

Abstract
Connecting tubule glomerular feedback (CTGF) is a mechanism in which Na reabsorption in the connecting tubule (CNT) causes afferent arteriole (Af-Art) dilation. CTGF is mediated by eicosanoids, including prostaglandins and epoxyeicosatrienoic acids (EETs), however their exact nature and source remain unknown. We hypothesized that during CTGF, the CNT releases prostaglandin E2 (PGE2), which binds EP4 receptors and dilates the Af-Art. Rabbit Af-Arts with the adherent CNT intact were microdissected, perfused, and preconstricted with norepinephrine. CTGF was elicited by increasing luminal NaCl in the CNT from 10 to 80 mmol/L. We induced CTGF with or without the EP4 receptor blocker ONO-AE3-208 added to the bath in the presence of the EET synthesis inhibitor MS-PPOH. ONO-AE3-208 abolished CTGF (control: 9.4±0.5, MS-PPOH+ONOAE3-208: −0.6±0.2 μm, p<0.001, n=6). To confirm these results we used a different, specific EP4 blocker, L161982 (10−5 mol/L) which also abolished CTGF (control: 8.5±0.9, MS-PPOH+L161982: 0.8±0.4μm, p<0.001, n=6). To confirm that the eicosanoids that mediate CTGF are released from the CNT rather than the Af-Art, we first disrupted the Af-Art endothelium with an antibody and complement. Endothelial disruption did not affect CTGF (7.9±0.9 vs. 8.6±0.6 μm, p=NS, n=7). We then added arachidonic acid (AA) to the lumen of the CNT while maintaining zero NaCl in the perfusate. AA caused dose-dependent dilation of the attached Af-Art (from 8.6±1.2 to 15.3±0.7 μm, p<0.001, n=6), and this effect was blocked by ONO-AE3-208 (10−7 mol/L). We conclude that during CTGF, the CNT releases prostaglandin E2, which acts on EP4 on the Af-Art inducing endothelium-independent dilation.
Keywords: arterioles; endothelium; prostaglandin E2; arachidonic acid PGE Receptor (EP4 Subtype); microcirculation; 11,12-epoxy-5,8,14-eicosatrienoic acid
Introduction
In the kidney, the afferent arteriole (Af-Art) is a major contributor to vascular resistance and thus it controls glomerular hemodynamics. At least three intrinsic mechanisms determine the tone of the Af-Art, namely the myogenic response, macula densa-mediated tubuloglomerular feedback (TGF), and connecting tubule-mediated glomerular feedback (CTGF). The mechanisms of myogenic response and TGF have been extensively studied; however, the newly discovered CTGF is not completely understood.
In rabbits, rats, mice, and humans, the CNT is in close contact with the Af-Art, 1–4. We have reported that CTGF regulates Af-Art tone both in vitro 4–7 and in vivo 8,9 by causing dilation of the Af-Art in response to increases in NaCl in the lumen of the attached CNT. In vivo, CTGF antagonizes the constrictor effect of TGF on the Af-Art8, and partly mediates the resetting of TGF induced by volume expansion9. In vitro, we studied the pathways that mediate CTGF, we found that Na entry into the CNT via the epithelial Na channel (ENaC) is required to induce CTGF and that eicosanoids derived from arachidonic acid (AA), including epoxyeicosatrienoic acids (EETs) and prostaglandins mediate CTGF 5. Possible sources for these eicosanoids include the CNT itself and/or the Af-Art endothelium.
Prostaglandins mediate about half of the CTGF response, while the other half is accounted for by EETs, however the exact identity of the prostaglandin and prostaglandin receptor involved remain unknown. PGE2 and PGI2, the major renal metabolites of cyclooxygenase, are both synthesized by the vascular endothelium 10,11. In addition, PGE2 is also synthesized in the nephron, with strong expression of microsomal PGE2 synthase in the CNT 12. Both PGI2 and PGE2 are vasodilators, with PGE2 being a more potent renal vasodilator compared with PGI213. The actions of PGE2 are due to its interaction with EP receptors (PGE2 receptors). There are four EP receptors, EP1, EP2, EP3 and EP4, however EP4 is the only vasodilator EP receptor expressed in the afferent arteriole 14.
We hypothesized that Na reabsorption by the CNT induces release of PGE2, which binds EP4 receptors and dilates the Af-Art in an endothelium-independent manner. CTGF was studied by using an in vitro preparation developed by us, in which an Af-Art and its adherent CNT are isolated and microperfused simultaneously. This approach avoids the confounding influence of the multiple systemic factors that regulate the renal microcirculation.
