

Abstract
Emerging evidence reveals that pattern-recognition receptors (PRR), Toll-like receptors (TLR) and Nucleotide-binding oligomerization domain proteins (NOD) mediate both infection-induced and sterile inflammation by recognizing pathogen-associated molecular patterns and endogenous molecules, respectively. PRR-mediated chronic inflammation is a determinant for the development and progression of chronic diseases including cancer, atherosclerosis and insulin resistance. Recent studies demonstrated that certain phytochemicals inhibit PRR-mediated pro-inflammation. Curcumin, helenalin, cinnamaldehyde and sulforaphane, containing α, β-unsaturated carbonyl or isothiocyanate group, respectively, that are known to interact with free SH groups in cysteine residues, but not resveratrol (with no unsaturated carbonyl group), inhibit TLR4 activation by interfering with TLR4 receptor dimerization. Similarly, curcumin, as well as parthenolide and helenalin, but not resveratrol and EGCG, also inhibits NOD2 activation by interfering with NOD2 dimerization. In contrast, resveratrol, EGCG, luteolin and structural analogs of luteolin, specifically inhibit TLR3 and TLR4 signaling by targeting TBK1 and RIP1 in TRIF complex. Together, these results suggest that PRRs and downstream signaling components are molecular targets for dietary strategies to reduce PRR-mediated chronic inflammation and consequent risks of chronic diseases.
Introduction
Pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and Nucleotide-binding oligomerization domain proteins (NODs) detect invading pathogens by recognizing pathogen-associated molecular patterns (PAMPs), and activate innate immune responses for host defense. However, it is now well documented that these PRRs can also be activated by a variety of endogenous molecules derived from tissue injury and elicit sterile inflammation to initiate wound-healing processes. Host defense and wound healing are the key biological processes for the survival of all multicellular organisms. Emerging evidence suggests that PRRs can detect metabolic disturbance and bridge immune responses to metabolic homeostasis [1]. Such functional diversity of PRRs may be achieved by the ability of PRRs to recognize a wide variety of agonists [2]. However, such promiscuous nature of agonist specificity can make PRRs vulnerable to dysregulation leading to chronic inflammation, which in turn can promote the development and progression of chronic diseases.
Many phytochemicals have been known for their anti-inflammatory, chemopreventive, and cardioprotective properties [3-5]. An increasing number of studies also indicates that diet rich in anti-inflammatory phytochemicals may have beneficial effects in ameliorating metabolic syndrome, a constellation of abnormal cardiometabolic factors that increase risk of cardiovascular disease and type 2 diabetes [6]. However, the mechanisms by which such beneficial effects are mediated is not well understood. Recent evidence revealed that certain phytochemicals inhibit PRR activation by targeting the receptor itself or their specific downstream signaling molecules [7-13]. These results suggest the possibility that PRR-mediated inflammation and consequent risk of development of chronic disease can be suppressed by diets we consume every day. This review summarizes and discusses the significance and implication of recent findings that certain phytochemicals inhibit the activation of PRRs.
Pattern recognition receptors (PRRs) and downstream signaling pathway
TLRs and NODs are two key PRRs involved in host defense. TLRs are type I transmembrane receptors composed of extracellular leucine-rich repeat (LRR) motifs, and a cytoplasmic Toll/interleukin-1 receptor (TIR) homology domain [14-15]. Thirteen TLRs have been identified in human and mouse, which are ubiquitously expressed in various tissues [16]. TLRs detect invading pathogens by recognizing PAMPs, and are responsible for the induction of innate immune responses for elimination of the pathogens.
NODs as cytoplasmic PRRs recognize conserved moieties of bacterial peptidoglycan and activate proinflammatory signaling pathways [17]. NODs are composed of a C-terminal leucine-rich repeat (LRR) domain for ligand recognition, a nucleotide-binding oligomerization domain (NOD), and an N-terminal Caspase-recruitment domain (CARD) for the initiation of downstream signaling. NOD1 recognizes a dipeptide, γ-D-glutamyl-mesodiaminopimelic acid (iE-DAP), from most Gram-negative bacteria and specific Gram-positive bacteria. NOD2 recognizes a muramyl dipeptide, MurNAc-L-Ala-D-isoGln (MDP) from both Gram-positive and Gram-negative bacteria [18-19].
PRRs can be also activated by endogenous molecules with non-microbial origin [20-29]. These molecules are generated during tissue injuries and cell death as well as from nutrient metabolism. The downstream signaling pathways of TLRs and NODs have been extensively reviewed [14, 17, 30] and are not the focus of this review. Downstream signaling pathways of PRRs and known targets of phytochemicals are depicted in Fig. 1.
Fig.1. Identified molecular targets of phytochemicals on PRR-mediated inflammatory signaling pathway.
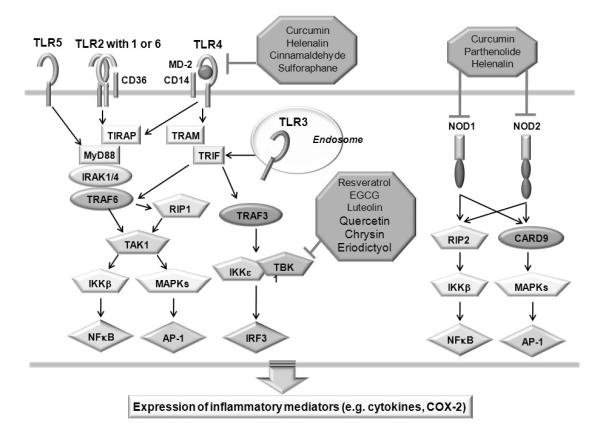
TLRs and NODs are pathogen recognition receptors (PRRs) that detect conserved molecules of pathogens. Stimulation of TLRs by ligands leads to the recruitment of adaptor molecules such as MyD88 and TRIF through the interaction of TIR domains, leading to MyD88 dependent and MyD88-independent (TRIF dependent) signaling pathway. The activation of NODs leads to the recruitment of RICK/RIP2 through CARD–CARD interactions, leading to activation of inflammatory signaling pathway. Curcumin, helenalin, cinnamaldehyde, and sulforaphane inhibit TLR4-mediated NF-κB activation by inhibiting TLR4 dimerization. Resveratrol, EGCG, luteolin, quercetin, chrysin, and eriodictyol directly inhibit TBK1 kinase activity. Similarly, curcumin, helenalin and parthenolide also inhibit NOD2 mediated NF-κB activation, presumably through inhibition of NOD2 dimerization.
