

Abstract
Ruthenium drugs are potent anti-cancer agents, but inducing drug selectivity and enhancing their modest activity remain challenging. Slow Ru ligand loss limits the formation of free sites and subsequent binding to DNA base pairs. Herein, we designed a ligand that rapidly dissociates upon irradiation at low pH. Activation at low pH can lead to cancer selectivity, since many cancer cells have higher metabolism (and thus lower pH) than non-cancerous cells. We have used the pH sensitive ligand, 6,6′-dihydroxy-2,2′-bipyridine (66′bpy(OH)2), to generate [Ru(bpy)2(66′(bpy(OH)2)]2+, which contains two acidic hydroxyl groups with pKa1 = 5.26 and pKa2 = 7.27. Irradiation when protonated leads to photo-dissociation of the 66′bpy(OH)2 ligand. An in-depth study of the structural and electronic properties of the complex was carried out using X-Ray crystallography, electrochemistry, UV/visible spectroscopy, and computational techniques. Notably, Ru-N bond lengths in the 66′bpy(OH)2 complex are longer (by ~0.3 Å) than in polypyridyl complexes that lack 6 and 6′ substitution. Thus, the longer bond length predisposes the complex for photo-dissociation and leads to the anti-cancer activity. When the complex is deprotonated, the 66′bpy(O−)2 ligand molecular orbitals mix heavily with the ruthenium orbitals, making new mixed metal-ligand orbitals that lead to a higher bond order. We investigated the anti-cancer activities of [Ru(bpy)2(66′(bpy(OH)2)]2+, [Ru(bpy)2(44′(bpy(OH)2)]2+, and [Ru(bpy)3]2+ (44′(bpy(OH)2 = 4,4′-dihydroxy-2,2′-bipyridine) in HeLa cells, which have a relatively low pH. It is found that [Ru(bpy)2(66′(bpy(OH)2)]2+ is more cytotoxic than the other ruthenium complexes studied. Thus, we have identified a pH sensitive ruthenium scaffold that can be exploited for photo-induced anti-cancer activity.
Keywords: prodrug, anti-cancer, ruthenium, polypyridyl, light-activated, pH-selective
1. Introduction
Metal-based anti-cancer agents owe both their efficacy and their troublesome side effects to their inherent reactivity with DNA. Of note are classic platinum complexes that can lead to cancer cell death. For example, cisplatin has been shown to form cross-links with nucleic acids to halt DNA replication and transcription [1, 2]. A major problem with these agents are so called “off-target” effects wherein non-cancer cells are effected by these DNA modification reactions [3, 4]. This problem generally leads to drug induced toxicity, side effects, and little margin for error between the therapeutic dose and the toxic dose. More recently novel strategies have attracted attention. In the platinum arena agents that induce large lesions without cross-link formation, platinum bound to targeting agents, as well as molecules with multiple platinum centers are being examined [5–7]. One path forward involves the use of ruthenium complexes [8]. Recently, two ruthenium complexes, NAMI-A and KP1019, have entered clinical trails [9]. Ruthenium has slow ligand exchange rates and therefore tends to have lower activity against cancer cells [10]. We hypothesize that a properly positioned pH switch can be used to overcome the slow exchange of ruthenium complexes as well as solve the off-target cytotoxicity problem. In this manuscript we rationally designed a ruthenium-based prodrug strategy where both light and pH are required to activate the Ru center towards reactivity. To our knowledge, this is the first example of a tumor activated prodrug made from a pH sensitive metal complex [11–17].
Photo-driven activation of a ruthenium complex is attractive and has already been demonstrated with platinum(IV) complexes [18, 19]. Recently, the Glazer lab has demonstrated light induced activity with ruthenium polypyridyl complexes [20, 21]. One key process in the design of the new prodrug strategy is that Ru(II)-polypyridyl complexes are relatively inert, however, ligand dissociation can occur in the excited state (Figure 1) [22–24]. In one study, Glazer utilized a 6,6′-dimethyl-2,2′-bipyridine (66′bpy(Me)2) ligand to enhance the photo-dissociation properties of the complex. The substitution at the 6,6′-position leads to a less strongly bound ligand that can exchange more readily upon excitation. While substitution in the 6,6′-position of a bipyridine ligand leads to an active anti-cancer agent upon the absorption of light, we were inspired to add an additional component to these Ru(II)-polypyridyl complexes that would instill a pH dependent feature that would effectively make them tunable and inherently cancer selective.
Figure 1.

Proposed mechanism for ligand dissociation upon absorption of light. R = CH3 for Glazer studies and R = OH for studies carried out in this work. * represents the excited state of the complex.
A burgeoning area of interest is the development of tunable catalysts and molecular switches utilizing ligands that are activated by changing the protonation state [25–34]. Recently, hydroxyl-substituted bipyridine metal complexes have been used to catalyze (de)hydrogenation reactions [27–31]; here the nature of the rate enhancement has been deprotonation yielding a stronger electron donor ligand and in some cases the resulting O− near the metal acts as a pendant base [27, 35, 36]. The water oxidation catalysts that use the same ligand set, [Ir(Cp*)(44′bpy(OH)2)Cl]+ and [Ir(Cp*)(66′bpy(OH)2)Cl]+ (Cp* = 1,2,3,4,5-pentamethylcyclopentadiene; 44′bpy(OH)2 = 4,4′-dihdroxy-2,2′-bipyridine; 66′bpy(OH)2 = 6,6′-dihydroxy-2,2′-bipyridine), have been used to show that ligand deprotonation enhances catalytic rates ~100 fold by facilitating Ir oxidation [37]. While the increased electron-donation upon deprotonation to the metal leads to enhanced catalytic activity, we hypothesized that we could also utilize this effect to alter the ligand exchange properties of complexes with the hydroxyl-substituted bipyridine ligands.
The structures of 44′bpy(OH)2 and 66′bpy(OH)2 and their deprotonated forms are depicted in Figure 2. Recently, we have characterized and studied the 44′bpy(OH)2 ligand on [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(44′bpy(OH)2)3]2+ to ascertain how the protonation state of the ligand affects the electronic and structural properties of the complex [38, 39]. With our understanding of ruthenium complexes containing the 44′bpy(OH)2 ligand, we envisioned that re-orienting the hydroxyl groups into the 6,6′-position could yield anti-cancer activity similar to the complex report by Glazer [20], yet the hydroxyl complex could be inherently more toxic in cancer cells due to pH switchable properties. Cancer cells are more acidic than normal cells due to their high metabolism [40], and a drug that is activated at low pH should selectively target cancer cells. Here we report [Ru(bpy)2(66′(bpy(OH)2)]2+ as a complex that only photo-dissociates at low pH and under blue light. This study illustrates how its structural, electronic, and anti-cancer properties change as a function of ligand protonation state.
Figure 2.
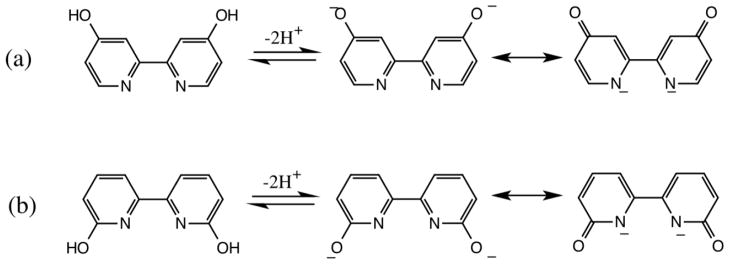
Structures of the protonated and deprotonated forms of the ligands: a) 4,4′-dihydroxy-2,2′-bipyridine (44′bpy(OH)2) and b) 6,6′-dihydroxy-2,2′-bipyridine 66′bpy(OH)2).