Methods
New Zealand White rabbits weighing 1.5–2 Kg (Myrtle's rabbitry, TN) were given standard chow (Ralston Purina, St. Louis, MO) and tap water ad libitum and anesthetized with ketamine (50 mg/kg i.m.), xylazine (10 mg/kg i.m.) and pentobarbital (25 mg/kg i.v.). All protocols were approved by Henry Ford Health System's Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. We used rabbits because their CNTs are well demarcated and microdissection of the CNT and attached Af-Art is easier than in rats or mice. To isolate and microperfuse the Af-Art and CNT, we used methods similar to those described previously 4,15. The kidneys were sliced along the corticomedullary axis and slices were placed in ice-cold minimum essential medium (MEM; Gibco Laboratories, Grand Island, NY) containing 5% bovine serum albumin (BSA; Sigma, St. Louis, MO). Using fine forceps, a single superficial Af-Art with its glomerulus intact was dissected together with the adherent CNT. Using a micropipette, the microdissected complex was transferred to a temperature-regulated perfusion chamber mounted on an inverted microscope with Hoffmann modulation. Both the Af-Art and CNT were cannulated with an array of concentric glass pipettes as described previously 16,17. This system allows us to exchange the perfusion solution in a few seconds, while keeping the holding and perfusion pipettes in place. The Af-Art was perfused with MEM containing 5% BSA gassed with room air. Intraluminal pressure was measured by Landis' technique and maintained at 60 mm Hg. The CNT perfusion solution contained (in mmol/L): 4 KHCO3, 10 HEPES, 0.5 Na acetate, 0.5 Na lactate, 0.5 K2HPO4, 1.2 MgSO4, 1 CaCO3, and 5.5 glucose, adding 1 mol/L NaCl to achieve the desired final NaCl concentration. Tubular perfusion was controlled by use of a syringe microperfusion pump (Harvard Apparatus Inc, Mass) set to 20 nL/min (calibration checked to be ≈ 20 nL/min), which is within the range of physiological flow rates 18,19. The bath was superfused with MEM containing 0.15% BSA at a rate of 1 mL/min.
Microdissection and cannulation of the Af-Art and CNT were completed within 90 min at 8°C, after which the temperature was gradually raised to 37°C. Once it was stable, a 30-min equilibration period was allowed before taking any measurements. Since isolated arteries have little or no tone and our preliminary studies showed that increasing NaCl in the CNT perfusate caused only modest dilation of the Af-Art unless it was preconstricted, we performed all CTGF experiments with Af-Arts preconstricted with norepinephrine (NE; 2–5 × 10−7 mol/L) to about half of its baseline diameter.
Experimental protocols
-
1)
Time control for experiments #2 and #3: three consecutive concentration-response curves were generated by increasing luminal NaCl in the CNT from 10 to 80 mmol/L.
-
2)
Effect of the EP4 antagonist ONO-AE3-208 on CTGF: three consecutive concentration-response curves were generated by increasing luminal NaCl in the CNT from 10 to 80 mmol/L. The EET synthesis inhibitor MS-PPOH (10−6 mol/L) was added to the second and third curves, and the EP4 antagonist ONO-AE3-208 (10−7 mol/L) to the third curve. This concentration of ONO-AE3-208 is 77 times its Ki for the EP4 receptor, and at least 100 times lower than its Ki for the EP2 receptor 20.
-
3)
Effect of the EP4 antagonist L161982 on CTGF: this experiment was similar to #2 but the EP4 antagonist L161982 (10−5 mol/L) was used instead of ONO-AE3-208. This concentration of L161982 is 312 times its Ki for the EP4 receptor, and 6 times lower than its Ki for the EP2 receptor 21.
-
4)
Effect of endothelium disruption on CTGF: CTGF was induced by increasing NaCl in the CNT from 10 to 80 mmol/L. Then a goat anti-human antibody against von Willebrand factor (14.29 mg/ml diluted 1:1000) plus 2% guinea pig complement were perfused into the lumen of the Af-Art for 10 minutes followed by a 20-minute wash-out period, and CTGF was induced again. To confirm complete functional removal of the endothelium, we added acetylcholine to the lumen of the Af-Art, 10−5 mol/L, a concentration we have repeatedly shown to be sufficient to dilate the Af-Art.
-
5)
Effect of exogenous arachidonic acid (AA) in the CNT: After the Af-Art was preconstricted with NE, AA was added to the lumen of the CNT at increasing concentrations from 10−7 to 10−5 mol/L in the absence of NaCl. At the end of the experiment, we removed AA and switched the CNT luminal perfusate to 80 mmol/L NaCl.
-
6)
Effect of an EP4 antagonist on CTGF induced by exogenous AA: this experiment was similar to #5, except that MS-PPOH was added to the lumen of the CNT and ONO-AE3-208 was added to the bath.
In all experiments, Af-Art diameter was measured in the region of maximal response to NE at three sites 3–5 μm apart and expressed as the average of these three measurements. Diameter was recorded at 5-second intervals with a video camera and measured with a computer equipped with Metavue image analysis software (MDS Analytical Technologies, Toronto, Ontario, Canada).
Chemicals
ONO-AE3-208 was a gift from Ono Pharmaceutical Co (Osaka, Japan), and MS-PPOH was a gift from Dr J.R. Falck (University of Texas Southwestern Medical Center, Dallas, Texas). L161982 was purchased from Tocris Bioscience (Ellisville, MO), norepinephrine and acetylcholine from Sigma-Aldrich (St Louis, MO), and arachidonic acid from Cayman Chemical (Ann Arbor, MI).