PRR-mediated inflammation and chronic diseases
TLRs and NODs recognize PAMPs and activate innate immune responses that are required for host defense against invading pathogens. In addition to infection-induced inflammation, these PRRs can also be activated by endogenous molecules of non-microbial origins, leading to sterile inflammation. Dysregulation of infection-induced or sterile inflammation can increase the risks of development and progression of many chronic diseases, including atherosclerosis, cancer and insulin resistance, as depicted in Fig. 2.
Fig. 2. Bioactive phytochemicals suppress PRR-mediated inflammation, leading to decreased risks of development and progression of chronic diseases.
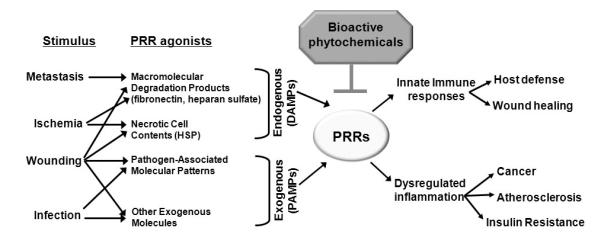
Both pathogen associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) from endogenous molecules from tissue injury and degradation can activate PRRs to culminate in innate immune responses, leading to host defense against invading pathogens, and promote tissue remodeling and wound healing. Dysregulated septic or sterile inflammation may increase the risks of chronic diseases. Bioactive phytochemicals may be beneficial in treating or preventing these chronic diseases through suppression of PRR activation.
Atheroslerosis is considered to be an inflammatory disease, in that inflammation drives the formation, progression and rupture of the atherosclerotic plaques [31]. Recent studies using atherosclerosis prone LDLr-/- or apoE-/- mice have shown that deficiency of TLR2, TLR4 or its immediate adaptor protein myeloid differentiation primary response gene 88 (MyD88) has resulted in reduction in plaque lesions [32-34]. On the other hand, recurrent administrations of TLR4 agonist lipopolysaccharide (LPS) [35] or TLR2 agonist, Pam3CSK4 (the synthetic bacterial lipoprotein mimetic) were shown to accelerate atherogenesis [32]. These results suggest that TLR4 and/or TLR2-mediated inflammation is involved in the development of atherosclerosis. Various endogenous molecules that activate TLR4 and might contribute to the development of atherosclerosis have been identified. For example, recent studies have shown that minimally oxidized LDL (mmLDL) and its active components, polyoxygenated cholesteryl ester hydroperoxides, are involved in activation of TLR4, which leads to cytoskeletal rearrangement and intracellular lipid accumulation through activation of spleen tyrosine kinase and its downstream signaling targets [36-39]. Extensive reviews on the activation of TLRs by their endogenous agonists, leading to atherosclerosis can be found elsewhere [2, 40-42]) and are not the focus of this review.
The continuous activation of TLRs by chronic infection, such as Helicobater pylori, can cause chronic inflammation leading to increased risk of gastric cancer [43-44]. TLRs are also responsible for recognizing microbial pathogens, such as Epstein-Barr virus [45], hepatitis B and C virus [46-47] and Helicobater pylori [48], which are important etiological agents of human cancer. In supporting of the roles in cancer, the association of many polymorphisms in TLRs with human cancer has been reported [49-50]. Moreover, dysregulation of sterile inflammation during wound healing can cause chronic inflammation providing the microenvironment that can promotes pathogenic processes including tumorigenesis. Certain types of cancer are considered as wounds that do not heal [51]. The activation of TLRs and development of cancer have also been extensively reviewed [52-54] and therefore are not the focus of this review.
Even in absence of infection or overt tissue injury, low grade chronic inflammation is known to be associated with obesity and insulin resistance. Many inflammatory signals are known to impair insulin receptor signaling leading to insulin resistance. Serine kinases Interleukin-1 receptor-associated kinase (IRAK-1), IkappaB kinase β (IKKβ) and c-Jun N-terminal kinase (JNK) inhibit insulin signaling by phosphorylating Insulin Receptor Substrate 1 (IRS-1) on serine residues, which in turn impedes the normal association of IRS-1 with insulin receptor, thereby impairing downstream propagation of insulin signaling [55-57]. Since IRAK-1, IKKβ and JNK are the downstream components of TLR signaling pathways, it is conceivable that the activation of TLRs would lead to impairment of insulin signaling. In supporting this notion, mice lacking TLR4 were protected from lipid infusion-induced suppression of whole body glucose metabolism and insulin signaling in muscle [58]. Mice lacking TLR2 were protected from high fat feeding-induced adiposity, insulin resistance and other metabolic abnormalities [59]. Saturated fatty acids (SFAs) have been suggested to be the endogenous agonists for TLR4 and TLR2 in vivo. Previous work have shown that SFAs activate TLR4 or TLR2 (dimerized with TLR6 or TLR1) in murine macrophage RAW264.7 cells [26-27], myotubes C2C12 cells [60] and 3T3-L1 adipocytes [58]. Together, these results support the role of TLRs in the development of obesity and insulin resistance. Therefore, the dietary factors which inhibit TLR activation may improve insulin sensitivity by alleviating TLR-mediated impairment of insulin signaling.