2. Experimental
2.1 General Procedures
Reagents were obtained from Aldrich Chemical Company and used without further purification. RuCl3•3H2O was purchased from Pressure Chemical Company. [Ru(bpy)2(Cl)2], [Ru(bpy)3][Cl]2, [Ru(bpy)2(CH3CN)2][PF6]2, and [Ru(bpy)2(44′bpy(OH)2)][PF6]2 were synthesized according to previously published methods [38, 41–43]. The 66′bpy(OH)2 ligand was synthesized according to previously published methods [27]. Elemental analysis was carried out by Atlantic Microlab Inc., Norcross, GA. For studies carried out in water, ruthenium hexafluorophosphate salts were converted to chloride salts by precipitation from acetone using tetrabutylammonium chloride dissolved in acetone. Aqueous solutions were prepared using a Millipore DirectQ UV water purification system.
1H-NMR spectra were collected on a Varian 300 MHz Fourier Transform spectrometer in deuterated acetonitrile (CD3CN). Infrared spectra were obtained on a Perkin Elmer Spectrum One FT-IR with Universal ATR sampling accessory. UV-visible absorption spectra were collected on a Scinco S-3100 diode-array spectrophotometer at a resolution of 1 nm. Luminescence data was collected on a Horiba Jobin Yvon Fluoromax 3. pH measurements were performed using a VWR SympHony pH meter, utilizing a three point calibration at pH = 4.0, 7.0, and 10.0.
Electrochemical measurements were carried out on a Bioanalytical Systems (BAS) CW-50 potentiostat. A standard three electrode setup with a Ag/Ag+ reference electrode, platinum wire auxiliary electrode and glassy carbon working electrode were used. All measurements were taken in 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in acetonitrile electrolyte solution. The solutions were degassed for approximately 20 minutes with argon before data collection. Ferrocene was used as an internal standard with E1/2 = +0.40 V vs. Saturated Calomel Electrode (SCE) [44].
2.2 Synthesis
[Ru(bpy)2(66′bpy(OH)2)][PF6]2•H2O
A round botton flask containing 30 mL of 1:1 ethanol:water was degassed with argon for 30 minutes. To the flask, 0.2260 g (1.201 mmol) 66′bpy(OH)2 and 0.4843 g (0.9999 mmol) Ru(bpy)2(Cl)2 were added. The reaction mixture was heated at 80 °C under argon for 12 h. The reaction mixture turned red in color. After heating, the reaction mixture was allowed to cool to room temperature and filtered to remove any insoluble, unreacted ligand. A few drops of concentrated HCl was added to the filtrate to ensure protonation and the solution was diluted to 200 mL with water. An aqueous solution of ammonium hexafluorophosphate was added to the filtrate to precipitate the complex as the hexafluorophosphate salt. The complex was filtered and rinsed with copious amounts of water and allowed to air dry overnight. Yield: 0.5738 g (0.6309 mmol), 63%. δH (300 MHz, CD3CN): δ 8.70 (broad), δ 8.50 (d, 2H), δ 8.35 (d, 2H), δ 7.95 (m, 10H), δ 7.55 (d, 2H), δ 7.45 (t, 2H), δ 7.20 (t, 2H), δ 6.70 (d, 2H). Elem. Anal: Found: C, 39.43; N, 9.23; H, 2.89%. Calc. for RuC30N6O2H24P2F12•H2O: C, 39.62; N, 9.24; H, 2.88%.
2.3 X-Ray structural analysis
[Ru(bpy)2(66′bpy(OH)2)][PF6]2
Crystals of [Ru(bpy)2(66′bpy(OH)2)][PF6]2 were grown by the slow diffusion of ether into a benzonitrile solution with dissolved complex. The solutions were shielded from the light using aluminum foil. A single red block (0.08 × 0.12 × 0.13 mm) was mounted using NVH immersion oil (Cargille Laboratories) onto a nylon fiber and cooled to the data collection temperature of 110(2) K. Data were collected on a Brüker-AXS Kappa APEX II CCD diffractometer with 0.71073 Å Mo-Ka radiation. Unit cell parameters were obtained from 60 data frames, 0.5° Φ, from three different sections of the Ewald sphere yielding a = 15.661(2), b = 16.821(2), c = 19.071(2) Å, α = 107.42(1), β = 100.60(1), γ = 105.36(1)°, V = 4429(1) Å3. 94387 reflections (Rint = 0.0534) were collected (33738 unique) over θ = 1.41 to 33.19°. The data was consistent with the centrosymmetric, triclinic space group P-1. The data-set was treated with SADABS absorption corrections based on redundant multi-scan data, Tmax/Tmin = 1.03. The asymmetric unit contains two [Ru(bpy)2(66′bpy(OH)2)]2+ cations, four [PF6]− anions, four molecules of diethylether solvent and 1/2 a molecule of benzonitrile solvent. Two of the ether molecules are disordered over two positions, which were located from the difference map and refined using SIMU, DELU, and SAME commands. The benzonitrile molecule is disordered about the inversion center. One of the two positions was located from the difference map and the occupancy of all atoms was set to 0.5 along. A PART-1 command was used to ignore the symmetry at this position. The O-H protons were originally located from the difference map but three of them would not survive a free refinement. They were refined using a riding model with O-H bond distance refinement for possible H-bonding interactions to be considered. There is some minor residual density remaining around one of the [PF6]− anions which is due to positional disorder. We have chosen not to model this in favor of a less restrained model. All non-hydrogen atoms were refined with anisotropic displacement parameters. All other hydrogen atoms were treated as idealized contributions. The goodness of fit on F2 was 1.019 with R1(wR2) 0.0551(0.1231) for [Iq>2(I)] and with largest difference peak and hole of 1.694 and −1.147 e/Å3 due to heavy atom noise around the ruthenium atom.
[Ru(bpy)2(66′bpy(O−)2)]
Crystals of [Ru(bpy)2(66′bpy(O−)2)] were grown by the slow diffusion of ether into a solution containing acetonitrile with dissolved complex and a few drops of aqueous tetrabutylammonium hydroxide to ensure deprotonation of the complex. A single red block (0.10 × 0.18 × 0.18 mm) was mounted using NVH immersion oil (Cargille Laboratories) onto a nylon fiber and cooled to the data collection temperature of 120(2) K. Data were collected on a Brüker-AXS Kappa APEX II CCD diffractometer with 0.71073 Å Mo-Ka radiation. Unit cell parameters were obtained from 60 data frames, 0.5° Φ, from three different sections of the Ewald sphere yielding a = 9.411(1), b = 12.287(1), c = 13.003(1) Å, α = 84.34(1), β = 88.75(1), γ = 73.88(1)°, V = 1436.6(2) Å3. 26434 reflections (Rint = 0.0293) were collected (9615 unique) over θ = 1.57 to 31.79°. The data was consistent with the centrosymmetric, triclinic space group P-1. The data-set was treated with SADABS absorption corrections based on redundant multi-scan data, Tmax/Tmin = 1.05. The asymmetric unit contains one [Ru(bpy)2(66′bpy(O−)2)] molecule, one molecule of acetonitrile solvent and one molecule of diethyl ether solvent. These solvent molecules were located from the difference map and were disordered over inversion centers. Attempts to model using PART-1 commands along with SIMU and DELU restraints resulted in unstable refinements so SQUEEZE was employed removing the electron density of the disordered solvent from the model. The molecular formula was augmented to include these solvent molecules. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were treated as idealized contributions. The goodness of fit on F2 was 1.024 with R1(wR2) 0.0368(0.0854) for [Iq>2(I)] and with largest difference peak and hole of 0.778 and −0.513 e/Å3.