Statistics
Values are expressed as mean ± SEM. Paired t-tests were used to compare CTGF (Δ Af-Art diameter) between the control and experimental periods. Hochberg's step-up procedure was used to adjust the P values for multiple comparisons so that the familywise type I error rate, predefined as 0.05, was controlled.
Results
To test whether CTGF is mediated by EETs and PGE2 produced endogenously in response to increases in luminal NaCl in the CNT, we conducted a series of experiments involving three consecutive CTGF responses. First, we performed a control experiment by preconstricting the Af-Art with norepinephrine and repeatedly switching NaCl in the tubular perfusate from 10 to 80 mmol/L. Increasing NaCl in the CNT to 80 mmol/L dilated the attached Af-Art, and switching NaCl back to 10 mmol/L returned the Af-Art diameter to preconstricted levels. Furthermore, all three CTGF responses were similar, indicating that CTGF is reproducible and stable over time (Fig 1).
Figure 1.
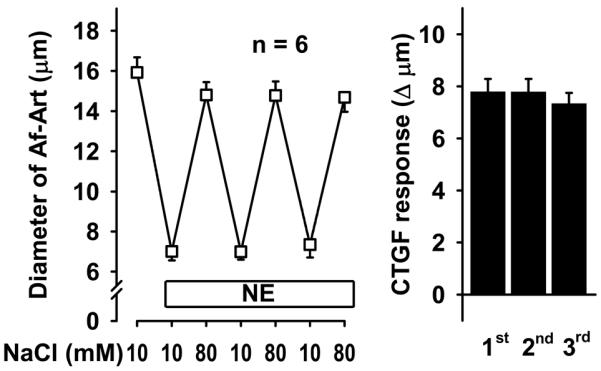
Repeatedly increasing NaCl concentration in the CNT dilated preconstricted Af-Art in a similar manner, indicating that CTGF is stable and reproducible over time.
To test whether PGE2, acting via EP4 receptors, mediates CTGF along with EETs, we again induced CTGF three consecutive times, first with vehicle, then adding the EET synthesis inhibitor MS-PPOH, and finally adding both MS-PPOH and the EP4 antagonist ONO-AE3-208. CTGF was attenuated by MS-PPOH, and abolished by the combination of MS-PPOH and ONO-AE3-208 (Fig 2). We confirmed these results with a second EP4 blocker, L161982. Again, CTGF was attenuated by inhibition of EET synthesis, and abolished by addition of the EP4 blocker (Fig 3). Taken together these data suggest that during CTGF, in addition to EETs, the CNT releases PGE2, which act on EP4 receptors in the Af-Art to cause vasodilation.
Figure 2.
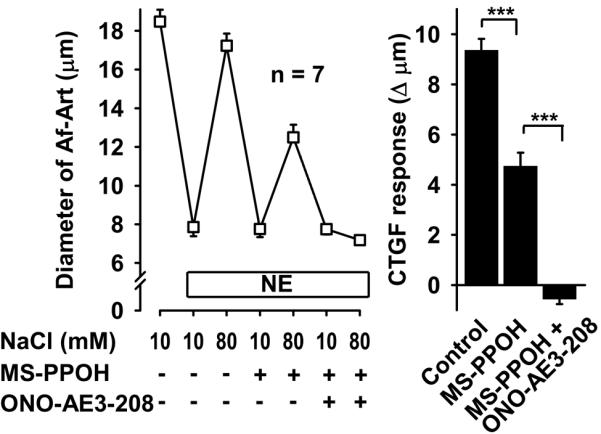
In the presence of the EET synthesis inhibitor MS-PPOH (10−6 mol/L), addition of the EP4 receptor blocker ONO-AE3-208 (10−7 mol/L) completely inhibited CTGF, suggesting that PGE2 acts on EP4 receptor on the Af-Art. *** P < 0.001.
Figure 3.
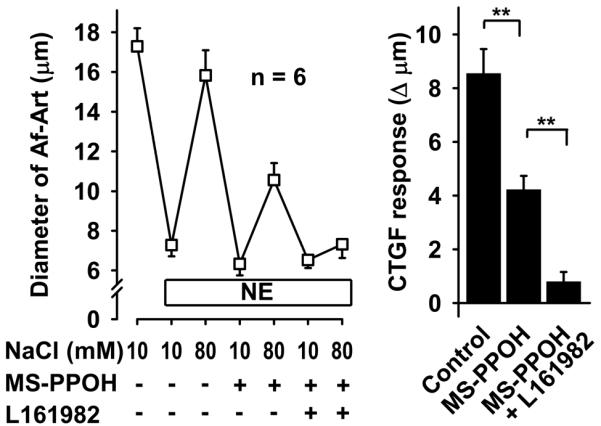
In the presence of the EET synthesis inhibitor MS-PPOH (10−6 mol/L), addition of the EP4 receptor blocker L161982 (10−5 mol/L) completely inhibited CTGF, suggesting that PGE2 acts on EP4 receptor on the Af-Art. ** P < 0.01.
