

Abstract
At the center of iron and oxidant metabolism is the ferritin superfamily: protein cages with Fe2+ ion channels and catalytic di- Fe/O redox centers that initiate formation of caged Fe2O3 • H2O. Ferritin nanominerals, initiated within the protein cage, grow inside the cage cavity (5 or 8 nm in diameter). Ferritins contribute to normal iron flow, maintenance of iron concentrates for iron cofactor syntheses, sequestration of iron from invading pathogens, oxidant protection, oxidative stress recovery and, in diseases where iron accumulates excessively, to iron chelation strategies. In eukaryotic ferritins, biomineral order/crystallinity is influenced by nucleation channels between active sites and the mineral growth cavity. Animal ferritin cages contain, uniquely, mixtures of catalytically active (H) and inactive (L) polypeptide subunits with varied rates of Fe2+/O2 catalysis and mineral crystallinity. The relatively low mineral order in liver ferritin, for example, coincides with a high % of L subunits, and, thus, a low % of catalytic sites and nucleation channels. Low mineral order facilitates rapid iron turnover and the physiological role of liver ferritin as a general iron source for other tissues. Here, current concepts of ferritin structure/function/genetic regulation are discussed and related to possible therapeutic targets such as mini-ferritin/Dps protein active sites (selective pathogen inhibition in infection), the nanocage pores (iron chelation in therapeutic hypertransfusion), the mRNA noncoding, IRE-riboregulator (normalizing ferritin iron content after therapeutic hypertransfusion, and as protein nanovessels to deliver medicinal or sensor cargo.
Keywords: Ferritin, iron, protein nanocage, antibacterials, drug delivery, riboregulator target
1. Introduction
Iron, essential for life, is mainly in proteins but recent studies show selective, coordination binding of Fe2+ to RNA; examples include enhanced ribozyme folding/activity 1 and changed RNA riboregulator conformation in the 5′untranslated region of animal mRNA that dissociated a protein repressor 2 and enhanced translation factor(eIF-4F) binding 3. The major iron proteins in humans are globins, hemoglobin and myoglobin, followed by ferritins and then by a variety of heme and iron-sulfur proteins and iron cofactors bound directly to protein, e.g. ribonucleotide reductase. Ferritin is a superfamily of protein-caged Fe2O3•H2O biominerals. They are ancient (in Archaea), ubiquitous (in marine and terrestrial organisms, both anaerobic and aerobic) and, have a rare quaternary structure: folded, polypeptide subunits (4 α-helix bundles) that self- assemble into hollow cages; interior cage spaces (biomineral growth cavities) are ~ 30% of the cage volume. The cage symmetry is 432 (24 subunit ferritins) or 32 (12 subunit ferritins, often called Dps proteins or mini-ferritins). The amino acid sequences of ferritins vary as much as 80% although subdomains, such as the Fe2+ entry/exit channels, contain highly conserved sequences. Multiple ferritin genes in eukaryotes create tissue-specific combinations of ferritin subunits in the cages, contrasting with bacterial ferritin genes, which encode ferritin subunits for homopolymeric cages synthesized at different times in the culture cycle4, 5. In animal tissues, including humans, both the ferritin mineral and the ferritin protein vary among each tissue and cell type. The crystallinity of ferritin biomineral coincides with variations in the structure of the protein cage6. Such cage variations include, in addition to amino acid sequence, the numbers of catalytically active subunits with nucleation channels (H subunits) and the iron ligands at the active sites, which emphasize the protein- based contributions to ferritin mineral structure 7. For decades, serum ferritin, normally 0.025% of the total body ferritin, has been used as a clinical marker of iron status; serum ferritn and likely originates from cell leakage. Recent awareness of the role of inflammation in altering serum ferritin concentrations complicated the interpretation of serum ferritn as a marker for body iron. The physiology and structure of serum ferritin remain poorly understood and will not be considered further. Briefly described here, are: ferritin protein structure and function, ferritin mineral structure and function, the regulation of ferritin biosynthesis, and the uses of ferritin as a therapeutic target in disease.
2. Ferritin protein structure
Ferritin subunits (Fig 1 A, C) spontaneously self-assemble into the native, protein nanocages (Fig. 1), although recent engineering approaches are now providing access to single ferritin subunits for study and modification before reassembly 8. Curvature of the subunit bundles creates a large hollow in the protein cage (Fig 1 A). It is the geometry of side chain interactions at the subunit dimer interfaces that controls ferritin cage self-assembly, based on recent data19. The use of a single Fe2+ channel to provide substrate to 3 di-iron catalytic sites, in the 3 subunits of ferritin cages that form the ion channels, is associated with a Hill coefficient of 3; the underlying protein: protein interaction remains unknown 10. Each ferritin subunit has an N-terminal peptide extension, which, in eukaryotic ferritins, is a gate for mineral dissolution/Fe2+ exit; it is held in place by conserved H bond interactions with residues in the subunit in channels. Gate function was recently identified through the structural disorder induced with amino acid substitution of conserved ion channel residues (Fig 1D) and the associated increase in rates of mineral dissolution and Fe2+ exit/chelation 11, 12.
Fig. 1. Ferritin protein.