2.4 Computational Studies
All calculations were performed using GAMESS [45]. Geometries were optimized using restricted B3LYP with the 6-31G* basis set for the main group elements. A scalar relativistic model core potential (first 30 electrons) was used for ruthenium, with the valence orbital set (5s and 4d) being of triple-zeta quality [46, 47]. Spherical harmonic d orbitals were used in all calculations and the default grid size was used for numerical integration in DFT. The maximum tolerance for any nuclear gradient component was set to 0.0005 hartrees/bohr, and the default RMS gradient maximum was used (0.00017 hartrees/bohr). The nature of each stationary point was determined by running frequency calculations at the Density Functional Theory (DFT) level. Numerical frequencies were calculated via central differences of analytically determined energy gradients. For both structures all vibrational modes were found to be real at the DFT determined stationary points. Vertical excitation energies were calculated using time-dependent DFT (TDDFT) with the same set of functionals and basis sets used to characterize the ground state structures. Solvent effects on the vertical excitation energies were evaluated using the PCM solvation model. The solvated energies were evaluated at gas-phase optimized geometries. Previous calculations on similar structures indicate that the PCM solvent plays a negligible role in structure determination but is necessary for more accurate determination of vertical excitation energies [38]. The solute cavity was determined using the simplified united atomic radii. The solvent considered in these calculations was water.
2.5 Anti-cancer activity
For cell culture, human cervical carcinoma (HeLa) cells were obtained from the ATCC. The cells were cultured at 37 °C in a 5% CO2, humidified atmosphere in DMEM medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Assays were accomplished by seeding cells at a density of 5,000 cells/well in a 96-well plate and incubated at 37 °C overnight [48]. Media was exchanged and then cells were treated with either 100 μM [Ru(bpy)2(66′bpy(OH)2)]2+, [Ru(bpy)2(44′bpy(OH)2)]2+, or [Ru(bpy)3]2+ for 48 h. The medium containing compounds was discarded, cells were washed, and fresh Hank’s Balanced Salt Solution was added. In the dark, cells were irradiated using a 450 nm light emitting diode (LED) flashlight positioned 4 cm from the plate for 1 h. The salt solution was exchanged with media and cells were allowed to grow for 24 h. Media containing 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,2-diphenyltetrazolium bromide (MTT) (5 mg/mL) was added to each well and incubated for an additional 3 h. The medium was removed. After adding 200 μL of DMSO to each well, the optical densities at 570 nm were determined. The p-values were calculated from triplicate data sets using KaleidaGraph software.
3. Results and Discussion
[Ru(bpy)2(66′bpy(OH)2)]2+ was synthesized by treating [Ru(bpy)2(Cl)2] with the 66′bpy(OH)2 ligand. We then carried out X-ray structural analysis, electrochemistry, UV/visible absorption spectroscopy, and computational studies to characterize the above complex in different protonation states and we monitored the kinetics of ligand loss at different pH values. These studies were designed to test the hypothesis that for [Ru(bpy)2(66′bpy(OH)2)]2+ ligand loss should be faster at lower pH and to elucidate what features of the bonding in the complex makes it a promising anti-cancer agent (vide infra). In addition, we compared the anti-cancer activity of the complex with two other complexes, [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(bpy)3]2+, which serve as controls. The use of these Ru complexes as active controls should elucidate whether or not hydroxyl groups are essential and if their placement near the metal matters.
3.1 Blue light driven anti-cancer activity
We set out to investigate the anti-cancer activity of [Ru(bpy)2(66′bpy(OH)2)]2+ as compared to [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(bpy)3]2+. We examined if [Ru(bpy)2(66′bpy(OH)2)]2+ could be especially cytotoxic to HeLa cells (relative to the other Ru complexes) due to the requirement for low pH to photo-dissociate the ligand. It has been shown that several cancer cell lines exhibit lower pH values due to their excessive metabolic activity, including HeLa cells [40]. HeLa cells are known to have a low pH of 6.5 in the golgi, when metabolizing glucose [49]. We therefore used HeLa as a model to demonstrate proof-of-concept for light-driven photo-dissociation of a ruthenium bound ligand. In addition, most Ru2+ and Pt2+ complexes are cytotoxic when a ligand is lost to form the di-aqua complex. This di-aqua complex can exchange the water ligands with biomolecules to induce toxicity [4, 50]. Significantly important within these ligand exchange reactions is the addition of the nucleobase guanine at the N7-positon. Nucleobase binding to metallodrugs can occur with both DNA and RNA [51]. Ru2+-polypyridyl complexes, with slower water exchange rates, require a means of actively dissociating the ligand. We examined if 66′bpy(OH)2 could be a trigger to allow for more rapid initiation of the exchange reactions upon excitation.
We explored anti-cancer activity of [Ru(bpy)2(66′bpy(OH)2)]2+, [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(bpy)3]2+ in HeLa cells, Figure 3. Upon irradiation the [Ru(bpy)2(66′bpy(OH)2)]2+ prodrug will be converted into a cytotoxic agent intracellularly. HeLa cells were plated, incubated with [Ru(bpy)2(66′bpy(OH)2)]2+, irradiated, and an MTT cell viability assay was used to assess cellular viability. Untreated cells that did not receive any irradiation are taken as 100% viability. When HeLa were treated with [Ru(bpy)2(66′bpy(OH)2)]2+ in the absence of light little cell death occurred. The average viability dropped to 93 +/− 6%. The change is not statistically significant with p greater than 0.05. Similar results are obtained when cells are not treated with the agent and only irradiated for one hour. The viability dropped to 88 +/− 9%. This shows that the ruthenium complex by itself is not cytotoxic. Importantly, when [Ru(bpy)2(66′bpy(OH)2)]2+ is incubated with HeLa cells and irradiated the viability drops to 47 +/− 12%. This is a statistically significant drop with p less than 0.004 and 0.02 for no ruthenium-complex and no irradiation respectively. Further experiments changing the concentration of [Ru(bpy)2(66′bpy(OH)2)]2+ in HeLa cells under irradiation allowed for measurement of an inhibitory concentration (IC50) of 88 μM for anti-cancer activity (Table 1), consistent with the data in Figure 3.
Figure 3.
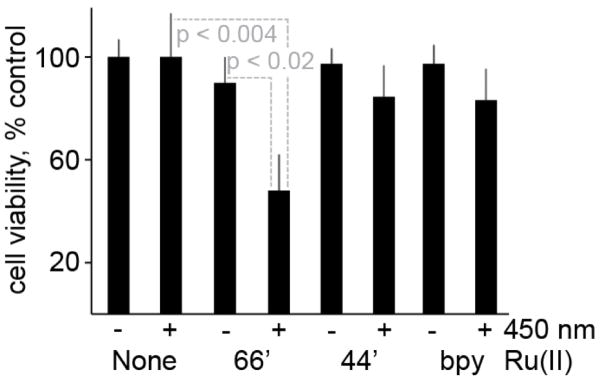
Light-induced HeLa cytotoxicity from ruthenium complexes. When HeLa cells were treated with [Ru(bpy)2(66′bpy(OH)2)]2+ or irradiated at 450 nm for 1 hour little cell death occurred. In contrast, when cells were treated with [Ru(bpy)2(66′bpy(OH)2)]2+ and 450 nm irradiation viability dropped to 47%. Controls using [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(bpy)3]2+ showed limited cell death, even with irradiation. (−) Indicates no irradiation and (+) indicates irradiation at 450 nm for 1 hour.
Table 1.