To test whether the eicosanoids that mediate CTGF are released from the Af-Art endothelium or the CNT, we studied CTGF before and after Af-Art endothelium removal with an antibody directed to von Willebrand factor (an antigen present in the endothelium, also referred to as factor VIII-related antigen) and complement, perfused into the lumen of the Af-Art. Endothelium removal did not alter CTGF (Fig 4). At the end of the experiment, we confirmed that endothelium was functionally removed by adding the endothelium-dependent vasodilator, acetylcholine, into the lumen of the Af-Art, which failed to dilate the preconstricted Af-Arts. These data suggest that during CTGF, eicosanoids are released from the CNT, rather than from the Af-Art endothelium.
Figure 4.
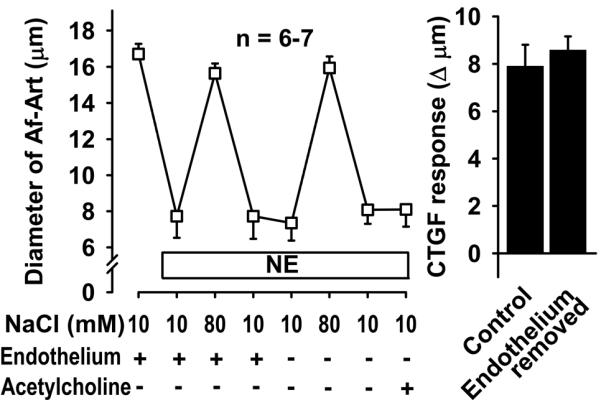
Removal of endothelium-dependent relaxation by perfusion of the Af-Art with an antibody (anti-von Willebrand factor) and complement did not alter CTGF. This suggests that the eicosanoids that mediate CTGF are released from the CNT, rather than the Af-Art endothelium. Acetylcholine failed to dilate preconstricted Af-Arts, suggesting successful endothelial removal with our antibody/complement method.
To confirm that the CNT produces the eicosanoids that mediate CTGF we added AA, the substrate for eicosanoids synthesis, into the lumen of the CNT while perfusing the tubule with zero NaCl. Addition of AA to the CNT dose dependently dilated the attached preconstricted Af-Art, returning the Af-Art diameter to its baseline, i.e. completely reversing norepinephrine-induced vasoconstriction. This is similar to what we observe when we induce CTGF by increasing luminal NaCl in the CNT (Fig 5). Because we reported that EETs mediate about half of CTGF, and we hypothesized the other half is due to PGE2 acting on EP4 receptors, we repeated this experiment in the presence of both MS-PPOH and ONO-AE3-208. Addition of AA to the CNT lumen failed to elicit Af-Art dilation in the presence of MS-PPOH and ONO-AE3-208 (Fig 6). These data suggest that the CNT is capable of producing EETs and PGE2, which cause dilation of its attached Af-Art.
Figure 5.
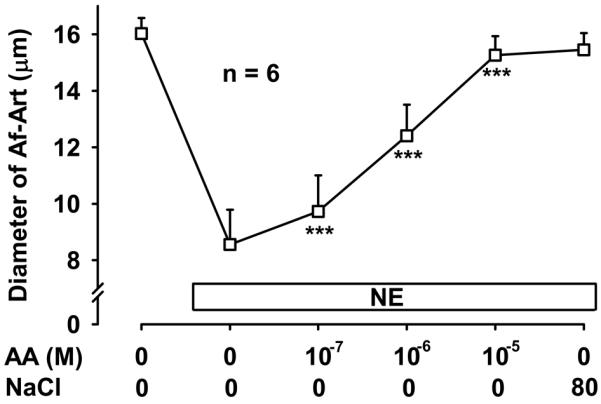
Addition of exogenous arachidonic acid (AA) to the CNT lumen in the absence of NaCl caused dose-dependent dilation of the attached Af-Art. *** P < 0.001 vs. preconstricted Af-Art diameter. The maximal vasodilation was similar to that achieved by CTGF induced with 80 mmol/L NaCl in the lumen of the CNT.
Figure 6.
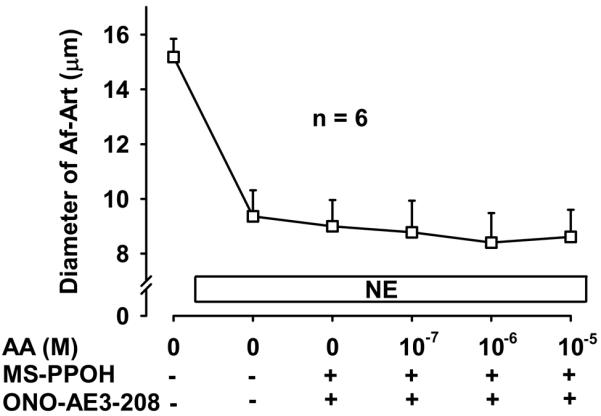
Addition of the EP4 blocker ONO-AE3-208 (10−7 mol/L) and the EET synthesis inhibitor MS-PPOH (10−6 mol/L) completely prevented the vasodilation induced by arachidonic acid (AA) added to the lumen of the CNT.