The significance of NOD1 and NOD2 in immune responses is evident from the linkage of their mutations with inflammatory diseases in humans and the increased susceptibility of NOD1-/- and NOD2-/- mice to gastrointestinal bacterial infections. While mutations in NOD1 have been associated with increased susceptibility to asthma and inflammatory bowel disease [61-62], Mutations in NOD2 have been linked to susceptibility to inflammatory granulomatous disorders, such as Crohn’s disease [63-64], Blau syndrome [65] and early on-set sarcoidoisis [66]. NOD1-/-mice are more susceptible to H. pylori oral infections with higher bacterial burden and mortality rates than wild-type [67]. On the other hand, NOD2-/- mice were shown to display increased bacterial loads when infected with L. monocytogenes via the oral route, compared to their wild-type counterparts [68]. Therefore, NODs play a critical role in host defense against bacterial infection. Dysregulation of NOD signaling pathways may lead to the development of certain chronic diseases. Recent study revealed that bioactive phytochemicals inhibit the activation of PRRs by targeting the receptors or their downstream signaling molecules [11] . How such inhibition of NODs by phytochemicals is related to their anti-inflammatory, chemopreventive, and cardioprotective properties is a very intriguing question that needs further investigation.
Inhibition of PRR activation by bioactive phytochemicals
Inhibition of PRR dimerization by phytochemicals
The conceptual clue for the phytochemicals to inhibit PRR activation was derived from the results demonstrating that the relative potency of different sesquiterpene lactones in inhibiting LPS-induced expression of cyclooxygenase-2 (COX-2; a TLR4 target gene) was directly related to the presence of α-methylene-gamma-lactone moiety that can confer a Michael addition to sulfhydryl group [69]. Furthermore, sulfhydryl compounds abolish the inhibitory effects of a sesquiterpene lactone on LPS-induced expression of COX-2 [69]. Subsequent studies showed that many phytochemicals with inhibitory effects on NF-κB activation or chemopreventive efficacy contain the structural motif conferring a Michael addition [70-71]. Recent studies have showed that the phytochemicals with the structural motif conferring a Michael addition inhibit PRR activation by interfering with the receptor dimerization [7, 9, 11, 13]. These have provided a new paradigm for understanding of the beneficial effects of phytochemicals on chronic diseases.
Curcumin, a polyphenol found in the plant Curcuma longa (Fig. 3), has been shown to suppress the activation of NF-κB induced by various pro-inflammatory stimuli, presumably through inhibition of IKKβ kinase activity or DNA binding of p65 [72-73]. A recent study revealed that curcumin inhibits MyD88-induced NF-κB activation. However, curcumin did not inhibit interferon regulatory factor 3 (IRF3) activation induced by another immediate TLR4 downstream component TIR-domain-containing adaptor inducing interferon-beta (TRIF), suggesting that the target of curcumin is the receptor itself, but not the downstream components of TRIF pathway [7]. Further studies indicate that curcumin and helenalin, which contain α, β-unsaturated carbonyl group, but not resveratrol (with no unsaturated carbonyl group, Fig. 3), inhibit TLR4 activation by interfering with receptor dimerization [7] (Fig. 4A). Such conclusion was further supported by the result that curcumin inhibits ligand-independent dimerization of constitutively active TLR4. The receptor dimerization is known to be the initial step of TLR4 activation [74-75]. Inhibition of receptor dimerization of TLR4 by phytochemicals with the structural motif conferring a Michael addition suggests that these phytochemicals might modify the free sulfhydryl groups of cysteine residues in TLR4, leading to interference of disulfide formation. Moreover, the activation loop of IKKβ containing cysteine residues has also been reported to be modified by Michael addition [76]. Similarly, cinnamaldehyde (Fig. 3), which also contains α, β-unsaturated carbonyl group, inhibited agonist-induced TLR4 dimerization [9]. The suppression of TLR4 dimerization by cinnamaldehyde was reversed by thiol donors, dithiothreitol and N-acetyl-L-cysteine, suggesting the involvement of cysteine modification in the inhibitory effects of anti-inflammatory phytochemicals [9]. Indeed, sulforaphane (SFN, Fig. 3), an isothiocyanate, formed adducts with cysteine residues in the extracellular domain of TLR4 as determined by micro LC-MS/MS analysis after incubation of recombinant TLR4 extracellular domain with SFN (Fig. 4C-D) [13]. This modification by SFN resulted in the blockade of TLR4 dimerization (Fig. 4B), which was reversed by thiol supplementation. Together, these data suggest that the inhibition of TLR activation by certain anti-inflammatory phytochemicals may be attributed to the modification of cysteines in TLR4 extracellular domain and the subsequent blockade of receptor dimerization through their electrophilic α, β-unsaturated carbonyl groups or isothiocyanate group (Fig. 3). Moreover, these results also suggest that one of targets of anti-inflammatory phytochemicals is TLR4 receptor complex and that the receptor dimerization can be an effective target for dietary and pharmacological agents to ameliorate chronic inflammatory diseases resulting from dysregulated TLR activation.
Fig.3. The chemical structures of phytochemicals curcumin, parthenolide, helenalin, cinnamaldehyde, sulforaphane, resveratrol, EGCG, and luteolin.
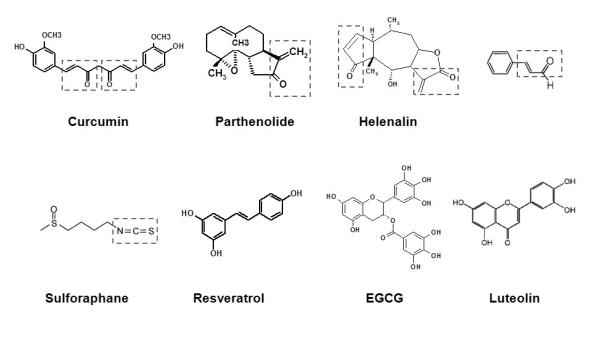
The α,β-unsaturated carbonyl groups in curcumin, parthenolide, helenalin, and cinnamaldehyde are boxed in red. An isothiocyanate group in sulforaphane is boxed in blue.