A. X-section of a eukaryotic ferritin protein cage, viewed with a 3-fold symmetry axis pore centered in the mineral growth cavityS. B. A ferritin protein subunit: metal ion traffic (white arrows); in a ; protein cocrystal with Mg2+ -green sphere; Co2+ -pink sphere; in channel at left. C. Progress curves of Fe2+/O2 catalysis: Fe3+-O-O-Fe3+ (DFP, λmax-650 nm-blue; Fe3+O broad absorbance, A350nm-red. D. N-terminal extension: R72D, left, N-terminus disordered; WT, right,-N-terminus-pink; helix numbers: 1,2,3,4. Fe2+ exit from ferritin mineral is accelerated in R72D11,12. Figure panels contributed by T. Tosha and R. K. Behera, using PDB 3KA4,PDB 3DE1 and Pymol.
Variations in ferritin primary structure between bacterial and eukaryotic ferritins can be huge, as much as 80%, even though the protein cage structures are similar. Among eukaryotic ferritins, however, the sequence divergence is only 30–40%, allowing animal sequences to identify plant genes13. The three dimensional structural similarities of 12 subunit mini-ferritins (Dps/DLP proteins) and 24 subunit maxi-ferritins escapes current bioinformatic analyses of linear ferritin sequences. The ferritin cage structure of Dps proteins was not recognized until the first protein crystal structure of a 12 subunit Dps protein was obtained in E. coli. 14 The N and C terminal extensions from ferritin 4-α-helix bundles vary in length and structure. In some mini-ferritins, N-termini are associated with DNA binding and DNA protection, hence the name Dps (DNA protection during stress/starvation). N-terminal extensions of some miniferritins contain short α-helices 15.
a. Protein cages
Ferritin protein cages in animals and plants are heteropolymers of ferritin subunits encoded in separate genes. The added level of complexity is critical physiologically, since different tissues e.g. root leaves, seeds in plants and liver spleen, heart, and intestine in animals, synthesize ferritins with different, specific mixtures of the subunits. In animals the different subunit mixtures coincide with different mineral structures 6 and with the accessibility of ferritin iron to iron chelators 16. A limitation of standard recombinant protein technology for ferritins from plants and animals is the fact that coexpression strategies are required to produce ferritin cages with mixtures of subunits 17, while most current research uses homopolymeric, ferritins. However, as more is learned about homopolymeric recombinant ferritin protein cages, especially of highly conserved sections, readily interpretable experiments with heteropolymeric cages can be designed and executed.
Protein crystallography has been the main source of structural information about ferritins since 1978 because suitable crystals of ferritin are obtained relatively easily. However, recently, a combination of solid state and solution 13C-13C-NOESY has provided new information on functional structure in eukaryotic ferritin protein cages18, 19. Currently, in the RCSB Protein Data Base, there are 177 structures of twenty-four subunit ferritins, 48 structures of twenty-four subunit heme bacterioferritins, and 6 structures of twelve subunit mini-ferritins.
The high α-helix content of ferritin protein cages (>80%) facilitates the use of circular dichroism to study the thermal and solvent stability of protein cages and subdomains in normal and variant cages 8, 20. Protein crowding (increasing osmolarity with soluble proteins to approximate cytoplasmic conditions) partly restores function in unfolded ferritin variants suggesting possible rescue pathways for abnormally unfolded ferritin in vivo11, 21. Several novel NMR approaches, both solution 19, 22, are being developed to solve problems of ferritin function, the first dealing with product release in catalysis and mineral nucleation.
Comparisons of the protein cages of ferritin family members show that they share multi-polypeptide, hollow cages, in spite of linear sequences that vary as much as 80%. The polypeptides spontaneously fold into bundles of 4 α-helices (Fig 1). Ferritin helix bundles deviate from classical polarity, helix 1- up/helix 2- down/helix3- up/helix4 -down, because of the very long loop connecting helices 2 and 3. In contrast, ferritin helix bundles are: 1- up/2-down/3-down/4-up (See Fig 1 D). Self-assembly of ferritin subunits depends on interfacial geometries conferred by hydrogen bonding that interdigitate, long, dimeric helical interfaces8. The increased stability gained by cage assembly is illustrated by the mid-point thermal unfolding transition at 40°C for each subunit bundle contrasting with 80°C for the assembled cage 8.
b. Ion channels-3 fold symmetry axes
Trimers of helix 4, form the walls of a single ion channel, around the cage 3-fold symmetry axes, which penetrate the ferritin protein cages. The eight ion channels in 24 subunit ferritins have an hour-glass shape, are 15Å long and assemble around the three-fold symmetry axes of the cage. Fe2+ ions traverse the ion channels during entry for mineral formation and during exit after mineral dissolution7, 15. Changing tertiary structural interactions in the 3-fold channel can alter ferritin function 4, 11, 12, 23–25
c. Catalytic sites
Ferritin catalytic sites are generally in the middle of each subunit with metal ligands from each of the 4 α helices in the bundle; exceptions occur in a few mini-ferritins 15. The amino acids at the active sites are similar among animals, plants, and microorganism, but not identical (Table 1). Ligand weakness at one of the active sites in most ferritins (Table 1) reflects to the release of the catalytic product, Fe3+O, An exception is bacterio ferritin where the ligand sets for each Fe at the di-iron catalytic centers are the same and like those in the di-iron oxygenases. In fact, the di-iron center in bacterioferritins, does, as in di-iron oxygenase, retain iron and acts as a cofactor 26. Given the variations in amino acid sequence of ferritin cages, the differences among ferritin catalytic site ligands (Table 1) and oxidant (O2 and/or H2O) (up 80%), attempts to fit one catalytic mechanism to data on all known ferritins 27 seem premature.