Discussion
We previously provided direct evidence of cross-talk between CNT and Af-Art4. It is initiated by increasing NaCl concentration in the lumen of the CNT, which stimulates Na transport via ENaC in the CNT and dilates the Af-Art. We also found that two classes of AA metabolites, EETs and PGs are involved. In this study, we demonstrated for the first time that the eicosanoids that mediate CTGF are produced in the CNT rather than in the Af-Art, that the prostaglandin that mediates CTGF is PGE2, and that the prostaglandin receptor involved is EP4.
We previously reported that addition of indomethacin to the CNT partially inhibits CTGF, suggesting that CTGF is mediated by a COX-derived metabolite such as PGE2 and PGI2, both of which are synthesized in the kidney and can regulate renal hemodynamics11,22,23. Here, we found that CTGF is mediated by PGE2. Our data is consistent with previous reports that PGE2 influences renal vascular resistance, causing an increase in renal vascular cAMP levels and inducing relaxation of preglomerular vessels24,25, while PGI2 was a less likely candidate to mediate CTGF because it is less potent as a renal vasodilator13,26. Furthermore, our finding that CTGF prostanoids are derived from the CNT rather than the Af-Art endothelium (see below), supports the hypothesis that PGE2 is the mediator of CTGF, since PCR studies have shown that connecting tubules express mPGES, the enzyme that synthesizes PGE2, while PGI2 synthase is not expressed in the nephron27. In addition, localization of mPGES by immunolabeling showed expression in the principal cells of the CNT (i.e. the cells that express ENaC and initiate CTGF)12, where it co-localizes with COX127, but not COX228.
PGE2 is a major renal cyclooxygenase metabolite of AA and interacts with four G protein-coupled receptors designated EP1, EP2, and EP3 and EP4. Acting through these receptors, PGE2 modulates renal hemodynamics as well as salt and water excretion. Generally, activation of EP1 and EP3 results in vasoconstriction29,30, while EP2 and EP4 receptors mediate vasodilation by activating Gs coupled to adenylate cyclase and elevating intracellular cAMP levels31,32 and could potentially mediate CTGF. Here we found that CTGF is mediated by EP4, since blockade of EP4 receptors with either of two different antagonists abolished CTGF. Our data is consistent with previous reports that butaprost, a PGE2 analog that preferentially activates EP2 is relatively ineffective as a vasodilator of the Af-Art, and that Af-Arts express EP4 but not EP214. Likewise, Purdy et al showed that EP4 is the major receptor in the preglomerular vasculature where it mediates the vasodilatory effects of PGE233. Furthermore, regulation of plasma renin activity and intrarenal renin mRNA is not different in wild-type and EP2-knockout mice34, thus the EP2 receptor does not appear to mediate the effects of PGE2 in the Af-Art and was therefore an unlikely candidate to mediate CTGF. Taken together, these previous reports and our present data indicate that CTGF is mediated by PGE2 acting on EP4 receptors.
The exact mechanism by which Na entry into the principal cells of the CNT can induce PGE2 production is unknown, however, because Na entry via ENaC leads to increases in intracellular Ca35, it is possible that Ca-dependent activation of phospholipases in the CNT cells initiate a cascade leading to PGE2 production.
The local concentration of PGE2 produced by the CNT during CTGF is not known, however, from a functional point of view, its effect is comparable to that of exogenous PGE2 when applied to isolated Af-Arts at a concentration of 10−7 mol/L (see Fig S1). Similar to endogenous CTGF, this concentration of PGE2 dilates the Af-Art to about 50% of its basal diameter from a preconstricted state. Likewise, the same concentration of the EP4 receptor antagonist L161982, that completely blocks endogenous CTGF can also completely block the vasodilatory effect of PGE2 10−7 mol/L. These data suggest that the local concentration of PGE2 in the CNT during CTGF may be approximately 10−7 mol/L.
We have previously reported that CTGF is mediated by EETs and prostaglandins, with each of these classes of eicosanoids accounting for about half of the vasodilatory effect of CTGF5. Here, we tested whether these eicosanoids were derived from CNT epithelial cells or from the Af-Art endothelium. For this, we took two approaches; first we studied CTGF in the absence of a functional endothelium. We found that endothelial removal did not affect CTGF, indicating that CTGF is endothelium-independent. Next, we tested the ability of the CNT to generate vasodilator eicosanoids when AA, the substrate for eicosanoid generation, was infused in the lumen of the tubule. AA dilated the attached Af-Art indicating the CNT is able to produce dilator eicosanoids that can act on the attached Af-Art and cause dilation. Prostanoids are generally considered to be locally acting factors10,11 that modulate cellular function in the vicinity of their site of generation. This means that prostaglandins can act within the same compartment, such as endothelium-derived prostaglandins causing relaxation of vascular smooth muscle cells in a vessel. However, it is also possible for prostaglandins to be generated in one structure and function in another. For example, inhibition of prostaglandin synthesis with indomethacin increased the sensitivity of the efferent arteriole to vasoconstrictors when the arteriole was perfused orthogradelly (through the glomerulus), but not when it was perfused retrogradelly, suggesting that prostaglandins produced in the glomerulus can act on the efferent arteriole36. Also in the outer medulla, PGE2 is produced in collecting duct epithelial and interstitial cells and causes dilation of neighboring descending vasa recta vessels37.