Fig. 4. Curcumin and sulforaphane inhibits TLR4 dimerization induced by LPS and sulforaphane binds to cysteines in TLR4 extracellular domain.
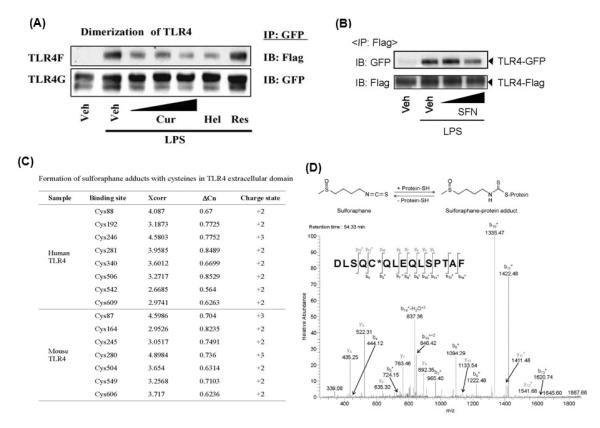
(A, B). Ba/F3 cells expressing TLR4-Flag (TLR4F), TLR4-GFP (TLR4G), MD2-Flag (MD2F), and CD14 were pre-treated with curcumin (10, 20, 50 μM), helenalin (5 μM), resveratrol (50 μM), or sulforaphane (10, 20 μM) for 1 h and then treated with LPS (50 ng/ml) for 20 min. Cell lysates were subjected to immunoprecipitation (IP) and immunoblotted (IB) with antibody as indicated. (C) Summary of cysteines in human and mouse TLR4, which bind to sulforaphane. Extracellular domain of human TLR4 (a.a. 27-631) or mouse TLR4 (a.a. 26-629) was incubated with sulforaphane and micro LC-MS/MS analysis was performed. (D) (Upper panel) Schematic diagram of reaction of sulforaphane with protein cysteine residue. (Lower panel) MS/MS spectrum of the SFN-cysteine adducts at human TLR4 (Asp502-Phe516; DLSQC506QLEQLSPTAF). * denotes fragment ions with one SFN. Cur, curcumin; Hel, helenalin; and Res, resveratrol; SFN, sulforaphane. Reproduced with permission from Biochemical Pharmacology and Journal of Immunology.
Similar to TLR4, NODs also contain LRR domain in their structure which may be involved in receptor dimerization and the dimerization of NODs is also the initial step of ligand-induced receptor activation [17]. Curcumin, parthenolide and helenalin (Fig. 3) were shown to inhibit NOD2 ligand MDP-induced NF-κB activation and target gene (IL-8) expression in a dose-dependent manner (Fig. 5A). In contrast, (-)-epigallocatechin-3-galate (EGCG, Fig. 3) and resveratrol, which do not contain the structural motif conferring Michael addition, exhibited no inhibitory effects (Fig. 5A) [11]. Moreover, curcumin inhibited NOD2 over-expression-induced NF-κB activation in 293T cells in a dose-dependent manner (Fig. 5B). However, curcumin did not inhibit NF-κB in 293T cells that over-express the immediate downstream signaling molecule Rip-like interactive clarp kinase (RICK), suggesting that the target of curcumin in inhibiting NOD2 signaling pathway must be located upstream of NOD2-RICK interaction, instead of downstream of RICK (Fig. 5B). Indeed, curcumin inhibited MDP-induced NOD2 dimerization in a dose-dependent manner. Curcumin also inhibited NOD2 dimerization induced by another NOD2 agonist, lauric acid (Fig. 5C) [11]. Similar inhibition of NOD2 dimerization was also observed with parthenolide with α, β-unsaturated carbonyl group [11]. It remains to be determined whether these phytochemicals prevent or alleviate NOD-mediated chronic inflammation and consequent risk of chronic diseases.
Fig. 5. Curcumin inhibits NOD2 dimerization induced by MDP and lauric acid.
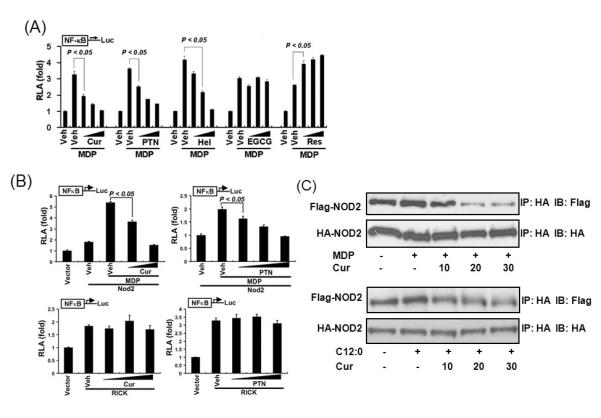
(A) HCT116 cells were transfected with NF-κB-luciferase and β-galactosidase reporters. The cells were pretreated with curcumin (10, 20, and 30 μM), parthenolide (5, 10, and 15 μM), helenalin (1, 3, and 5 μM), EGCG (10, 30, and 50 μM), or resveratrol (10, 30, and 50 μM) for 1 h and then coincubated with MDP (50 μM) for additional 6 h. (B) HEK293T cells were cotransfected with NF-κB-luciferase and β-galactosidase reporter and either Nod2 expression vector (top panels) or RICK expression vector (bottom panels). The cells were pretreated with curcumin (10, 20 μM) or parthenolide (5, 10, and 15 μM) for 1 h and then co-incubated with MDP (200 ng/ml) for an additional 6 h (top panels) or were treated with curcumin (10, 20, and 30 μM) or parthenolide (5, 10, and 15 μM) for 6 h (bottom panels). Relative luciferase activity (RLA) was normalized with β-galactosidase activity. Values are mean ± S.E.M (n = 3). Statistically significant difference (p < 0.05) is indicated. (C) HEK293T cells were co-transfected with HA-Nod2 and Flag-Nod2 cDNA expression vectors. The cells were pretreated with curcumin (10, 20, and 30 μM) for 1 h and then co-incubated with MDP (200ng/ml) (upper) or lauric acid (C12:0; 100 μM) (lower) for 15 min. Nod2 proteins from cell lysates were immunoprecipitated (IP) with anti-HA affinity matrix, and HA-Nod2 and Flag-Nod2 proteins were detected by Western blotting (IB) using anti-HA and anti-Flag antibodies. Cur, curcumin; PTN, parthenolide; Hel, helenalin; and Res, resveratrol. Reproduced with permission from Molecular Pharmacology.