Table 1.
Examples of Ligands at 24 subunit Ferritin Di-iron Catalytic Centers (n= 4–24/cagea)
| Fe-1 | Fe-2 | Function | Reference | |
|---|---|---|---|---|
| Eukaryotic maxi-ferritin: H. sapiens/R. catesbeiana/G. max | E, ExxH | E, QxxA/D/S | Substrate; DFPb | 10, 28–33 |
| Bacterial maxi-ferritin: E. coli BFR | E, ExxH | E, ExxH | Cofactor & Substrate | 34, 35 |
| Archeael maxi-ferritin: P. furiosus | E, ExxH E | QxxE | Cofactor& substrate | 27, 36 |
| Bacterial mini-ferritinc, B. anthracis Dps-1 | H, HxxEc | E, DxxxE | Substrate | 37–39 |
In animals, ferritin from different tissues combine different numbers of catalytically inactive (L) and active (H) subunits/cage. In plants, ferritins have multiple types H subunits with different amino acid sequences 40. In bacteria, ferritins contain only on type of catalytically active (H) subunit at a time, but different ferritins, encoded in separate genes, are synthesized under different conditions 5, 26.
DFP, diferric peroxo (Fe3+-O-O-Fe3+) forms as a transient intermediate at eukaryotic ferritin catalytic centers.
In the absence of oxidant, the Fe1 site is empty.
Only in animal ferritins is the number of catalytic sites less than the number of cage subunits, because only animals produce catalytically inactive L subunits, lacking both a functional catalytic site and nucleation channel residues. Tissue specific difference in the H and L subunits content of ferritin protein cages coincide with tissue specific differences in ferritin mineral crystallinity6.
d. Postoxidation/Nucleation channels and the 4 fold symmetry axes
The ferritin Fe3+O channels in 24 subunit ferritins, which terminate around the inner surfaces of the 4-fold cage symmetry axes (Fig. 3), were discovered by monitoring the pathway of Fe3+ after catalytic reactions with O2, from the active sites to the mineral growth cavities, with 13C-13C solution NOESY (Fig. 3) 19; the amino acids along the post-oxidation channel alter DFP kinetic when they are replaced 4. Restoration rates of active sites for Fe2+ binding are slow and heterogeneous, (6–24 hours) 41, 42, and contrast with the simple, rapid decay of DFP (seconds) (Fig. 1). However, the long periods of time required for site restoration (turnover) match that required for fluctuations in NMR spectra to return to the original steady state, after the addition of Fe2+ in air19 and likely reflect the slow movement of Fe3+O catalytic products through the subunit helices to the mineral growth cavity. Movement of Fe3+O multimers through the stable subunit α-helices in eukaryotic ferritins requires significant conformational change. Such changes will be slow in such a large protein assemblage (480 kDa). The contrast between rates of active site turnover (hours) and rapid catalytic coupling of 2 Fe and O (msec) is enormous.
Fig 3. Differences mineral order among human tissue ferritins6 coincide with differences in the numbers of catalytic centers and nucleation channels/cage7.
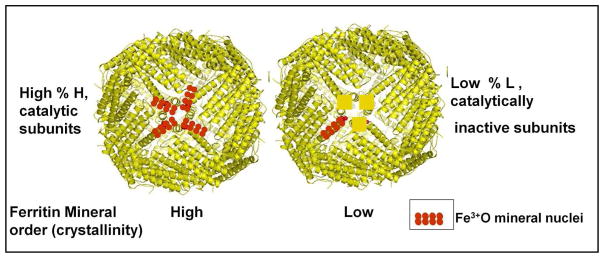
Ferritin protein cages rich in H subunits and with highly ordered ferritin minerals, are found in tissues with high oxygenase, such as heart, while ferritin protein cages rich in L subunits and with relatively disordered minerals, are found in human liver. The supply of stored iron to other tissues is a major function of liver ferritin, which is facilitated by the high surface/volume of the more disordered ferritin mineral.
In 24 subunit ferritin protein cages, contrasting with the 12 subunit mini-ferritins, a non-bonded, four α-helix polypeptide bundle forms around each four-fold symmetry axis. The bundles form through the alignment of helix 5, extending from each of the 4 α-helix bundles. No function has been definitively established for the structure around the 4-fold symmetry axes, but, deletion of helix 5 creates a protein cage that exposes the ferritin mineral to reductants and Fe2+ leakage25. The exits of the post-oxidation channels, through which mineral nuclei travel between the active sites and the mineral growth cavity, are symmetrically placed around the 4-fold cage axes19
3. Ferritin Function
Ferritin function can be divided into two parts: 1. Fe2+ entry, catalytic redox with O2, Fe3+O multimer formation (nucleation and mineral growth) and 2. Mineral reduction/dissolution with Fe2+ exit/chelation. Each part occurs in different physical parts of the protein/mineral complex (channel transit, cage-based catalysis and nucleation, or the caged mineral surface) and in different time regimes: catalytic coupling of 2 Fe-/O2 (msec) and nucleation/mineralization (minutes to hours). Since iron and dioxygen will react to form mineralized, hydrated ferric iron without the ferritin protein cage, the complexity of ferritin function must reflect the importance of rates and control over Fe2+ distribution.
