutations removed (e.g., replaced by an insertional marker), since CDH1 is non-essential.
CLB3-db
Deletion of the destruction box from the coding sequence of CLB2, the major yeast mitotic cyclin, is lethal (Wäsch and Cross, 2002). Clb3 is almost as abundant as Clb2, and these cyclins are at least partially redundant in function. Therefore, it seemed plausible that deletion of the CLB3 destruction box might also be lethal. Thus, direct selection of CLB3-db replacements might either be simply unsuccessful, or if recombinants were obtained they might also contain unidentified second-site suppressors. We applied the HO cut site strategy to address this problem. A fragment containing the promoter and 5′ end of the CLB3 coding sequence including a destruction box deletion, appended to an HO cut site, was integrated adjacent to wild-type CLB3 (Fig. 5), in a MATa-inc GAL-HO strain. These initial transformants should produce only normal Clb3, and a truncated non-functional version of Clb3-db. As with the CDH1-11m experiments above, GAL-HO induction gave rapid cleavage of the CLB3-associated HO cut site (Fig. 5). Priming PCR with one oligo upstream of the destruction box deletion and one oligo in the 3′ region of CLB3 coding sequence initially amplified only the wild-type copy (as detected by restriction enzyme digestion of PCR products to yield a small band whose size was diagnostic of presence or absence of the destruction box sequence), but following galactose induction, a high yield of CLB3-db mutant sequence was detected by this PCR assay (data not shown). As with the CDH1-11m experiments, >80% of cells formed Ura- colonies when plated on YEPD after ~4 hrs of galactose induction. Across many experiments, 50–80% of these viable Ura- colonies proved to contain the CLB3 destruction box deletion when tested using allele-specific primers (Fig. 5), in most cases in pure form (a minority of colonies amplified with both allele-specific primers, most likely indicative of late switching and colony sectoring, as was observed microscopically with the lethal generation of cdc28 and CDH1-m11 mutant sectored colonies above). Single cell micromanipulation after brief galactose incubation showed no preferential inviability or slow growth of colonies that proved to contain the CLB3-db mutation compared to CLB3-wt recombinants (data not shown). Thus, CLB3-db apparently results in no growth defect, even without allowing any time for accumulation of second-site suppressor mutations.
Figure 5.
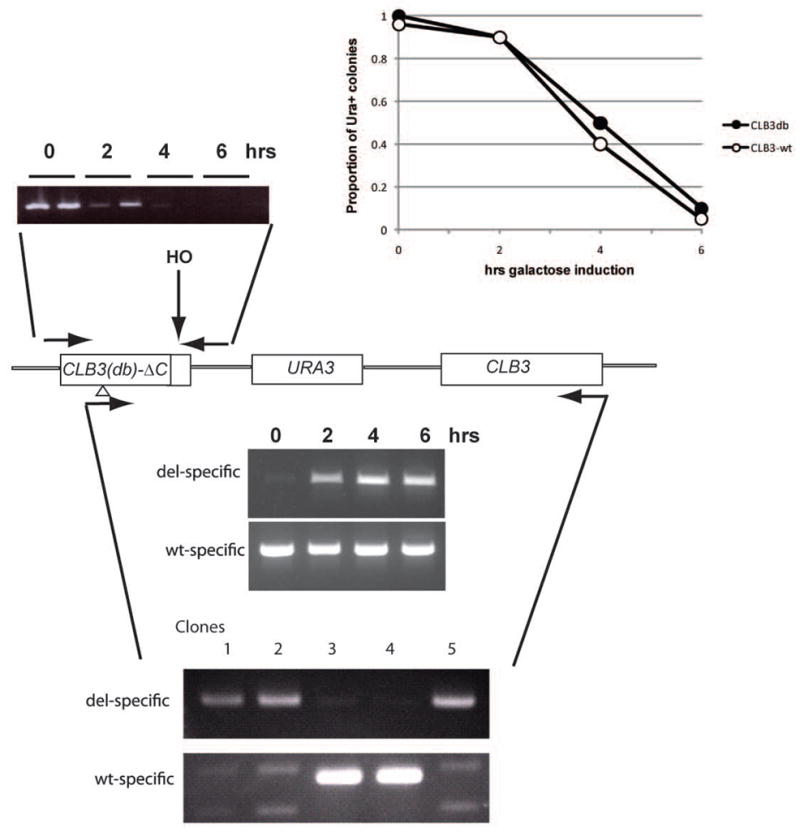
Analysis as in Figure 2, with a CLB3db-delC- HOcs-URA3-CLB3 strain and a CLB3wt-delC- HOcs-URA3-CLB3 control. PCR: top: rapid cleavage of the HO cut site, as in Fig. 5; middle: timecourse of recombination with CLB3db-delC- HOcs-URA3-CLB3 strain, using left-hand allele-specific ‘db’ or ‘wt’ oligonucleotides (priming across the 27-nucleotide db deletion, or within the deleted sequences, respectively), and a right-hand oligonucleotide in C-terminal CLB3 coding sequence not present in the duplicated sequences. Recovery of CLB3-db recombinants started at 2 hrs of galactose incubation; bottom: viable colonies recovered from the CLB3db-delC- HOcs-URA3-CLB3 strain tested for presence of CLB3db or CLB3wt using the same oligonucleotides, demonstrating recovery of pure CLB3-wt or CLB3db colonies respectively. Typically, ~80% of plated single cells were viable, and ~70% of viable colonies contain the CLB3-db mutation. There was no evident growth disadvantage of CLB3-db colonies compared to wild-type. Further characterization of CLB3-db will be reported elsewhere.