The studies with curcumin, cinnamaldehyde, and sulforaphane, which have structural motifs that can interfere with receptor dimerization, have provided a paradigm that PRR dimer assembly could be a novel target for the anti-inflammatory strategies to ameliorate chronic inflammation resulting from dysregulated PRR activation.
Inhibition of MyD88-independent signaling pathways downstream of TLR3 and TLR4 by phytochemicals
Phytochemicals without the structural motif conferring Michael addition(e.g., resveratrol, EGCG) did not inhibit TLR4 dimerization; however, they specifically inhibited the MyD88-independent TRIF complex containing serine kinases TANK binding kinase 1 (TBK1) and Receptor –interacting protein 1 (RIP1), the downstream signaling components of TLR3 and TLR4 [8, 10, 12]. Despite numerous reports demonstrating the inhibitory effects of resveratrol on NF-κB activation and target gene expression induced by various proinflammatory stimuli [77-78], the direct molecular target has not been fully identified. Although NF-κB can be stimulated by multiple signaling pathways derived from the activation of different types of receptors including TLRs and NODs [79], the inhibition by resveratrol of NF-κB activated by different agonists suggests that the targets of resveratrol are likely to be downstream signaling components responsible for the activation of the transcription factor, rather than the receptors themselves. Resveratrol suppressed NF-κB activation and COX-2 expression following TLR3 and TLR4 stimulation, but not TLR2 or TLR9 in RAW264.7 cells (Fig. 6A) [8]. Moreover, resveratrol inhibited NF-κB activation induced by stimulation of TRIF, but not by MyD88. Furthermore, the suppressive effect of resveratrol on LPS-induced NF-κB activation was abolished in TRIF-deficient mouse embryonic fibroblasts, but not in MyD88-deficient macrophages (Fig. 6B-C), suggesting that resveratrol specifically inhibits MyD88-independent signaling pathways downstream of TLR3 and TLR4. In further delineating the target of resveratrol, it was found that resveratrol inhibited the kinase activity of TBK1 and the NF-κB activation induced by RIP1 [8] (Fig. 6D). Together, these results demonstrate that resveratrol specifically inhibits TLR3 and TLR4 signaling pathway by targeting TBK1 and RIP1 in TRIF complex. Additional studies showed that EGCG, luteolin and structural analogs of luteolin such as quercetin, chrysin, and eriodictyol, also inhibited TRIF signaling pathway by targeting TBK1 kinase [10, 12]. Together, the studies suggest that TBK1 in TRIF pathway, can be targeted by phytochemicals to suppress the activation of TLR3 and TLR4 and their functional consequences. Future studies will identify additional phytochemicals that can target TBK1
Fig.6. Resveratrol suppresses the functional activity of TBK1.
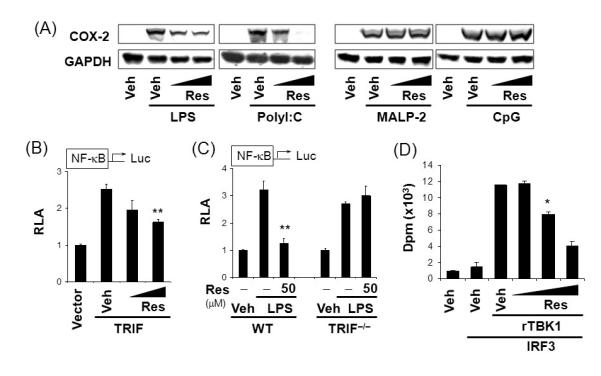
(A) RAW264.7 cells were treated with resveratrol (30, 50 μM) for 1 hr and further stimulated with LPS (5 ng/ml), polyI:C (10 μg/ml), MALP-2 (2 ng/ml), or CpG DNA (ODN1668, 0.2 μM) for 18 hrs. Cell lysates were analyzed for COX-2 and GAPDH immunoblots. (B) RAW264.7 cells were transfected with NF-κB-luciferase reporter plasmid and the TRIF expression plasmid. Cells were further treated with resveratrol (30, 50 μM) for 18 hrs. (C) TRIF-deficient (TRIF-/-) or wild-type mouse embryonic fibroblasts were transfected with NF-κB-luciferase reporter plasmid. Cells were treated with resveratrol for 1 hr and further stimulated with LPS (100 ng/ml) for 18 hrs. Relative luciferase activity (RLA) was determined. Values are mean±S.E.M. (n=3). (D) In vitro TBK1 kinase assay was performed using recombinant active TBK1 (rTBK1) with IRF3 in the presence of resveratrol (20, 50, 100 μM). Values are mean ± S.E. (n = 2). **, Significantly different from (B) TRIF plus vehicle or (C) LPS alone, p<0.01. *,, Significantly different from (D) vehicle plus rTBK1 plus IRF3, p < 0.05. Veh, vehicle. Res, resveratrol. WT, wild-type. Reproduced with permission from Journal of Immunology.