Of note, our finding that AA added to the lumen of the CNT dilates the Af-Art cannot be explained by diffusion of the AA to act directly on the Af-Art, because AA, when added to isolated Af-Arts is a vasoconstrictor, rather than a vasodilator38, thus the CNT is necessary to convert AA to vasodilator eicosanoids. In addition, the setting of our preparation makes direct diffusion from the tubular perfusate to the Af-Art very unlikely, since the effluent from the tubular perfusate is diluted 50,000 times in the bath before it can act on the Af-Art.
The interaction between prostaglandins and angiotensin II (Ang II) plays a critical role in the modulation of the renal microcirculation. Specifically, Ang II causes constriction of the renal vasculature, and decreases renal blood flow. However, Ang II also increases the production of prostaglandins by the kidney11,39, and these prostaglandins partially buffer the decrease in renal blood flow induced by Ang II13,40. Microdissected superficial Af-Arts demonstrate an enhanced responsiveness to Ang II during COX inhibition41,42. This likely to be due to the fact that Ang II induces the expression of COX2 in some nephron segments, such as the thick ascending limb and macula densa43. However, it is also likely that it involves CTGF. We recently reported that Ang II acting in the lumen CNT enhances CTGF6. Taken together with our present findings that PGE2 mediates CTGF, it is likely that Ang II, by enhancing CTGF, increases PGE2 and partially counters its direct constrictor effect on the Af-Art.
Perspectives
We report here for the first time that CTGF is an endothelium-independent vasodilator mechanism of the Af-Art, that the CNT is capable of producing and releasing vasodilator eicosanoids that act in a paracrine manner on the Af-Art, and that during CTGF, increases in NaCl in the lumen of the CNT trigger CTGF which is mediated by EETs and also by PGE2, which acts on EP4 receptors in the Af-Art to cause vasodilation. Our findings shed light on the physiological regulation of renal hemodynamics by the nephron, as well as its modification by pharmacological agents. For example, non-steroidal anti-inflammatory drugs are widely used medications with well-known adverse effects on the kidney function. Of relevance, they inhibit the production of vasodilatory prostaglandins such as PGE2, thus decreasing renal blood flow44,45. Our studies suggest that part of this detrimental effect may be due to blockade of CTGF.
Novelty and Significance.
What is New?
Connecting tubule glomerular feedback, or CTGF, is a novel mechanism of regulation of the microcirculation in the kidney. This mechanism is activated by the presence of increased concentrations of sodium chloride (salt) in a segment of the kidney tubules known as the connecting tubule, and causes the afferent arteriole (a vessel that is right next to the connecting tubule) to relax.
Our data are the first to show that CTGF is mediated by small lipid molecules known as eicosanoids, including epoxyeicosatrienoic acids and prostaglandin E2. We also show for the first time that said eicosanoids are produced by the connecting tubule.
What is Relevant?
The kidney plays a key role in high blood pressure. Kidney diseases cause hypertension, and hypertension can in turn damage the kidney.
Much of the renal function is controlled by the blood supply through the afferent arteriole, which in turn is in part controlled by CTGF.
Our studies will help to better understand the control of the renal microcirculation and function.
Summary
We found that during CTGF, the connecting tubule produces eicosanoids, including epoxyeicosatrienoic acids and prostaglandin E2, that dilate the afferent arteriole. We also found that prostaglandin E2 does this by activating its type 4 receptor.
Acknowledgements
None.
Source(s) of Funding: This work was supported by a grant from the National Institutes of Health (HL28982).
Footnotes
Conflict(s) of Interest/Disclosure(s): None.
References
- 1.Barajas L, Powers K, Carretero O, Scicli AG, Inagami T. Immunocytochemical localization of renin and kallikrein in the rat renal cortex. Kidney Int. 1986;29:965–4. doi: 10.1038/ki.1986.94. [DOI] [PubMed] [Google Scholar]
- 2.Vio CP, Figueroa CD, Caorsi I. Anatomical relationship between kallikrein-containing tubules and the juxtaglomerular apparatus in the human kidney. Am J Hypertens. 1988;1:269–4. doi: 10.1093/ajh/1.3.269. [DOI] [PubMed] [Google Scholar]