The reason for the surprising full viability of CLB3-db exact gene replacements obviously cannot be determined without further work; for the present, the point is that the very high and efficient recovery of these replacements shows they are fully viable without any second-site suppressors; this conclusion would be difficult to reach by other means. In principle, this determination might be made by tetrad analysis crossing CLB3-db replacements to wild-type, to try to follow segregation of suppressors from CLB3-db; however, CLB3-db dominantly prevents effective meiosis, yielding spores that are nearly all inviable (data not shown). The present method allows easy combination of CLB3-db with other mutations, though, since a simple cross can recombine some mutation of interest with the CLB3-db-delC-HO-CLB3 cassette, GAL-HO, and MATa-inc, and recombination then induced with galactose to generate full-length CLB3db. This method has allowed us to determine genes that are required for full viability of the CLB3db background (F.C., K.P. and A. Singhal, unpublished data).
Discussion
We characterize a method for exact endogenous gene replacement that allows recovery of replacements at efficiencies of ~50–80%, without the need for selection or long incubations. The method allows direct determination of phenotypes of lethal replacements without the need to devise a strategy for conditional growth, even for dominant lethal gain of function mutations such as CDH1-11m. In our previous work, we found that GAL-SIC1 expression could make CLB2-db replacements viable, and similarly for GAL-ESP1 and PDS1-db, and GAL-ACM1 for CDH1-11m ((Lu and Cross, 2009; Robbins and Cross, 2010; Wäsch and Cross, 2002). However, finding such conditional constructs required considerable prior knowledge of the function of the stabilized lethal protein, and further, the conditional strains were not wild-type under permissive conditions (e.g., elongated G1 for GAL-SIC1, and poor viability for GAL-ESP1 cells unless a very attenuated GAL1 promoter was used).
Viable exact replacements have been constructed by integration-excision, selecting the excision events using 5-FOA to kill URA3+ cells and allow colony formation by rare ura3- popouts. This strategy has the defect that since popouts are rare and of uncertain initial frequency, one can only analyze clones that emerge, and the possibility exists that these have accumulated modifiers to allow enhanced growth. This idea can be tested genetically but in general will remain a worrisome background issue for such replacements. The present strategy eliminates this problem, at least for analysis of phenotypes that can be scored microscopically. The increasing utility of fluorescent proteins has widely broadened the number of such phenotypes. For example, we have found in preliminary experiments that the spindle phenotype resulting from depletion of essential kinetochore components can be analyzed with this method, by replacing the kinetochore component with a null allele using GAL-HO induction while carrying out time-lapse microscopy on the microcolonies monitoring GFP-tubulin to detect spindles (J. Rosenberg, unpublished).