Fe2+ entry into ferritin cages occurs spontaneously in solution, but in vivo is likely dependent on transport by a protein “chaperone”43, except during stress when normal regulatory/transport mechanisms are saturated. Fe2O3•H2O mineral reduction and dissolution occurs in response to physiological signals of iron need, in vivo, often referred to as the “size of the labile iron pool”. In solution the addition of reductant triggers the mineral reduction and dissolution process, in which is measured as the formation of Fe2+-chelate complexes outside the protein cage (Fig. 2). Ferritin functions are separated in space (within and inside the protein cage) and time (msec to hours).
Fig. 2. Protein control of ferritin mineral dissolution/Fe2+chelation rates.
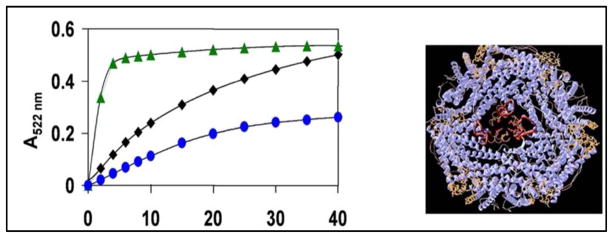
Two heptapeptides selectively targeted to ferritin pore structure were isolated from a heptapetide library (109 peptides) and altered WT mineral dissolution and Fe2+ exit rates20. Left: Fe2+ exit/chelation progress curves: green=peptide 1; blue-peptide 2; black-no peptide. Right: Structures of ferritin protein cage- blue: pore sequences: WT, folded-tan; L134P-unfolded-red. View: 3-fold symmetry axis is centered. Ferritin mineral dissolution : NADH/FMN – reductant; Fe2+ bipyridyl- rate of Fe2+ exit. This research was originally published in the Journal of Biological Chemistry 2007, volume 282, page 31821, 2007.
a. Fe2+ entry and ion channels
Fe2+ entry into ferritin protein cages is through the 8 Fe2+ ion channels, at the 3-fold symmetry axes of 24 subunit ferritins. (There are 4 Fe2+ channels in the 12 subunit mini-ferritins). The channels also influence Fe2+ exit after mineral reduction and issolution.
Fe2+ entry through ferritin iron channels is currently measured though effects on rates of enzymatic, Fe2+/O redox reactions, which are also called “ferroxidase” or “Fox” reactions. The reactions, which occur at di-iron sites with some properties shared with di-iron cofactor dioxygenases {Liu, 2005 #107}, cause spectral changes in the broad UV-vis absorbance range 310–420 nm. However, it has not yet been possible to find distinct UV-vis spectral signatures that distinguish among all the multiple ferric species, which include: include Fe3+O(H)-Fe3+, the catalytic product/mineral precursor, mineral nuclei and the mineral itself. Thus, the absorbance changes, in solutions of mineralizing ferritin protein cages, continue for many hours. In eukaryotic ferritins, Fe2+ entry and catalysis are monitored as the diferric peroxo (DFP_intermediate (λ650nm) that forms rapidly (msec) from Fe2+/O2 and decays quickly (sec) (Fig. 1C). DFP measurements allow the determination of the effects of engineered amino acid variations in the Fe2+ entry ion channels, and elsewhere, on Fe2+/O2 reaction kinetics. Fe2+/O redox reactions in ferritins are also studied by Mossbauer, resonance Raman, and XAS /EXAFS absorbance and EPR spectroscopies 27, 44–47. Direct binding of each Fe2+, and the ligand structure, occurred at the di-iron catalytic centers before O2 binding in eukaryotic maxi-ferritins, incidicated by VTVH MCD/CD analysis {Schwartz, 2008 #19}. By contrast, only a single Fe2+ binds at the catalytic centers of mini-ferritins, in the absence of O2/H2O238. Fe2+ entry through the ferritin ion channels has also been studied as function of the Fe2+/O2 reaction or tryptophan fluorescence quenching, e.g. 12, 48. Isothermal calorimetry, which is less specific, given the size of ferritin and the multiple channels and active sites, is also used to assess Fe2+/ferritin protein interactions 8. Intermediate stages in the movement of Fe3+ away from the catalytic sites to the mineral growth cavity in eukaryotic ferritins were studied for the first time, after site –specific residues assignments from combined solid and solution NMR, by comparing 13C-13C NOESY ± Fe 19.
b. Catalysis
Ferritin proteins catalyze the reaction between Fe2+ and O2 (or H2O2 in Dps proteins). Each subunit has one, di-iron active site, with ligands from each of 4 α –helices in each bundle; there is some variability in the iron ligands among different species and, in organisms with multiple tissues, among ferritins in different tissues (Table 1). In some 12 subunit Dps proteins, components of the active sites are donated by each of two subunits along the subunit dimer interfaces Table 1 15. Catalysis is monitored using progress curves of Fe3+O formation (red line, Fig.1C), which has the disadvantage of spectral overlap between the early species, such as diferric peroxo and later species of diferric oxo/hydroxo and mineral. In animal ferritins, by contrast, the initial reaction intermediate, blue diferric peroxo, can be selectively measured 42, 44–46 (Fig 1 C, blue line), using rapid mixing methods.