Future directions
Excessive generation of stimulatory endogenous molecules, or the presence of insufficient amounts of endogenous molecules that inhibit the activation of PRRs, can increase the propensity to PRR activation. Although PRR-mediated innate immune responses are a part of the essential elements for host defense and wound healing, the termination of PRR-mediated innate immune responses is required to prevent tissue damage and chronic inflammation. Currently, the mechanisms by which PRR-mediated inflammatory responses are resolved are not well understood. It has been reported that negative regulation of TLRs can be achieved at multiple levels [80-81]. MicroRNA-mediated RNA interference is also emerging as an important regulatory mechanism that operates at the translation level to modulate TLR-mediated immune responses [82-85]. For example, the production of miR-146 was up-regulated by TLR4 activation. The expression of IRAK and Tumor necrosis factor receptor associated factor 6 (TRAF6) proteins, the downstream signaling components of TLR4, was suppressed by the miR-146 family [82]. On the other hand, NOD2 agonists activated PI3K pathways, which negatively regulate NOD-induced NF-κB activation [86]. These results suggest that the activation of PRRs stimulates both pro-inflammatory and intrinsic resolution pathways, probably with different time course. Therefore, the duration and intensity of PRR-induced inflammation can be modulated not only by the strength of initial activation but also by timely resolution of the inflammation. The findings that the activation of PRRs is inhibited by certain phytochemicals suggest that these dietary components can act as extrinsic resolution factors for PRR-mediated inflammation. Future studies will reveal whether the intrinsic resolution pathways of PRR-induced inflammation can also be modulated by dietary factors, including bioactive phytochemicals.
The fact that the activation of these PRRs is dynamically modulated by dietary factors [2] suggests that PRRs can act as the sensor for disturbance in metabolic homeostasis. Thus, PRRs act as the fulcrum for Yin and Yang of inflammation modulated by dietary factors (Fig. 7). Identifying molecular targets by which dietary factors modulate PRR-mediated signaling pathways and target gene expression would provide new opportunities to reduce the risk and to manage chronic inflammatory diseases resulting from the dysregulation of PRR-mediated inflammatory responses.
Fig. 7. TLRs and NODs are the fulcrum in the Yin-Yang of inflammation.
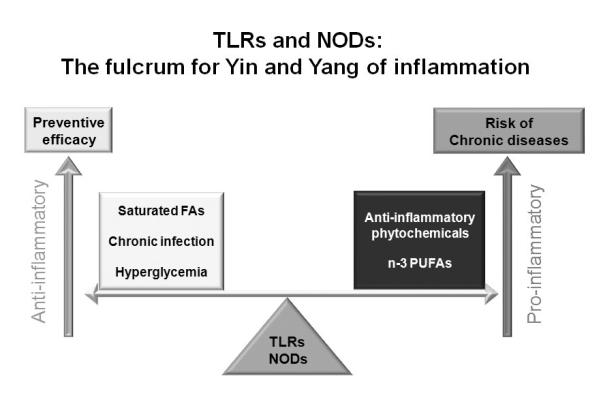
Activation of these PRRs is dynamically modulated by dietary factors suggesting that PRRs can act as a sensor for disturbance in metabolic homeostasis. Identifying molecular targets by which dietary factors modulate PRR-mediated signaling pathways and target gene expression would provide new opportunity to reduce the risks and to manage chronic inflammatory diseases resulting from the dysregulation of PRR-mediated inflammatory responses.
Abbreviations
- CARD
caspase-recruitment domain
- COX
cyclooxygenase
- DAMPs
danger-associated molecular patterns
- DTT
dithiothreitol
- IKK
IkappaB kinase
- EGCG
(-)-epigallocatechin-3-gallate
- IRAK
Interleukin-1 receptor-associated kinase
- IRF3
Interferon regulatory factor 3
- IRS-1
Insulin receptor substrate-1
- JNK
c-Jun N-terminal kinase
- LDL
low density lipoprotein
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- MDP
muramyl dipeptide, MurNAc-l-Ala-disoGln
- MyD88
myeloid differentiation primary response gene 88
- NAC
N-acetyl-L-cysteine
- NF-κB
nuclear factor kappa B
- NODs
nucleotide-binding oligomerization domain proteins
- PAMPs
pathogen- associated molecular patterns
- PI3K
phosphoinositide-3-kinase
- PRRs
pattern recognition receptors
- RICK
Rip-like interactive clarp kinase
- SFA
Saturated fatty acid
- TBK1
TANK binding kinase 1
- TIR
Toll/interleukin-1 receptor
- TLR
Toll-like receptor
- TRAF
Tumor necrosis factor receptor associated factor
- TRIF
TIR-domain-containing adaptor inducing interferon-beta
Footnotes
This work was supported by grants DK064007, DK41868 and CA75613 from the National Institutes of Health, grant 2001-35200-10721 from the United States Department of Agriculture (USDA), grant 01A095Rev from the American Institutes for Cancer Research, and program funds from the Western Human Nutrition Research Center/ARS/USDA (D.H.H.) and grants from Cell Dynamics Research Center, the Korea Science and Engineering Foundation (KOSEF) (R11-2007-007-02003-0), and KOSEF, Korea government (MEST) (No. 20090080185) (J.Y.L.).
Declaration of interest All authors have no interest(s) to declare.