IC50 Values for Ruthenium Complexes.
| Complex | IC50 (error), μM |
|---|---|
| [Ru(bpy)2(66′bpy(OH)2)]2+ | 88 (9) |
| [Ru(bpy)2(44′bpy(OH)2)]2+ | >100 |
| [Ru(bpy)3]2+ | 152 (18) |
We then investigated the importance of the hydroxyls at the 6 and 6′ position of the bipyridine ligand by both re-orienting the hydroxyl groups to the 4 and 4′ position of the bipyridine or removing them completely. When the hydroxyl groups are removed the complex, [Ru(bpy)3]2+ is formed. When [Ru(bpy)3]2+ is incubated with HeLa cells the viability is 91 +/− 4%. The [Ru(bpy)3]2+ complex once incubated with HeLa cells, followed by irradiation results in a viability drop to 84 +/− 5%. When the hydroxyl groups are moved to the 4 and 4′ position, the viability is 91 +/− 4% without irradiation and 86 +/− 6% with irradiation. Thus, only [Ru(bpy)2(66′bpy(OH)2)]2+ possesses high anti-cancer cell activity upon irradiation. Importantly, when the anti-proliferative effects of both [Ru(bpy)2(44′bpy(OH)2)]2+ and [Ru(bpy)3]2+ on HeLa cells were investigated no significant effects were found for [Ru(bpy)2(44′bpy(OH)2)]2+ while [Ru(bpy)3]2+ had an IC50 value of 152 μM. Furthermore, the work described bellow will show that the 66′bpy(OH)2 ligand allows for both light triggered ligand release and pH sensitivity that can allow for selective toxicity towards more acidic (cancerous) cells.
3.2 X-ray structural analysis shows that light induces ligand displacement
The structures of [Ru(bpy)2(66′bpy(OH)2)][PF6]2 and the deprotonated form of the complex, [Ru(bpy)2(66′bpy(O−)2)] were determined by X-ray diffraction. The structure helps explain the propensity for 66′bpy(OH)2 ligand dissociation. Relevant bond lengths and angles for these complexes are reported in Table 2 and the structures are depicted in Figure 4. Early attempts to grow crystals suitable for X-Ray analysis were carried out by dissolving [Ru(bpy)2(66′bpy(OH)2)][PF6]2 in acetonitrile with slow diffusion of ether without protection from light. Crystal formation took several weeks and upon analysis, yielded the substituted complex, [Ru(bpy)2(CH3CN)2][PF6]2 whereby two solvent molecules displaced the bidentate 66′bpy(OH)2 ligand. The [Ru(bpy)2(CH3CN)2][PF6]2 complex structure has been previously reported in the literature [52, 53]. Other crystals were also obtained from the crystallization that were clearly of a different type, but not resolvable. There is precedence for the photolysis of [Ru(bpy)3]2+ to replace one of the bpy ligands with a coordinating ligand if present in solution [23]. In addition, photo-dissociation of a ligand with ruthenium polypyridyl complexes has been studied in the literature [43, 54, 55]. We hypothesized that a lack of protection from light led to photo-substitution of ligand with solvent, which yielded the corresponding crystals with solvent replacing the 66′bpy(OH)2. Most importantly, this result supports the hypothesis that the [Ru(bpy)2(66′bpy(OH)2)]2+ complex does readily lose the 66′bpy(OH)2 ligand upon irradiation (vide infra) and thus could be used as an anti-cancer prodrug [20, 21].
Table 2.
Selected Bond Lengths (Å) and Bond Angles (deg) for Ruthenium Complexes
| [Ru(bpy)2(66′bpy(OH)2)]2+ | [Ru(bpy)2(66′bpy(O−)2)] | |||
|---|---|---|---|---|
|
| ||||
| Crystal | Theoretical | Crystal | Theoretical | |
| Bond Lengths | ||||
| Ru(1)-N(1) | 2.091(2) | 2.156 | 2.0969(15) | 2.100 |
| Ru(1)-N(2) | 2.094(2) | 2.171 | 2.1020(16) | 2.093 |
| Ru(1)-N(3) | 2.046(2) | 2.098 | 2.0439(14) | 2.076 |
| Ru(1)-N(4) | 2.043(2) | 2.086 | 2.0567(15) | 2.094 |
| Ru(1)-N(5) | 2.066(2) | 2.091 | 2.0511(14) | 2.061 |
| Ru(1)-N(6) | 2.053(2) | 2.083 | 2.0467(16) | 2.095 |
| C(1)-O(1) | 1.340(3) | 1.347 | 1.249(3) | 1.247 |
| C(10)-O(2) | 1.329(3) | 1.346 | 1.251(2) | 1.248 |
| Bond Angles | ||||
| N(1)-Ru(1)-N(2) | 77.37(9) | 76.96 | 77.83(7) | 78.03 |
| N(1)-Ru(1)-N(3) | 96.25(9) | 96.13 | 95.85(6) | 94.20 |
| N(1)-Ru(1)-N(4) | 173.93(9) | 173.58 | 173.38(6) | 171.22 |
| N(1)-Ru(1)-N(5) | 86.64(9) | 89.05 | 88.00(6) | 89.79 |
| N(1)-Ru(1)-N(6) | 96.64(8) | 100.66 | 99.00(7) | 98.74 |
Figure 4.
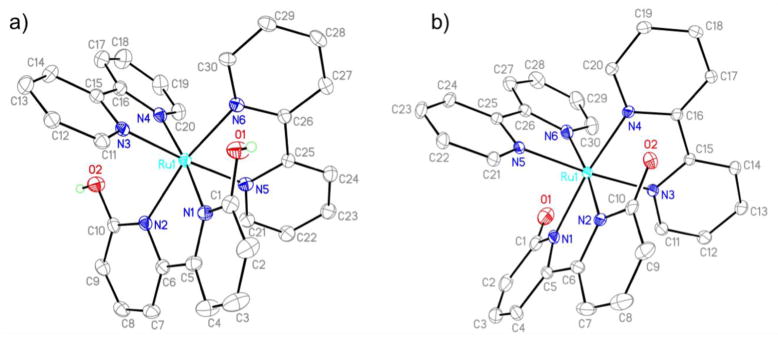
Crystal structures of a) [Ru(bpy)2(66′bpy(OH)2)][PF6]2 and b) [Ru(bpy)2(66′bpy(O−)2)]. Counter ions are omitted for clarity.
To determine if light was causing ligand dissociation, crystals were grown by keeping the solutions in the dark and resulted in the desired complex, [Ru(bpy)2(66′bpy(OH)2)]2+ with two PF6− counter ions. The complex takes on a distorted octahedral geometry with adjacent N-Ru-N bond angles ranging from 77.37(9)° to 98.95(9)°. This geometry is in accordance with other ruthenium hydroxyl-substituted-bipyridine complexes reported previously as well as the parent complex, [Ru(bpy)3]2+ [38, 39, 56]. The Ru-N bond lengths associated with the unsubstituted bipyridine ligands range from 2.043(2) Å to 2.066(2) Å, which are also close in length to those bonds previously reported. However, the Ru-N bond lengths are elongated on average by ~ 0.04 Å to 2.091(2) Å and 2.094(2) Å when the ligand is 66′bpy(OH)2. This length is ~0.3 Å longer than the Ru-N bond lengths for the 44′bpy(OH)2 ligand substituted complex, [Ru(bpy)2(44′bpy(OH)2)][PF6]2 [38]. This longer bond length is most likely due to the orientation of the hydroxyl groups closer to the ruthenium center of the complex causing steric clashes that do not exist when the hydroxyl groups are oriented away from the metal center as with the 44′bpy(OH)2 ligand. The longer and weaker bond between the Ru center and the 66′bpy(OH)2 ligand explains why this ligand is the one substituted in the complex upon light absorption.