Ferritin di-iron ligands at the catalytic sites have been examined by a variety of methods. Direct identification of Fe2+ ligands at the active sites in ferritin was achieved in a eukaryotic ferritin and a bacterial mini-ferritin (Dps protein), using VTVH MCD/CD10, 38. For eukaryotic ferritins, the results confirm many of the inferences from effects of deletional 49 and insertional mutagenesis 29on solution kinetics, using nonspecific (Fe3+O) and specific (Fe3+-O-O-Fe3+) spectral probes(Fig 1C), and from protein crystallography with proxy ions such as Zn2+, Ca2+, Mg2+, and Co2+ 28, 30, 33, 50. Ferritin Fe3+- ferritin protein crystals, obtained by soaking crystals with Fe2+ in air, showed differences from the Fe2+ sites in eukaryotic ferritins 51 likely representing the conformational differences between Fe2+ substrate and Fe3+ O product binding at the active sites.
Bacterial ferritin active sites, like those in eukaryotic ferritins, have been studied by crystallography and deletional mutagenesis for FTNA 35 and for the heme containing BFR. Unlike di-iron oxygenases, and BFR, where iron remains in the di-iron site throughout catalysis, in eukaryotic ferritins iron is only transiently bound to the active site. Thus, in all crystallographic studies of eukaryotic ferritins, in contrast to the bacterial ferritin cofactor sites, proxy metals such as Co2+, Cu2+ or Zn2+ are observed in the active sites. Both bacterial ferritins, the heme and non-heme carieties, have an extra metal binding site C (Fe-3) that contributes to transfer of the catalytic products to the mineral growth cavity and to active site regeneration. In archaeal ferritins, where there have been fewer opportunities for site direct mutagenesis combined with solution kinetic studies and where monitoring the catalytic intermediates appears to be complex, the ferritin active site ligands are still changing 27, 36. While the understanding of ferritin catalytic mechanisms is more advanced in eukaryotic ferritins compared to the ferritins of bacterial adversaries in disease, much remains to be learned for all. Host/pathogen battles over iron involve host sequestration of iron in macrophage ferritin and the release of oxidants as antibacterial agents. Microbial virulence is associated with the production of miniferritins (Dps proteins 15, 38, 52, 53), with H2O2 as the oxidant The differences between active site structure, substrates (H2O2 vs. O2) and mechanisms in host and pathogen ferritins, provide a tempting target for the development of antibacterial agents targeted at selective inhibition of bacterial mini-ferritin active sites.
c) Ferritin mineral nucleation
Ferritin minerals will nucleate at different places in the protein cage, which reflects the combination of ligands at the catalytic di-iron sites, location of the catalytic sites and the number of active sites per cage. In 12 subunit mini-ferritins, the catalytic centers are most often on the inner surface of the mineral growth cavity 15, 39, 54, suggesting that after oxidation and coupling the di-iron products of catalysis are simply released into the mineral growth cavity. Difference in H:L subunit ratios in heart and liver coincide with differences in mineral order. Liver ferritin (H:L ~ 1:4) has much more disordered mineral than heart ferritin (H:L~ 1:1)6, 55. An explanation is the number of Fe3+O nuclei that emerge near each other, around the 4-fold symmetry axes of the protein cage (Fig. 3). On average, Fe3+O multimers in heart ferritin have a high probability of interacting with other Fe3+O multimers emerging from adjacent channels around the 4 fold symmetry axes to form large mineral nuclei and highly ordered minerals. By contrast, in liver ferritin (H:L ~ 1:4), more solitary Fe3+O multimers will enter the mineral growth cavity, leading to less ordered mineral growth. Other than assessing the numbers of Fe atoms that can be accommodated in mini-ferritin cages there has been little direct study of mineral structure in mini-ferritins.
Among the 24 subunit mini-ferritins, the large number of carboxylate ligands on and near the interior surface of ferritins also led to hypotheses that mineral nucleation began at such carboxylates and mineral growth simply extended from such protein sites into the mineral growth cavity. Such conjectures were supported by the inhibition of mineral formation caused by replacement of surface carboxylates on mineral nucleation in catalytically inactive L ferritins lacking cavity surface carboxylates E55, E60, E6356, 57. (Residue numbers are the classical ferritin sequence that applies to all eukaryotic subunits; for human H ferritin numbering, add +4). In ferritins without catalytic sites, Fe2+ can enter the ferritin protein cage through the 8 ion channels at the three fold symmetry axes, exit directly into the mineral growth cavity and be oxidized by dioxygen dissolved in the cavity fluid since there is no competition for Fe2+ by the di-iron centers. However, when catalytic centers are present (H ferritin), replacement of nucleation center residues (E60 and E63) had no effect on catalysis or mineral growth 58, presaging the identification of the nucleation channels 19.