Reference
- 1.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8(12):923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JY, Zhao L, Hwang DH. Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr Rev. 2010;68(1):38–61. doi: 10.1111/j.1753-4887.2009.00259.x. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71(10):1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Riccioni G, Mancini B, Di Ilio E, Bucciarelli T, D’Orazio N. Protective effect of lycopene in cardiovascular disease. Eur Rev Med Pharmacol Sci. 2008;12(3):183–190. [PubMed] [Google Scholar]
- 5.Liu RH. Potential synergy of phytochemicals in cancer prevention: mechanism of action. J Nutr. 2004;134(12 Suppl):3479S–3485S. doi: 10.1093/jn/134.12.3479S. [DOI] [PubMed] [Google Scholar]
- 6.Minich DM, Bland JS. Dietary management of the metabolic syndrome beyond macronutrients. Nutr Rev. 2008;66(8):429–444. doi: 10.1111/j.1753-4887.2008.00075.x. [DOI] [PubMed] [Google Scholar]
- 7.Youn HS, Saitoh SI, Miyake K, Hwang DH. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol. 2006;72(1):62–69. doi: 10.1016/j.bcp.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 8.Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific Inhibition of MyD88-Independent Signaling Pathways of TLR3 and TLR4 by Resveratrol: Molecular Targets Are TBK1 and RIP1 in TRIF Complex. J Immunol. 2005;175(5):3339–3346. doi: 10.4049/jimmunol.175.5.3339. [DOI] [PubMed] [Google Scholar]
- 9.Youn HS, Lee JK, Choi YJ, Saitoh SI, Miyake K, Hwang DH, Lee JY. Cinnamaldehyde suppresses toll-like receptor 4 activation mediated through the inhibition of receptor oligomerization. Biochem Pharmacol. 2008;75(2):494–502. doi: 10.1016/j.bcp.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 10.Youn HS, Lee JY, Saitoh SI, Miyake K, Kang KW, Choi YJ, Hwang DH. Suppression of MyD88- and TRIF-dependent signaling pathways of Toll-like receptor by (-)-epigallocatechin-3-gallate, a polyphenol component of green tea. Biochem Pharmacol. 2006;72(7):850–859. doi: 10.1016/j.bcp.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 11.Huang S, Zhao L, Kim K, Lee DS, Hwang DH. Inhibition of Nod2 signaling and target gene expression by curcumin. Mol Pharmacol. 2008;74(1):274–281. doi: 10.1124/mol.108.046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JK, Kim SY, Kim YS, Lee WH, Hwang DH, Lee JY. Suppression of the TRIF-dependent signaling pathway of Toll-like receptors by luteolin. Biochem Pharmacol. 2009;77(8):1391–1400. doi: 10.1016/j.bcp.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, Seo JA, Lim KM, Kwak MK, Hwang DH, et al. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol. 2010;184(1):411–419. doi: 10.4049/jimmunol.0803988. [DOI] [PubMed] [Google Scholar]
- 14.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 15.O’Neill LA. How Toll-like receptors signal: what we know and what we don’t know. Curr Opin Immunol. 2006;18(1):3–9. doi: 10.1016/j.coi.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168(2):554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 17.Inohara, Chamaillard, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 18.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 19.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 20.Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277(23):20847–20853. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 21.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 22.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177(2):1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 23.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164(2):558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 24.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276(13):10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 25.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195(1):99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 27.Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279(17):16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- 28.Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57(11):3090–3098. doi: 10.2337/db08-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao L, Kwon MJ, Huang S, Lee JY, Fukase K, Inohara N, Hwang DH. Differential modulation of Nods signaling pathways by fatty acids in human colonic epithelial HCT116 cells. J Biol Chem. 2007;282(16):11618–11628. doi: 10.1074/jbc.M608644200. [DOI] [PubMed] [Google Scholar]
- 30.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 31.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74(2):213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 32.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115(11):3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Ukai T, Yumoto H, Davey M, Goswami S, Gibson FC, 3rd, Genco CA. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis. 2008;196(1):146–154. doi: 10.1016/j.atherosclerosis.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101(29):10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehr HA, Sagban TA, Ihling C, Zahringer U, Hungerer KD, Blumrich M, Reifenberg K, Bhakdi S. Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation. 2001;104(8):914–920. doi: 10.1161/hc3401.093153. [DOI] [PubMed] [Google Scholar]
- 36.Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278(3):1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- 37.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25(6):1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 38.Harkewicz R, Hartvigsen K, Almazan F, Dennis EA, Witztum JL, Miller YI. Cholesteryl ester hydroperoxides are biologically active components of minimally oxidized low density lipoprotein. J Biol Chem. 2008;283(16):10241–10251. doi: 10.1074/jbc.M709006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104(12):1355–1363. doi: 10.1161/CIRCRESAHA.108.192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol. 2006;6(2):130–135. doi: 10.1016/j.coph.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.den Dekker WK, Cheng C, Pasterkamp G, Duckers HJ. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis. 2010;209(2):314–320. doi: 10.1016/j.atherosclerosis.2009.09.075. [DOI] [PubMed] [Google Scholar]
- 42.Miller YI, Choi SH, Fang L, Harkewicz R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. 2009;19(7):227–232. doi: 10.1016/j.tcm.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Houghton J, Wang TC. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology. 2005;128(6):1567–1578. doi: 10.1053/j.gastro.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimura T, Shimoyama T, Tanaka M, Sasaki Y, Fukuda S, Munakata A. Gastric mucosal inflammation and epithelial cell turnover are associated with gastric cancer in patients with Helicobacter pylori infection. J Clin Pathol. 2000;53(7):532–536. doi: 10.1136/jcp.53.7.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaudreault E, Fiola S, Olivier M. Gosselin J: Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol. 2007;81(15):8016–8024. doi: 10.1128/JVI.00403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, Gerken G, Schlaak JF. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48(6):914–922. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 47.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46(6):1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 48.Ferrero RL. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol Immunol. 2005;42(8):879–885. doi: 10.1016/j.molimm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 49.El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27(2):244–252. doi: 10.1038/sj.onc.1210912. [DOI] [PubMed] [Google Scholar]