This method could have considerable utility in characterizing the results of blocking post-translational modifications of many proteins, without detailed knowledge of the protein’s function and without overexpression. This could be very useful for the large number of Cdk phosphorylation target proteins recently identified, as well as for the diverse set of proteins subject to APC-mediated ubiquitination and subsequent proteolysis. The consequence of blocking these modifications on most individual targets is unknown; if the modification is required for viability, the present method could provide a rapid first look at the phenotype, with detailed characterization possible if single-cell microscopic assays can be devised.
This method allows easy manipulation and phenotypic characterization of lethal loss-of-function mutations as well as lethal gain-of-function mutations such as CDC1-11m (for which exact gene replacement is generally not feasible by conventional methods), and dominant sporulation-blocking mutations such as CLB3-db (obviously, a dominant sporulation block is a serious obstacle to conventional genetic analysis).
This method is certainly not high-throughput, but it appears highly reliable (since it worked with high efficiency with all three of the genes we tested), and avoids multiple ambiguities and difficulties associated with more standard methods. The method relies on well-understood mechanisms of gene conversion repair after double-stranded break induction, so it is very likely that the structure of the resulting gene replacements will almost always be a high-fidelity replacement of exactly the desired structure.
Plasmids containing the 110-bp HO cut site fragment in a multiple cloning site will be deposited with Addgene, to make this technology generally available.
Acknowledgments
Thanks to J. Nickoloff for strains, and to J. Rosenberg and A. Singhal for initial testing of the system. This work was supported by PHS grants GM47238 to F.C.
References
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- Cross FR, Archambault V, Miller M, Klovstad M. Testing a mathematical model of the yeast cell cycle. Mol Biol Cell. 2002;13:52–70. doi: 10.1091/mbc.01-05-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. Mating-type gene switching in Saccharomyces cerevisiae. Trends Genet. 1992;8:446–452. doi: 10.1016/0168-9525(92)90329-3. [DOI] [PubMed] [Google Scholar]
- Levine K, Oehlen LJ, Cross FR. Isolation and characterization of new alleles of the cyclin-dependent kinase gene CDC28 with cyclin-specific functional and biochemical defects. Mol Cell Biol. 1998;18:290–302. doi: 10.1128/mcb.18.1.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz AT, Reed SI. Sequence analysis of temperature-sensitive mutations in the Saccharomyces cerevisiae gene CDC28. Mol Cell Biol. 1986;6:4099–4103. doi: 10.1128/mcb.6.11.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cross F. Mitotic exit in the absence of separase activity. Mol Biol Cell. 2009;20:1576–1591. doi: 10.1091/mbc.E08-10-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JS, Jeong DE, Choi E, Billings BM, Hall MC. Acm1 is a negative regulator of the CDH1-dependent anaphase-promoting complex/cyclosome in budding yeast. Mol Cell Biol. 2006;26:9162–9176. doi: 10.1128/MCB.00603-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioli DW, Haber JE. A CIS-Acting Mutation within the MATa Locus of SACCHAROMYCES CEREVISIAE That Prevents Efficient Homothallic Mating-Type Switching. Genetics. 1980;94:341–360. doi: 10.1093/genetics/94.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott JR, Rai R, Carter BL. A bifunctional gene product involved in two phases of the yeast cell cycle. Nature. 1982;298:391–393. doi: 10.1038/298391a0. [DOI] [PubMed] [Google Scholar]
- Reed SI. The selection of S. cerevisiae mutants defective in the start event of cell division. Genetics. 1980;95:561–577. doi: 10.1093/genetics/95.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JA, Cross FR. Requirements and reasons for effective inhibition of the anaphase promoting complex activator CDH1. Mol Biol Cell. 2010;21:914–925. doi: 10.1091/mbc.E09-10-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin N, Haber JE. Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol. 1988;8:3918–3928. doi: 10.1128/mcb.8.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci U S A. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, Nasmyth K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 1991;65:145–161. doi: 10.1016/0092-8674(91)90416-v. [DOI] [PubMed] [Google Scholar]
- Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA. Fine-resolution mapping of spontaneous and double-strand break-induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol Cell Biol. 1994;14:3863–3875. doi: 10.1128/mcb.14.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Wäsch R, Cross FR. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418:556–562. doi: 10.1038/nature00856. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]