The Fe3+ O nucleation channels (Fig 1 B, Fig 3) in eukaryotic ferritins, as identified by the paramagnetic effects of Fe3+ on 13C-13C NOESY spectra, at ~ 50Å´ are longer than the Fe2+ ion entry channels, and pass through the center of the 4 α helix bundles of each subunit to connect catalytic centers and mineral growth cavity in eukaryotic ferritins. Movement of the Fe3+O multimers will be slowed by the rigidity of the 4 α helix bundles, which likely explains to the long periods of time required for turnover of the catalytic sites 41, 42. Co-variation between active site and nucleation channel amino acids 4 emphasizes the interdependence between catalysis and transfer of the Fe3+O products to the mineral growth cavity of eukaryotic ferritins 4. Coupled with the contributions of ion channel residues to catalysis 4, 12 integrated function throughout each ferritin subunit, over 50 Å´, becomes clear. Still to be understood are the persistent observations of protein: protein co-operatively related to cage self-assembly and function, e.g. 8, 10. Ferritin nucleation channels, and their relation to the presence of ferritin catalytic sites and mineral with different crystalline order 6 (Fig. 3) can be exploited to control product structures when ferritin protein cages are used as nanovessels and nanotemplates for drug delivery and nanosensors 8, 59–63.
The later stages in ferritin iron mineralization remain poorly understand, in part because of the lack of easily accessible spectroscopic markers that resolve the products of catalysis into one more intermediate. Mössbauer analysis of multimeric Fe3+O intermediates in mineralization becomes very complex beyond iron dimers 64. The effects of replacing key amino acids on the ferritin Fe3+O spectrum (A 310-42- nm) 31 suggest that such goals may be are accessible. Experimental study of ferritin protein cages composed of defined mixtures of heterogeneous subunits, either natural 17 or chemically produced 8, is key to full understanding of Nature’s array of tissue-specific differences in ferritin protein cages and minerals.
d) Reduction/dissolution of ferritin protein- caged mineral
Sometimes called “iron release” or “Fe2+ exit”, the reduction and dissolution of caged ferritin mineral is studied by adding a reductant to solutions of ferritin (caged mineral + protein) and trapping mineral reduced and dissolved as an Fe2+ - chelator complex. In solution, the reductants most often used are [S2O4]2– or the biological mixture of NADH and FMN. Dithiothreitol is also used, even though it is slow, since dithionite can modify the proteins (Y. Kwak, T. Tosha, E.C. Theil and E.I. Solomon, 2011 unpublished observations, 65). Some the reductants appear to enter the protein cage 65 and, thus, Fe2+ exits after mineral dissolution, and iron chelation rates are controlled by the dynamics of opening and closing the protein cage pores, as well as by the surface effects of mineral dissolution ; see Fig. 2, 20, 23. When iron chelator complexes are colored, the measurement of ferritin iron mineral dissolution of the iron derivative is color facilitated; filtration studies show that the Fe2+-chelates (bipyridyl) are outside the protein cage 66. The Fe2+-bipyridyl complex is measured by UV-vis spectrometry. If Fe3+ chelators are used, the kinetics of Fe2+ removal from ferritin minerals is complicated by the oxidation to Fe3+.
Ferritin function is sometimes studied in cultured, living cells rather than in solution. The cells grow on a complex medium, e.g. DMEM (Dulbecco’s modified Eagle’s Medium), which is a neutral solution containing vitamins, amino acids, glucose, phosphate and other salts; DMEM is usually mixed with 10% serum. Thus, only 10% of the normal transferrin is present and, unless extra iron is added, only 10% of the environmental iron normal for mammalian cells. Cultured mammalian cells are usually incubated in an atmosphere of 5 % CO2 in air, which at 20% O2 is much more concentrated than oxygen dissolved in blood (~5% O2); dissolved CO2, provides HCO3-1 buffer and the synergistic anion for transferrin-Fe binding. In some studies 59 Fe-transferrin was used to examine ferritin iron turnover/chelator binding 24, while in the peptide uptake studies described here, cells were incubated with or without 125I-tyrosine labeled peptide; labeling used solid-phase 1,3,4,6-tetrachloro-3a,6a-diphenyl-glycoluril (Thermo Pierce) reagents with a peptide previously identified as unfolding surface pores in ferritin protein pores 20. Recovered cells were washed with buffer containing unlabeled peptide to measurement of specifically bound peptide in whole cells and in cellular proteins precipitated with 5% trichloroacetic acid).
Changes in cellular amounts of ferritin protein, which are increased by iron (mRNA target)67, 68 or heme and oxidative stress (DNA target)69, 70, or both, is measured using immunological reactions (immunoprecipitation or immunoblots, often called, “Western blots”). The intracellular distribution of mineralized ferritin is assessed by microscopy of the mineral itself. For exogenous ferritin proteins, fluorescent-labels can be coupled with electron microscopy. The key to opening iron chelation therapies equally to heart and liver ferritin iron, in humans at risk for lethal iron accumulation in Thalassemia and Sickle cell disease16, 71, may well be understanding the tissue specificity of ferritin protein cage control over ferritin mineral order, crystallinity, and chelator access.