Adding a few drops of aqueous tetrabutylammonium hydroxide to the crystallization solution yielded crystals of the corresponding deprotonated complex (Figure 4b). The overall structure of the complex does not change significantly upon deprotonation. The Ru-N bond lengths to both the unsubstituted bpy ligand and the 66′bpy(O−)2 ligand do not vary significantly on average from the protonated form. The most noticeable bond length distance changes occurs as the C-O bonds decrease from 1.340(3) Å and 1.329(3) Å to 1.249(3) Å and 1.251(2) Å upon deprotonation. This decrease in bond length can be explained by the resonance structure wherein the C-O bonds take on double bond character upon deprotonation, Figure 2A. This decrease in bond length of ~0.08 Å is slightly larger than the ~0.05 Å bond length decrease observed in deprotonating [Ru(bpy)2(44′bpy(OH)2)]2+ [38]. However, in the deprotonated [Ru(bpy)2(44′bpy(O−)2)] complex there are several hydrogen bonded water molecules that most likely result in an elongation of the C-O bond, that is absent in the crystal structure of [Ru(bpy)2(66′bpy(O−)2)] where no hydrogen bonded solvent molecules are present. Although the Ru-N bond lengths remain longer for the Ru to 66′bpy(O−)2 ligand, the complex does not appear to undergo as rapid photo-dissociation when exposed to 450 nm blue light compared to the protonated form, vide infra. This result is most likely due to the significantly enhanced electron-donating effects of the ligand to the metal upon deprotonation. These effects were examined by computational means to help explain the lack of photo-dissociation of the deprotonated form upon irraditation, vide infra.
3.3 Cyclic voltammetry studies to determine ligand effects on metal
Cyclic voltammetry studies can help elucidate how the ligand interacts with the metal d orbitals in different protonation states, and can therefore explain why the protonated 66′bpy(OH)2 ligand is more labile in the above anti-cancer studies. Cyclic voltammetry data was collected on [Ru(bpy)2(66′bpy(OH)2)]2+ in acetonitrile solvent with 0.1 M TBAPF6, Figure 5. The RuIII/II reduction wave is reversible and occurs at a potential of 1.12 V vs. SCE. This potential is similar to the 1.16 V vs. SCE RuIII/II potential observed for [Ru(bpy)2(44′bpy(OH)2)]2+ studied previously, indicating the ortho-substituted 66′bpy(OH)2 ligand has similar electronic influences on the metal center to the para-hydroxy-substituted 44′bpy(OH)2 ligand [38]. In addition, both complexes scale with the electron-donation properties of the ligand compared to [Ru(bpy)3]2+ at 1.30 V vs. SCE and [Ru(44′bpy(OH)2)3]2+ at 0.88 V vs. SCE [39]. By increasing electron-donation to the metal center the RuIII state is stabilized, thus making the complex easier to oxidize and decreasing the reduction potential. The [Ru(bpy)2(66′bpy(OH)2)]2+ complex has several reductive and oxidative waves between −1.3 V and −2.1 V vs. SCE associated with ligand redox processes, however, there is significant overlap and individual redox steps cannot be distinguished. There are clear irreversible reductions that have also been observed for [Ru(bpy)2(44′bpy(OH)2)]2+. These processes contrast with ruthenium complexes containing only methoxy-substituted-bipyridine and unsubstituted-bipyridine ligands which have reversible cyclic voltammograms associated with ligand redox processes [38, 39].
Figure 5.
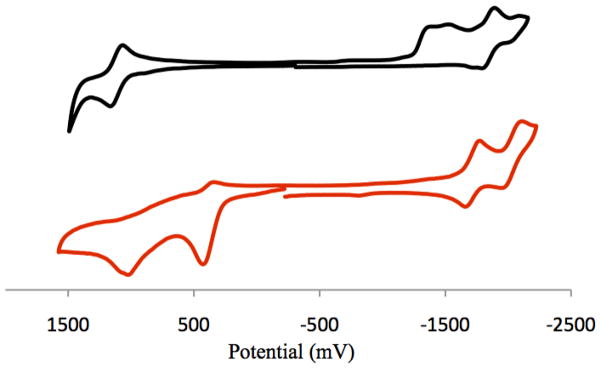
Cyclic Voltammograms of (black line, top) 1.3 mM [Ru(bpy)2(66′bpy(OH)2)]2+ and (red line, bottom) 1.1 mM [Ru(bpy)2(66′bpy(O−)2)] in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate at 25 °C. Tetrabutylammonium hydroxide was used for deprotonation. Scan rates were 200 mV/s. Data reported versus SCE.
In order to obtain cyclic voltammetry data of the deprotonated [Ru(bpy)2(66′bpy(O−)2)] complex, a 5:1 ratio of tetrabutylammonium hydroxide to complex was used. The reductive region of the cyclic voltammogram for [Ru(bpy)2(66′bpy(O−)2)] becomes reversible upon deprotonation (also observed with the corresponding 4,4′-substituted complex). Two reversible ligand reductions are observed at −1.72 V and −2.04 V vs. SCE and assigned to the unsubstituted bipyridine ligands. These ligand reduction potentials are approximately 0.2 V lower than the potentials observed for deprotonated [Ru(bpy)2(44′bpy(O−)2)]. Two irreversible oxidative peaks at 0.45 V and 1.03 V vs. SCE were also observed. The tetrabutylammonium hydroxide base is redox active in this region and attempts to subtract the oxidative wave of the base were unsuccessful. As a result it was difficult to determine which of the oxidative waves were associated with the complex in this region. However, it is clear that the metal-centered RuIII/II reversible wave that appears in the protonated form of the complex is absent as there is no evidence of reversible oxidative processes. This result is indicative of the fact that the metal-centered orbitals mix with the ligand orbitals upon deprotonation, and this was studied by theoretical methods to examine the nature of this mixing, vide infra. This mixing upon deprotonation, leads to the more stable metal to ligand bond in the deprotonated from and slows down photo-dissociation compared to the protonated form.
3.4 pH-dependent changes in absorbance spectroscopy
UV/visible absorbance data was collected for [Ru(bpy)2(66′bpy(OH)2)]2+ in aqueous buffers ranging from pH = 1 to pH = 13, Figure 6. The deprotonated form of [Ru(bpy)2(66′bpy(OH)2)]2+ is red shifted compared to the protonated form of the complex. The pH titration data showed two distinct events that yielded a pKa1 = 5.26 and pKa2 = 7.27. The titration data is reported in the supplementary materials. This result is in contrast to previous studies carried out with hydroxyl-substituted polypyridyl complexes of ruthenium, whereby the individual deprotonations are not observed and only an average pKa value can be reported [27, 37–39, 57].
Figure 6.

UV/Visible spectra of 75 μM [Ru(bpy)2(66′bpy(OH)2)]2+ in aqueous buffers ranging from pH = 1 to pH = 13 at 25 °C. The arrows indicate the directions of absorbance change with increasing pH.
Further analysis of the absorbance spectra in aqueous solution reveal that the lowest energy MLCT band for the protonated complex is λmax = 461 nm at pH = 1 and shifts approximately 1800 cm−1 to λmax = 504 nm at pH = 13 where the complex is completely deprotonated. As a comparison, the protonated [Ru(bpy)2(44′bpy(OH)2)]2+ complex has a λmax = 462 nm in water and λmax = 493 nm when deprotonated, a shift of approximately 1300 cm−1 [38]. As an additional comparison, another ruthenium complex containing hydroxyl-substituted phenanthroline ligands, [Ru(bpy)2(47phen(OH)2)]2+ (47phen(OH)2 = 4,7-dihydroxy-1,10-phenanthroline) shifts approximately 1500 cm−1 upon deprotonation of the ligand in aqueous solution [58]. All three of complexes contain two deprotonatable hydroxyl groups.