- 3.Sipos A, Toma I, Kang JJ, Rosivall L, Peti-Peterdi J. Advances in renal (patho)physiology using multiphoton microscopy. Kidney Int. 2007;72:1188–4. doi: 10.1038/sj.ki.5002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int. 2007;71:1116–4. doi: 10.1038/sj.ki.5002190. [DOI] [PubMed] [Google Scholar]
- 5.Ren Y, D'Ambrosio MA, Garvin JL, Wang H, Carretero OA. Possible mediators of connecting tubule glomerular feedback. Hypertension. 2009;53:319–4. doi: 10.1161/HYPERTENSIONAHA.108.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren Y, D'Ambrosio MA, Garvin JL, Carretero OA. Angiotensin II enhances connecting tubule glomerular feedback. Hypertension. 2010;56:636–4. doi: 10.1161/HYPERTENSIONAHA.110.153692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren Y, D'Ambrosio MA, Wang H, Peterson EL, Garvin JL, Carretero OA. Mechanisms of angiotensin II-enhanced connecting tubule glomerular feedback. Am J Physiol Renal Physiol. 2012;303:F259–F265. doi: 10.1152/ajprenal.00689.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Garvin JL, D'Ambrosio MA, Ren Y, Carretero OA. Connecting tubule glomerular feedback antagonizes tubuloglomerular feedback in vivo. Am J Physiol Renal Physiol. 2010;299:F1374–F1378. doi: 10.1152/ajprenal.00403.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, D'Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback mediates acute tubuloglomerular feedback resetting. Am J Physiol Renal Physiol. 2012;302:F1300–F1304. doi: 10.1152/ajprenal.00673.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlondorff D, Ardaillou R. Prostaglandins and other arachidonic acid metabolites in the kidney. Kidney Int. 1986;29:108–4. doi: 10.1038/ki.1986.13. [DOI] [PubMed] [Google Scholar]
- 11.Navar LG, Inscho EW, Majid DS, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–4. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- 12.Fuson AL, Komlosi P, Unlap TM, Bell PD, Peti-Peterdi J. Immunolocalization of a microsomal prostaglandin E synthase in rabbit kidney. Am J Physiol Renal Physiol. 2003;285:F558–F564. doi: 10.1152/ajprenal.00433.2002. [DOI] [PubMed] [Google Scholar]
- 13.Chatziantoniou C, Arendshorst WJ. Prostaglandin interactions with angiotensin, norepinephrine, and thromboxane in rat renal vasculature. Am J Physiol. 1992;262:F68–F76. doi: 10.1152/ajprenal.1992.262.1.F68. [DOI] [PubMed] [Google Scholar]
- 14.Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole : role of EP(3) and EP(4) receptors. Circ Res. 2000;86:663–4. doi: 10.1161/01.res.86.6.663. [DOI] [PubMed] [Google Scholar]
- 15.Ren Y, Carretero OA, Garvin JL. Mechanism by which superoxide potentiates tubuloglomerular feedback. Hypertension. 2002;39:624–4. doi: 10.1161/hy0202.103299. [DOI] [PubMed] [Google Scholar]
- 16.Burg MB. Perfusion of isolated renal tubules. Yale J Biol Med. 1972;45:321–4. [PMC free article] [PubMed] [Google Scholar]
- 17.Ito S, Carretero OA. An in vitro approach to the study of macula densa-mediated glomerular hemodynamics. Kidney Int. 1990;38:1206–4. doi: 10.1038/ki.1990.335. [DOI] [PubMed] [Google Scholar]
- 18.Good DW, Wright FS. Luminal influences on potassium secretion: sodium concentration and fluid flow rate. Am J Physiol. 1979;236:F192–F205. doi: 10.1152/ajprenal.1979.236.2.F192. [DOI] [PubMed] [Google Scholar]
- 19.Khuri RN, Strieder WN, Giebisch G. Effects of flow rate and potassium intake on distal tubular potassium transfer. Am J Physiol. 1975;228:1249–4. doi: 10.1152/ajplegacy.1975.228.4.1249. [DOI] [PubMed] [Google Scholar]
- 20.Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–4. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machwate M, Harada S, Leu CT, Seedor G, Labelle M, Gallant M, Hutchins S, Lachance N, Sawyer N, Slipetz D, Metters KM, Rodan SB, Young R, Rodan GA. Prostaglandin receptor EP(4) mediates the bone anabolic effects of PGE(2) Mol Pharmacol. 2001;60:36–4. doi: 10.1124/mol.60.1.36. [DOI] [PubMed] [Google Scholar]
- 22.Schlondorff D. Renal prostaglandin synthesis. Sites of production and specific actions of prostaglandins. Am J Med. 1986;81:1–4. doi: 10.1016/0002-9343(86)90903-4. [DOI] [PubMed] [Google Scholar]
- 23.Chaudhari A, Gupta S, Kirschenbaum MA. Biochemical evidence for PGI2 and PGE2 receptors in the rabbit renal preglomerular microvasculature. Biochim Biophys Acta. 1990;1053:156–4. doi: 10.1016/0167-4889(90)90008-2. [DOI] [PubMed] [Google Scholar]
- 24.Breyer MD, Breyer RM. Prostaglandin receptors: their role in regulating renal function. Curr Opin Nephrol Hypertens. 2000;9:23–4. doi: 10.1097/00041552-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Imig JD, Breyer MD, Breyer RM. Contribution of prostaglandin EP2 receptors to renal microvascular reactivity in mice. Am J Physiol Renal Physiol. 2002;283:F415–F422. doi: 10.1152/ajprenal.00351.2001. [DOI] [PubMed] [Google Scholar]