3. Ferritin mineral
a. Mineral Structure
X-ray diffraction, X-ray absorption, electron microscopy (STEM, TEM, etc.), density (usually sedimentation/(ultracentrifugation, often through sucrose gradients), are all used to examine ferritin minerals. The low phosphate form of ferritin minerals that are found in animals, is hydrated ferric oxide/hydrous ferric oxyhydroxide and is most similar to ferrihydrite, a nanomineral: 5Fe2O3•9H2O; such minerals have 20% tetrahedrally and 80% octahedrally coordinated Fe3+. Ferritin minerals vary in size, limited by both the wall of the spherical central cavity (5 or 8 nm diameter) and the availability of Fe2+ and oxidant. The use of techniques provided single molecule information on ferritins more than half a century before the single molecule experiments of contemporary science.
b) Mineral function
Monitoring the fate of 59Fe or 55Fe-radiolabelled ferritin in different tissues is common method for studying the function of ferritin minerals. Whole body, tissue or cell radioactivity is measured in samples of tissue; measuring red blood cell 59Fe or 55Fe radioactivity is another common technique used. In situations where a tissue biopsy is performed for other purposes, radiolabelled iron in the tissue samples can also be analyzed. Such studies have been used to study the absorption of iron from different sources 72–75 and the redistribution in a legume of nodule iron (nitrogenase and leghemoglobin ) to seeds (ferritin mineral) 76.
Isotope experiments have the requirement that the “reporter” isotope or “tracer” must equilibrate with the material being traced. An example of the difficulties that occur when this condition is not met is experiments about the bioavailability of iron in beans. Experiment carried out thirty years, where the tracer did not equilibrate with bean ferritin iron reached the conclusion that bean iron was not bioavailable. In the experiment, 59FeCl3 was added to ground beans before consumption. The 59FeCl3 did not equilibrate with the ferritin mineral iron and worse, was chelated by a normal bean component, phytic acid, which is poorly absorbed77. The conclusion was reached that anemia, prevalent in many populations (30% worldwide), would not be cured by increasing the consumption of iron-rich beans77. Ten years later, the experiments were repeated in beans from plants cultivated with 59Fe-EDTA during growth and seed formation. In these experiments, equilibration of the label with the iron in bean ferritin mineral was possible, and the results showed clearly that bean iron is bioavailable both in model animal studies and in humans78, 79. However, even today the issue is still being examined experimentally80.
b. Mineral dissolution and Fe2+ exit
How iron is recovered from the ferritin mineral in vivo has been little studied. But iron chelation therapies in human disease would be facilitated by such information. Often, in studying ferritin, extra iron was added to increase the amount of ferritin protein and facilitate ferritin detection. However, apparently the added iron was high enough to be toxic, and cells responded by engulfing the extra ferritin in an intracellular compartment, the lysosome. The observation was interpreted to mean that the normal pathway for recovering iron from ferritin was destruction by lysosomal enzymes. If this were so, there would be no evolutionary advantage to the complex genetic regulatory system that controls ferritin biosynthesis. Moreover, enormous amounts of cell energy would be consumed (1 GTP for each of the > 4000 peptide bonds) in the synthesis of a protein where the only function is to be degraded with the generation of exposed, reactive iron mineral! Ferritin protein is degraded, in a regulated manner, i.e., only when the cell is iron deficient and the ferritin iron content is low 81, 82. The ferritin protein degradation site is the proteasome in the cell cytoplasm 83. The degradation signal for low iron ferritin is not known but after multiple cycles of electron transfers in the Fe2+/O2 catalytic reaction, and Fe2O3•H2O synthesis, peptide bond breakage or amino acid side chain oxidation may reach a level sensed as “excessive”. Ferritin iron can be recovered by adding external reductants and chelators in solution or injection/absorption chelators in vivo, but the process is slow because most of the time ferritin protein cages block reductant access to ferritin mineral.
Ferritin protein cages are very stable, resisting 6 M urea at pH 7 or >80°C, pH 7 in solution. Nevertheless, regions of local instability in the protein cage unfold at 56°C or 1 mM urea23. They are at the external pores of the ion channels in ferritin protein cages and essentially “open” the ferritin cage pores. Opening/unfolding the pores increases rates of ferritin mineral dissolution (Fe2+ exit). Many of the pore residues are highly conserved; substitution of channel residues also “open” the pores. Recently, when the ferritin dimer interface was modified so single, folded ferritin subunits could be produced and studied, the subunits unfolded 40°C below that of the cage, Tm=80°C8, showing the enormous stabilization conferred on the protein cage by intersubunit interactions. In cultured human cells, when ferritin pore unfolding was increased by mutation, iron retention by the altered ferritin was significantly lower than in wild type protein under the same conditions 24.
During iron toxic states created by modern transfusion therapies, which bypass the homeostatic control mechanisms for iron absorption in the intestine, increased ferritin protein synthesis cannot keep up with the increased iron entering the body. As a result, the iron content of each ferritin protein increases above normal (3–4000 Fe/protein cage compared to ~2000 Fe atoms/protein cage). Eventually, the ferritin protein cages are damaged, which exposes ferritin iron mineral to cytoplasmic reductants and initiates redox chemistry involving free radicals and protein damage. Damaged ferritin is called hemosiderin, which is functionally defined as insoluble cellular iron. (Native ferritin is very soluble >100 mg/ml). The cell response to ferritin protein cage damage and hemosiderin formation is autophagy, explaining iron mineral accumulation into lysosomes of cells grown with high concentrations of iron; the result is to deprive the cell cytoplasm of the antioxidant effect of ferritin protein.