3.5 Computational analysis of electronic transitions
Analysis of the electronic transitions in [Ru(bpy)2(66′bpy(OH)2)]2+ and [Ru(bpy)2(66′bpy(O−)2)] was carried out by computational methods using water as the solvent. The energies of the transitions and oscillator strengths for each complex are reported in Tables 3 and 4. In addition, the four highest occupied molecular orbitals for each complex are depicted in Figure 7. For the protonated complex, the two lowest energy transitions occur at 411 nm and 405 nm, and are assigned as Metal to Ligand Charge Transfer (MLCT) from a filled metal d orbital to π* orbitals on all three ligands. There is no clear distinction between the bpy and 66′bpy(OH)2 ligands within these transitions. Following these transitions, there is a gap before two sharp electronic transitions appear between 329 nm and 323 nm. The two sharp electronic transitions are observable in the experimental spectrum of the protonated complex, Figure 6. These electronic transitions are localized π to π* and Ligand to Ligand Charge Transfer (LLCT) transitions occurring from π molecular orbitals on the 66′bpy(OH)2 ligand to π* molecular orbitals on both the unsubstituted bpy and 66′bpy(OH)2 ligands.
Table 3.
Electron Transitions for Protonated [Ru(bpy)2(66′bpy(OH)2)]2+ at the TDDFT level.
| energy (eV) | λ (nm) | Oscillator strength |
|---|---|---|
| 3.02 | 411 | 0.108 |
| 3.06 | 405 | 0.127 |
| 3.77 | 329 | 0.060 |
| 3.84 | 323 | 0.233 |
Table 4.
Electronic Transitions for Deprotonated [Ru(bpy)2(66′bpy(O−)2)] at the TDDFT level (OS greater than 0.025)
| energy (eV) | λ (nm) | Oscillator strength |
|---|---|---|
| 2.60 | 477 | 0.075 |
| 2.76 | 449 | 0.052 |
| 3.09 | 401 | 0.049 |
| 3.13 | 396 | 0.055 |
| 3.16 | 392 | 0.027 |
| 3.18 | 390 | 0.044 |
| 3.26 | 380 | 0.046 |
| 3.28 | 378 | 0.029 |
| 3.44 | 360 | 0.042 |
| 3.56 | 348 | 0.089 |
| 3.65 | 340 | 0.082 |
Figure 7.

Highest occupied molecular orbitals and relative energies involved in electronic transitions for a) [Ru(bpy)2(66′bpy(OH)2)]2+ and b) [Ru(bpy)2(66′bpy(O−)2)].
Upon deprotonation, computational modeling predicts a red shift as is apparent in the experimental data. The data indicates numerous transitions within the range of 477 nm to 340 nm, Table 4. These results are apparent in the experimental spectrum, Figure 6, for [Ru(bpy)2(66′bpy(O−)2)] which appears as broad over a significant portion of the visible region, with a lowest energy wavelength maxima appearing in the range of 504 nm to 546 nm depending upon the solvent (see supplementary materials). These transitions are all widely varied originating from pure metal d orbitals, deprotonated ligand orbitals, and orbitals consisting of a mixture metal and deprotonated ligand, Figure 7. The highest occupied orbitals are higher in energy when compared to the corresponding protonated complex. Most notably, the Highest Occupied Molecular Orbital (HOMO) and HOMO-1 of the deprotonated complex are mixed metal-deprotonated ligand orbitals, which are in contrast to the protonated complex where no mixing is observed between the metal d orbitals and 66′bpy(OH)2 ligand. The transitions occur to π* molecular orbitals on the unsubstituted bpy ligand at lower energy and π* molecular orbitals on the deprotonated 66′bpy(O−)2 ligand at higher energy. The electronic transitions to the substituted 66′bpy(O−)2 ligand occur at higher energy due to the more electron-rich nature of the ligand when compared to bpy. The several transitions consist of MLCT, LLCT, and mixed Metal-Ligand to Ligand Charge Transfer (MLLCT). Similar MLLCT transitions upon changing protonation states with mixed ligand complexes have also been observed in the literature [38, 59, 60]. The implications of the significant metal-66′bpy(O−)2 ligand molecular orbital mixing upon deprotonation cannot be understated. From simple bond order analysis, there is a slight increase in the bond order between the metal and 66′bpy(O−)2 ligand that occurs upon deprotonation of the complex (0.4 to 0.5). These new molecular orbitals shared between the metal and ligand result in stronger bonding character between the metal center and the ligand that is not observed in the protonated state where molecular orbital mixing is absent. This result helps support why the complex would photo-dissociate the ligand to a larger extent in the protonated state compared to the deprotonated state.
3.6 Kinetics of photo-dissociation reactions
The rate of ligand loss from [Ru(bpy)2(66′bpy(OH)2)]2+ to give the active anti-cancer agent ([Ru(bpy)2(solvent)2]2+) depends greatly on pH and the solvent composition. At pH 5 in aqueous solution, [Ru(bpy)2(66′bpy(OH)2)]2+ undergoes blue light induced ligand loss which was monitored by UV/visible spectroscopy over one hour (Figure 8a). At this pH, which is below pKa1 and pKa2, the dihydroxybipyridine ligand should be in its native (protonated) state and should correspond to weaker Ru-N bonds (cf. deprotonated ligand), due to a less electron rich ligand as corroborated by crystallographic and computational results described above. Analysis of λmax = 461 nm showed that the decrease in absorbance appears consistent with a zero order process (kobs = 1.7(2) × 10−8 M/s, R2 = 0.986) with an initial rate (0–300 s) that is independent of ruthenium starting material concentration. This A → B process is consistent with the clean isosbestic point observed, and furthermore, a first or second order process can be ruled out due to nonlinear (curved) plots for these rate laws (see supplementary materials). Note, that this reaction does appear to slow down slightly from 300–3600 s, and this most likely suggests that an equilibrium is being established due to appreciable back reaction to reform [Ru(bpy)2(66′bpy(OH)2)]2+.
Figure 8.

UV/Visible spectra of a) protonated 50 μM [Ru(bpy)2(66′bpy(OH)2)]2+ at pH 5 in aqueous media and b) deprotonated 50 μM [Ru(bpy)2(66′bpy(O−)2)] at pH 7.5 in aqueous media irradiated with 450 nm blue light from (red = —) 0 min to (purple = —) 60 min. c) protonated 50 μM [Ru(bpy)2(66′bpy(OH)2)]2+ in acetonitrile irradiated with 450 nm blue light from (red = —) 0 min to (purple = —) 20 min. The vertical arrows indicate the directions of absorbance changes with time. All spectra were collected at 25 °C.
In contrast, at pH 7.5, there is no measurable photodecomposition of [Ru(bpy)2(66′bpy(OH)2)]2+ to give the active anti-cancer agent (Figure 8b). At this pH, which is above pKa1 and pKa2, the 6,6′-dihydroxybipyridine ligand should be fully deprotonated, which corresponds to stronger Ru-N bonds. Slight fluctuations in the absorbance values can be attributed to noise (see supplementary materials for details), and at this pH the rate of photodecomposition appears to be zero. Thus, the deprotonated, more strongly donating ligand (see computational bond orders above) appears to block photodecomposition. Thus, this establishes the theoretical framework for how a metal-based prodrug can be selectively activated in cancer cells, which have a lower pH as compared to normal cells.