- 26.Villa E, Garcia-Robles R, Haas J, Romero JC. Comparative effect of PGE2 and PGI2 on renal function. Hypertension. 1997;30:664–4. doi: 10.1161/01.hyp.30.3.664. [DOI] [PubMed] [Google Scholar]
- 27.Vitzthum H, Abt I, Einhellig S, Kurtz A. Gene expression of prostanoid forming enzymes along the rat nephron. Kidney Int. 2002;62:1570–4. doi: 10.1046/j.1523-1755.2002.00615.x. [DOI] [PubMed] [Google Scholar]
- 28.Campean V, Theilig F, Paliege A, Breyer M, Bachmann S. Key enzymes for renal prostaglandin synthesis: site-specific expression in rodent kidney (rat, mouse) Am J Physiol Renal Physiol. 2003;285:F19–F32. doi: 10.1152/ajprenal.00443.2002. [DOI] [PubMed] [Google Scholar]
- 29.Imig JD. Eicosanoid regulation of the renal vasculature. Am J Physiol Renal Physiol. 2000;279:F965–F981. doi: 10.1152/ajprenal.2000.279.6.F965. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy I, Coleman RA, Humphrey PPA, Levy GP, Lumley P. Studies on the characterisation of prostanoid receptors: a proposed classification. Prostaglandins. 1982;24:667–4. doi: 10.1016/0090-6980(82)90036-3. [DOI] [PubMed] [Google Scholar]
- 31.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. 2000;279:F12–F23. doi: 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- 32.Breyer MD, Zhang Y, Guan Y-F, Hao C-M, Hebert RL, Breyer RM. Regulation of renal function by prostaglandin E receptors. Kidney Int. 1998;54(Suppl 67):S-88–S-94. doi: 10.1046/j.1523-1755.1998.06718.x. [DOI] [PubMed] [Google Scholar]
- 33.Purdy KE, Arendshorst WJ. EP(1) and EP(4) receptors mediate prostaglandin E(2) actions in the microcirculation of rat kidney. Am J Physiol Renal Physiol. 2000;279:F755–F764. doi: 10.1152/ajprenal.2000.279.4.F755. [DOI] [PubMed] [Google Scholar]
- 34.Tilley SL, Audoly LP, Hicks EH, Kim H-S, Flannery PJ, Coffman TM, Koller BH. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–4. doi: 10.1172/JCI6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandit MM, Strait KA, Matsuda T, Kohan DE. Na delivery and ENaC mediate flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol. 2012;302:F1325–F1330. doi: 10.1152/ajprenal.00034.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arima S, Ren Y, Juncos LA, Carretero OA, Ito S. Glomerular prostaglandins modulate vascular reactivity of the downstream efferent arterioles. Kidney Int. 1994;45:650–4. doi: 10.1038/ki.1994.87. [DOI] [PubMed] [Google Scholar]
- 37.Silldorff EP, Yang S, Pallone TL. Prostaglandin E2 abrogates endothelin-induced vasoconstriction in renal outer medullary descending vasa recta of the rat. J Clin Invest. 1995;95:2734–4. doi: 10.1172/JCI117976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imig JD, Navar LG. Afferent arteriolar response to arachidonic acid: involvement of metabolic pathways. Am J Physiol. 1996;271:F87–F93. doi: 10.1152/ajprenal.1996.271.1.F87. [DOI] [PubMed] [Google Scholar]
- 39.Purdy KE, Arendshorst WJ. Prostaglandins buffer ANG II-mediated increases in cytosolic calcium in preglomerular VSMC. Am J Physiol. 1999;277:F850–F858. doi: 10.1152/ajprenal.1999.277.6.F850. [DOI] [PubMed] [Google Scholar]
- 40.Chatziantoniou C, Arendshorst WJ. Renal Vascular Reactivity to Vasodilator Prostaglandins in Genetically Hypertensive Rats. Am J Physiol. 1992;262:F124–F130. doi: 10.1152/ajprenal.1992.262.1.F124. [DOI] [PubMed] [Google Scholar]
- 41.Juncos LA, Ren Y, Arima S, Garvin J, Carretero OA, Ito S. Angiotensin II action in isolated microperfused rabbit afferent arterioles is modulated by flow. Kidney Int. 1996;49:374–4. doi: 10.1038/ki.1996.55. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida H, Tamaki T, Aki Y, Kimura S, Takenaka I, Abe Y. Effects of angiotensin II on isolated rabbit afferent arterioles. Jpn J Pharmacol. 1994;66:457–4. doi: 10.1254/jjp.66.457. [DOI] [PubMed] [Google Scholar]
- 43.Ichihara A, Imig JD, Inscho EW, Navar LG. Cyclooxygenase-2 participates in tubular flow-dependent afferent arteriolar tone: interaction with neuronal NOS. Am J Physiol. 1998;275:F605–F612. doi: 10.1152/ajprenal.1998.275.4.F605. [DOI] [PubMed] [Google Scholar]
- 44.Cheng HF, Harris RC. Renal effects of non-steroidal anti-inflammatory drugs and selective cyclooxygenase-2 inhibitors. Curr Pharm Des. 2005;11:1795–4. doi: 10.2174/1381612053764922. [DOI] [PubMed] [Google Scholar]
- 45.Bennett WM, Henrich WL, Stoff JS. The renal effects of nonsteroidal anti-inflammatory drugs: summary and recommendations. Am J Kidney Dis. 1996;28:S56–S62. doi: 10.1016/s0272-6386(96)90570-3. [DOI] [PubMed] [Google Scholar]