- 50.Forrest MS, Skibola CF, Lightfoot TJ, Bracci PM, Willett EV, Smith MT, Holly EA, Roman E. Polymorphisms in innate immunity genes and risk of non-Hodgkin lymphoma. Br J Haematol. 2006;134(2):180–183. doi: 10.1111/j.1365-2141.2006.06141.x. [DOI] [PubMed] [Google Scholar]
- 51.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 52.Fukata M, Abreu MT. Pathogen recognition receptors, cancer and inflammation in the gut. Curr Opin Pharmacol. 2009;9(6):680–687. doi: 10.1016/j.coph.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirsch I, Caux C, Hasan U, Bendriss-Vermare N, Olive D. Impaired Toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol. 2010;31(10):391–397. doi: 10.1016/j.it.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 54.Kluwe J, Mencin A, Schwabe RF. Toll-like receptors, wound healing, and carcinogenesis. J Mol Med. 2009;87(2):125–138. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mussig K, Fiedler H, Staiger H, Weigert C, Lehmann R, Schleicher ED, Haring HU. Insulin-induced stimulation of JNK and the PI 3-kinase/mTOR pathway leads to phosphorylation of serine 318 of IRS-1 in C2C12 myotubes. Biochem Biophys Res Commun. 2005;335(3):819–825. doi: 10.1016/j.bbrc.2005.07.154. [DOI] [PubMed] [Google Scholar]
- 56.Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277(50):48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 57.Kim JA, Yeh DC, Ver M, Li Y, Carranza A, Conrads TP, Veenstra TD, Harrington MA, Quon MJ. Phosphorylation of Ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by Mouse Pelle-like kinase/interleukin-1 receptor-associated kinase: cross-talk between inflammatory signaling and insulin signaling that may contribute to insulin resistance. J Biol Chem. 2005;280(24):23173–23183. doi: 10.1074/jbc.M501439200. [DOI] [PubMed] [Google Scholar]
- 58.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Himes RW, Smith CW. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010;24(3):731–739. doi: 10.1096/fj.09-141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281(37):26865–26875. doi: 10.1074/jbc.M513304200. [DOI] [PubMed] [Google Scholar]
- 61.Hysi P, Kabesch M, Moffatt MF, Schedel M, Carr D, Zhang Y, Boardman B, von Mutius E, Weiland SK, Leupold W, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. 2005;14(7):935–941. doi: 10.1093/hmg/ddi087. [DOI] [PubMed] [Google Scholar]
- 62.McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A, Cookson WO, Jewell DP. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet. 2005;14(10):1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 63.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 64.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 65.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, Chamaillard M, Zouali H, Thomas G, Hugot JP. CARD15 mutations in Blau syndrome. Nature genetics. 2001;29(1):19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 66.Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, Fuji A, Yuasa T, Manki A, Sakurai Y, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195–1197. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 67.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5(11):1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 69.Hwang D, Fischer NH, Jang BC, Tak H, Kim JK, Lee W. Inhibition of the expression of inducible cyclooxygenase and proinflammatory cytokines by sesquiterpene lactones in macrophages correlates with the inhibition of MAP kinases. Biochem Biophys Res Commun. 1996;226(3):810–818. doi: 10.1006/bbrc.1996.1433. [DOI] [PubMed] [Google Scholar]
- 70.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci U S A. 2001;98(6):3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siedle B, Garcia-Pineres AJ, Murillo R, Schulte-Monting J, Castro V, Rungeler P, Klaas CA, Da Costa FB, Kisiel W, Merfort I. Quantitative structure-activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-kappaB. J Med Chem. 2004;47(24):6042–6054. doi: 10.1021/jm049937r. [DOI] [PubMed] [Google Scholar]
- 72.Pan MH, Lin-Shiau SY, Lin JK. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem Pharmacol. 2000;60(11):1665–1676. doi: 10.1016/s0006-2952(00)00489-5. [DOI] [PubMed] [Google Scholar]
- 73.Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, Sartor RB. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol. 1999;163(6):3474–3483. [PubMed] [Google Scholar]
- 74.Zhang H, Tay PN, Cao W, Li W, Lu J. Integrin-nucleated Toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Lett. 2002;532(1-2):171–176. doi: 10.1016/s0014-5793(02)03669-4. [DOI] [PubMed] [Google Scholar]
- 75.Saitoh S, Akashi S, Yamada T, Tanimura N, Kobayashi M, Konno K, Matsumoto F, Fukase K, Kusumoto S, Nagai Y, et al. Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int Immunol. 2004;16(7):961–969. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]
- 76.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403(6765):103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 77.Holmes-McNary M, Baldwin AS., Jr. Chemopreventive properties of trans resveratrol are associated with inhibition of activation of the IkappaB kinase. Cancer Res. 2000;60(13):3477–3483. [PubMed] [Google Scholar]
- 78.Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164(12):6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- 79.Li ZW, Rickert RC, Karin M. Genetic dissection of antigen receptor induced-NF-kappaB activation. Mol Immunol. 2004;41(6-7):701–714. doi: 10.1016/j.molimm.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 80.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5(6):446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 81.Wang J, Hu Y, Deng WW, Sun B. Negative regulation of Toll-like receptor signaling pathway. Microbes Infect. 2009;11(3):321–327. doi: 10.1016/j.micinf.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 82.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen XM, Splinter PL, O’Hara SP, LaRusso NF. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem. 2007;282(39):28929–28938. doi: 10.1074/jbc.M702633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai R, Phillips RA, Zhang Y, Khan D, Crasta O, Ahmed SA. Suppression of LPS-induced Interferon-gamma and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood. 2008;112(12):4591–4597. doi: 10.1182/blood-2008-04-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benakanakere MR, Li Q, Eskan MA, Singh AV, Zhao J, Galicia JC, Stathopoulou P, Knudsen TB, Kinane DF. Modulation of TLR2 protein expression by a miR-105 in human oral keratinocytes. J Biol Chem. 2009 doi: 10.1074/jbc.M109.013862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao L, Lee JY, Hwang DH. The phosphatidylinositol 3-kinase/Akt pathway negatively regulates Nod2-mediated NF-kappaB pathway. Biochem Pharmacol. 2008;75(7):1515–1525. doi: 10.1016/j.bcp.2007.12.014. [DOI] [PubMed] [Google Scholar]