Studies in acetonitrile were done to monitor the process that gave rise to ([Ru(bpy)2(CH3CN)2]2+) and investigate the influence of solvent on the rates. UV/visible studies showed that [Ru(bpy)2(66′bpy(OH)2)]2+ is stable in acetonitrile solution when protected from light or when exposed to 720 nm red light (supplementary materials). In contrast, upon irradiation with blue light (450 nm), [Ru(bpy)2(66′bpy(OH)2)]2+ in acetonitrile undergoes decomposition to give [Ru(bpy)2(CH3CN)2)]2+ and the free ligand, 66′bpy(OH)2 (λmax = 343 nm, 421 nm). These products have been prepared independently and their spectra are consistent with those shown for the final result of photodecomposition (supplementary materials and Figure 8c). The observed zero order rate constant, kobs = 2.6 × 10−7 M/s, was determined by fitting an A → B process at 457 nm (see supplementary materials for details). The large changes in light absorption show a clean isosbestic point at λ = 422 nm and is consistent with an A → B process. Furthermore, the light induced loss of a ligand is consistent with a zero order process that is independent of concentration. The observed rate constant, kobs = 2.6(1) × 10−7 M/s, reflects that photodecomposition is nearly complete (87%) in 5 minutes, and it is entirely complete within 20 minutes, Figure 9c. Thus, the reaction proceeds 15 times more quickly in acetonitrile vs. pH 5 aqueous media. This could be attributed to decreased back reaction in acetonitrile, or enhanced stability of the product ligand in acetonitrile since it is better solvated. The product complex is analogous to the likely product of photodecomposition in vivo, namely [Ru(bpy)2(H2O)2)]2+.
Labile ligands, like acetonitrile or water, make the products of [Ru(bpy)2(66′bpy(OH)2)2)]2+ photodecomposition potentially useful for DNA binding. In a similar fashion, Glazer proposed that [Ru(bpy)2(66′bpy(Me)2)]2+ loses its bulky 6,6′-dimethyl-bipyridine ligand to form the active drug that binds DNA [20]. Studies carried out with [Ru(bpy)2(44′bpy(OH)2)]2+ reveal no photo-dissociation of ligand under conditions of blue light, demonstrating the 6 and 6′ substitution on the photo-dissociating ligand is critical for this mechanism (supplementary materials). Furthermore, the above pH dependent UV-Vis kinetics of photo-dissociation for [Ru(bpy)2(66′bpy(OH)2)2)]2+ in blue light clearly show that at pH 7.5 in water the rate of ligand loss is measured as zero, whereas at pH 5 the ligand readily dissociates. Thus we have demonstrated proof of concept for a prodrug that is selective for more acidic (i.e. cancerous) cells.
4. Conclusion
The design of prodrugs that can be activated selectively are extremely useful in treating diseases such as cancer. The ability to discriminate between healthy cells and cancer cells could limit some of the adverse effects patients normally suffer from when being treated. We have synthesized a new pH sensitive photo-dissociating agent, [Ru(bpy)2(66′bpy(OH)2)]2+. Selective targeting of cancer cells can theoretically be achieved with this prodrug because cancer cells are typically more acidic than normal cells. Thus we have demonstrated a new approach to selectivity: a tumor activated prodrug wherein ligand protonation is the activation event. The photo-dissociating ligand is bound more weakly to the metal center when protonated, and this ensures that the active cell-killing agent should be made in greater quantities in cancerous cells. This strategy will be the foundation for the development of a new class of pH sensitive, tumor activated metallo-prodrugs in which, in future studies, we can vary both the pH and the wavelength at which anti-cancer activity is induced.
Supplementary Material
Highlights.
A pH-sensitive, blue light induced ruthenium complex that is a prodrug for anti-cancer activity has been synthesized.
The metal complex photo-dissociates a ligand to make the anti-cancer agent only under acidic conditions.
The metal complex properties are studied utilizing X-ray crystallography, electrochemistry, UV/visible spectroscopy, and theoretical methods.
Acknowledgments
We would like to thank Nicholas Piro for help with Figure 4. JJP thanks the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research and support from the College of Liberal Arts and Sciences at Villanova University is gratefully acknowledged. ETP thanks NSF CAREER for generous financial support. EJM thanks the UC Technology Accelerator for funding as well as an Institutional Clinical and Translational Science Award, NIH/NCR R Grant Number 1UL1RR026314-01. This article is dedicated to Michael Paul, who lost his battle with cancer early this year. This article is also dedicated to Jessica Bongiovanni, a former undergraduate student in the Papish group, whose diagnosis and death from cancer in 2012 helped inspire this project.
5. Abbreviations
- 66′bpy(OH)2
6,6′-dihydroxy-2,2′-bipyridine
- bpy
2,2′-bipyridine
- 44′bpy(OH)2
4,4′-dihydroxy-2,2′-bipyridine
- 66′bpy(Me)2
6,6′-dimethyl-2,2′-bipyridine
- Cp*
1,2,3,4,5-pentamethylcyclopentadiene
- CH3CN
acetonitrile
- PF6
hexafluorophosphate
- TBA
tetrabutylammonium
- SCE
Saturated Calomel Electrode
- DFT
Density Functional Theory
- TDDFT
time-dependent DFT
- LED
light emitting diode
- 47phen(OH)2
4,7-dihydroxy-1,10-phenanthroline
- MLCT
metal to ligand charge transfer
- LLCT
ligand to ligand charge transfer
- HOMO
highest occupied molecular orbital
- MLLCT
mixed metal-ligand to ligand charge transfer
Footnotes
Infrared spectroscopy, 1H-NMR spectroscopy, cyclic voltammetry, pH titration, UV-visible spectroscopy, kinetics, and crystallographic data in CIF format. Cartesian coordinates for each species and GAMESS input parameters are also included.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yarema KJ, Lippard SJ, Essigmann JM. Nucleic Acids Res. 1995;23:4066–4072. doi: 10.1093/nar/23.20.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ang WH, Myint M, Lippard SJ. J Am Chem Soc. 2010;132:7429–7435. doi: 10.1021/ja101495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li G, Bell T, Merino EJ. Chem Med Chem. 2011;6:869–875. doi: 10.1002/cmdc.201100014. [DOI] [PubMed] [Google Scholar]
- 4.Jones AR, Bell-Horwath TR, Li G, Rollmann SM, Merino EJ. Chem Res Toxicology. 2012;25:2542–2552. doi: 10.1021/tx300337j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandlin RD, Starling MP, Williams KM, Inorg J. Biochemistry. 2010;104:214–216. doi: 10.1016/j.jinorgbio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci USA. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheung-Ong K, Song KT, Ma Z, Shabtai D, Lee AY, Gallo D, Heisler LE, Brown GW, Bierbach U, Giaever G, Nislow C. ACS Chem Biology. 2012;7:1892–1901. doi: 10.1021/cb300320d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotsova I. Curr Med Chem. 2006;13:1085–1107. doi: 10.2174/092986706776360941. [DOI] [PubMed] [Google Scholar]
- 9.Bergamo A, Gaiddon C, Schellens JHM, Beijnen JH, Sava G. J Inorg Biochemistry. 2012;106:90–99. doi: 10.1016/j.jinorgbio.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 10.Antonarakis ES, Emadi A. Cancer Chemotherapy and Pharmacology. 2010;66:1–9. doi: 10.1007/s00280-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murdter TE, Firedel G, Backman JT, McClellan M, Schick M, Gerken M, Bosslet K, Fritz P, Toomes H, Kroemer HK, Sperker B. J Pharm and Exptl Therapeutics. 2002;301:223–228. doi: 10.1124/jpet.301.1.223. [DOI] [PubMed] [Google Scholar]
- 12.Denny WA. Eur J Med Chem. 2001;36:577–595. doi: 10.1016/s0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 13.Tietze LF, Neumann M, Mollers T, Fischer R, Glusenkamp KH, Rajewsky MF, Jahde E. Cancer Research. 1989;49:4179–4184. [PubMed] [Google Scholar]
- 14.Gillies ER, Goodwin AP, Frechet MJ. Bioconjugate Chem. 2004;15:1254–1263. doi: 10.1021/bc049853x. [DOI] [PubMed] [Google Scholar]
- 15.Tietze LF, Lieb M, Herzig T, Haunert F, Schuberth I. Bioorg Med Chem. 2001;9:1929–1939. doi: 10.1016/s0968-0896(01)00098-0. [DOI] [PubMed] [Google Scholar]
- 16.Kuin A, Aalders M, Lamfers M, van Zuidam DJ, Essers M, Beijnen JH, Smets LA. Brit J Cancer. 1999;79:793–801. doi: 10.1038/sj.bjc.6690127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tietze LF, Hannemann R, Buhr W, Logers M, Menningen P, Lieb M, Starck D, Grote T, Doring A, Schuberth I. Angew Chem Int Ed. 1996;35:2674–2677. [Google Scholar]
- 18.Farrer NJ, Woods JA, Munk VP, Mackay FS, Sadler PJ. Chem Res Toxicology. 2010;23:413–421. doi: 10.1021/tx900372p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pracharova J, Zerzankova L, Stepankova J, Novakova O, Farrer NJ, Sadler PJ, Brabec V, Kasparkova J. Chem Res Toxicology. 2012;25:1099–1111. doi: 10.1021/tx300057y. [DOI] [PubMed] [Google Scholar]
- 20.Howerton BS, Heidary DK, Glazer EC. J Am Chem Soc. 2012;134:8324–8327. doi: 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- 21.Wachter E, Heidary DK, Howerton BS, Parkin S, Glazer EC. Chem Comm. 2012;48:9649–9651. doi: 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]
- 22.Durham B, Caspar JV, Nagle JK, Meyer TJ. J Am Chem Soc. 1982;104:4803–4810. [Google Scholar]
- 23.Durham B, Walsh JL, Carter CL, Meyer TJ. Inorg Chem. 1980;19:860–865. [Google Scholar]
- 24.Van Houten J, Watts RJ. J Am Chem Soc. 1976;98:4853–4858. [Google Scholar]
- 25.Hashiguchi BG, Young KJH, Yousufuddin M, Goddard WA, III, Periana RA. J Am Chem Soc. 2010;132:12542–12545. doi: 10.1021/ja102518m. [DOI] [PubMed] [Google Scholar]
- 26.Crabtree RH. Science. 2010;330:455–456. doi: 10.1126/science.1197652. [DOI] [PubMed] [Google Scholar]
- 27.Nieto I, Livings MS, Sacci Jb, III, Reuther LE, Zeller M, Papish ET. Organometallics. 2011;30:6339–6342. [Google Scholar]
- 28.Kawahara R, Fujita K-i, Yamaguchi R. J Am Chem Soc. 2012;134:3643–3646. doi: 10.1021/ja210857z. [DOI] [PubMed] [Google Scholar]
- 29.Himeda Y, Onozawa-Komatsuzaki N, Sugihara H, Kasuga K. Organometallics. 2007;26:702–712. [Google Scholar]
- 30.Himeda Y. Eur J Inorg Chem. 2007:3927–3941. [Google Scholar]
- 31.Himeda Y, Onozawa-Komatsuzaki N, Miyazawa S, Sugihara H, Hirose T, Kasuga K. Chem Eur J. 2008;14:11076–11081. doi: 10.1002/chem.200801568. [DOI] [PubMed] [Google Scholar]
- 32.Constable EC, Housecroft CE, Thompson AC, Passaniti P, Silvi S, Maestri M, Credi A. Inorg Chim Acta. 2007;360:1102–1110. [Google Scholar]
- 33.Gao F, Chen X, Sun Q, Cao JN, Lin JQ, Xian QZ, Ji LN. Inorg Chem Comm. 2012;16:25–27. [Google Scholar]
- 34.Thompson AMWC, Smailes MCC, Jeffery JC, Ward MD. Dalton Trans. 1997:737–744. [Google Scholar]
- 35.Hull JF, Himeda Y, Wang WH, Hashiguchi B, Periana R, Szalda DJ, Muckerman JT, Fujita E. Nature Chem. 2012;4:383–388. doi: 10.1038/nchem.1295. [DOI] [PubMed] [Google Scholar]
- 36.Wang WH, Hull JF, Muckerman JT, Fujita E, Himeda Y. Energy Environ Sci. 2012;5:7923–7926. [Google Scholar]
- 37.DePasquale J, Nieto I, Reuther LE, Herbst-Gervasoni CJ, Paul JJ, Mochalin V, Zeller M, Thomas CM, Addison AW, Papish ET. Inorg Chem. 2013;52:9175–9183. doi: 10.1021/ic302448d. [DOI] [PubMed] [Google Scholar]
- 38.Klein S, Dougherty WG, Kassel WS, Dudley TJ, Paul JJ. Inorg Chem. 2011;50:2754–2763. doi: 10.1021/ic1017054. [DOI] [PubMed] [Google Scholar]
- 39.Fuentes MJ, Bognanno RJ, Dougherty WG, Boyko WJ, Kassel WS, Dudley TJ, Paul JJ. Dalton Trans. 2012;41:12514–12523. doi: 10.1039/c2dt31706k. [DOI] [PubMed] [Google Scholar]
- 40.Singh Y, Palombo M, Sinko PJ. Curr Med Chem. 2008;15:1802–1826. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broomhead JA, Young CG. Inorg Synthesis. 1982;21:127–128. [Google Scholar]
- 42.Ji Z, Huang SD, Gaudalupe AR. Inorg Chim Acta. 2000;305:127–134. [Google Scholar]
- 43.Liu Y, Turner DB, Singh TN, Angeles-Boza AM, Chouai A, Dunbar KR, Turro C. J Am Chem Soc. 2009;131:26–27. doi: 10.1021/ja806860w. [DOI] [PubMed] [Google Scholar]
- 44.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA. J Comput Chem. 1993;14:1347–1363. [Google Scholar]
- 46.Sakai Y, Miyoshi E, Klobukowski M, Huzinaga S. J Comput Chem. 1987;8:256–264. [Google Scholar]
- 47.Lovallo CC, Klobukowski M. J Comput Chem. 2004;25:1206–1213. doi: 10.1002/jcc.20044. [DOI] [PubMed] [Google Scholar]
- 48.Bell-Horwath TR, Vadukoot AK, Thowkeik FS, Li G, Wunderlich M, Mulloy JC, Merino EJ. Bioorg Med Chem Lett. 2013;23:2951–2954. doi: 10.1016/j.bmcl.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Proc Natl Acad Sci USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solivio M, Nemera DB, Sallans L, Merino EJ. Chem Res Toxicology. 2012;25:326–336. doi: 10.1021/tx200376e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park GY, Wilson JJ, Song Y, Lippard SJ. Proc Natl Acad Sci USA. 2012;109:11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heeg MJ, Kroener R, Deutsch E. Acta Crystallogr, Sect C. 1985;41:684–686. [Google Scholar]
- 53.Xu F, Huang W. Acta Crystallogr, Sect E. 2007;63:m2114. [Google Scholar]
- 54.Zayat L, Salierno M, Etchenique R. Inorg Chem. 2006;45:1728–1731. doi: 10.1021/ic0512983. [DOI] [PubMed] [Google Scholar]
- 55.Cruz AJ, Kirgan R, Siam K, Heiland P, Rillema DP. Inorg Chim Acta. 2010;363:2496–2505. [Google Scholar]
- 56.Rillema DP, Jones DS. Chem Commun. 1979:849–851. [Google Scholar]
- 57.Zakeeruddin SM, Fraser DM, Nazeeruddin MK, Gratzel M. J Electroanal Chem. 1992;337:253–283. [Google Scholar]
- 58.Giordano PJ, Bock CR, Wrighton MS. J Am Chem Soc. 1978;100:6960–6965. [Google Scholar]
- 59.Lancaster KM, Gerken JB, Durrell AC, Palmer JH, Gray HB. Coord Chem Rev. 2010;254:1803–1811. [Google Scholar]
- 60.Das S, Saha D, Karmakar S, Baitalik S. J Phys Chem A. 2012;116:5216–5226. doi: 10.1021/jp300820p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.