4. Regulated Ferritin protein biosynthesis (role of mRNA structure and protein binding)
In health, ferritins supply metabolic iron concentrates for cellular syntheses of iron proteins in the cytoplasm and mitochondria, making them particularly important preceding mitochondrial division and cell growth. The ability of ferritins to scavenge free iron and to consume dioxygen makes them effective response proteins after normal or pathological oxidative damage.
a) Ferritin mRNA biosynthesis-transcriptional control
The transcriptional regulation of ferritin genes is coordinated with a large group of proteins that are important in restoring the cytoplasm of a cell to normalcy after oxidant stress (thioredoxin reductase, heme oxygenase, NADPH- quinone reductase, etc). The genes are regulated, in part, through interactions of the ARE promoter and Bach1 repressor protein, a DNA binding protein that binds the ARE DNA sequence until heme binds to Bach 1; each ARE has different binding affinities for Bach 170, which allows a set of quantitatively different response among the genes for each biological signal. These proteins all contribute to reestablishing normal redox in cells. The consumption of Fenton chemistry reactants, Fe2+ and O2 in making protein-caged biominerals, makes ferritin an antioxidant, illustrating its ARE (antioxidant response element) gene regulation.
b) Ferritin protein cage biosynthesis-translational control
Ferritin biosynthesis has been most studied in animals. When cellular iron concentrations are low, ferritin mRNA is stored in the cytoplasm as complex with IRP1 or IRP2 bound to the IRE-RNA, noncoding riboregulator. When cellular iron concentrations are high, IRE1 or IRP2, Fe2+ binds to the IRE and changes theRNA conformation, which causes IRP dissociation to increase2, 3. As a result, IRP dissociates from the IRE-RNA, eIF4F binds and ferritin mRNA translation increases2, 3. Ferritin protein subunits are always found in assembled cages.
c) The ferritin protein cage/Fe2+/O feedback loop
Regulated ferritin protein biosynthesis is unusual because DNA (transcription) and mRNA (translation are regulated) and the regulatory signals are both inorganic and organic [heme, Fe2+, oxidants (O)] and are related to the substrates used by the encoded protein (ferritin cages), Fe2+ and O2 or H2O2. When heme binds to Bach1, transcription of ferritin genes/ferritin mRNA biosynthesis, increases. When cells have oxidant damage or large excesses of iron ferritin gene transcription also increases. As a result, Fe and O (O2, H2O2, and “Oxidant”) are part of a feedback loop involving DNA, mRNA, and the gene product (ferritin protein cages) that consume Fe2+ and O2 to synthesize the caged ferritin mineral, Fe2O3•H2O. Fe2+ and O2 or oxidant, are genetic “signals” and for Fe2+ and O2, protein substrates. Three protein regulators in the feedback loop are known to date: Bach 1, a DNA or heme binding transcriptional repressor, IRPs, (two mRNA binding translation repressor protein, and eIF-4F an RNA binding translational enhancer that facilitates ribosome binding to mRNA3, 70.
Excess Fe or O activate ferritin DNA and mRNA to synthesize more ferritin protein. Di-iron catalytic sites in ferritin protein cages consume the Fe and O substrates that are also the genetic signals, shutting down ferritin gene transcription and ferritin mRNA translation/protein synthesis 84. When plants, animals or people are iron deficient ferritin synthesis is diminished, but in plants ferritin iron-dependent ferritin regulation is restricted to mRNA synthesis 85. The critical nature of ferritin to animal life is emphasized by the unique metabolite signal/gene/protein substrate feedback loop and by the embryonic lethality of gene deletions in mice 86. Heme-Fe is the signal for DNA regulation by the Bach1 transcription regulatory protein 70 while Fe2+ binding to an mRNA regulator (IRE) is the signal for DNA-independent regulation of protein biosynthesis 2, 3.
5. Ferritin as a therapeutic target and delivery agent in Disease
Iron/ferritin interactions contribute to several disease responses. First, ferritins decrease iron available to pathogens by decreasing serum iron that normally is derived from the recycling of old red blood cells. The result is “the anemia of chronic disease” 67. Second, when hosts release damaging oxidants, successful pathogens synthesize mini-ferritins (u proteins). Dps proteins are mini-ferritins (12 subunit protein cages) that use H2O2 to make ferritin mineral, thereby resisting the antibacterial H2O2 released by the host. Finally, when tissue damage in disease is extensive, as in the lungs in cystic fibrosis, ferritin escaping from damaged cells is absorbed by opportunistic pathogens 87. Targets in ferritin protein cages for pharmacological exploitation are the selective properties of mini-ferritin (Dps protein) active sites, pores around the 3-fold axes of the protein cages, the ferritin IRE-mRNA riboregulator and the dimer interface of the subnit helix bundle, where geometric interdigitation of amino acid side chains control cage assembly.
a) Selective properties of mini-ferritin (Dps protein) catalytic sites as a target for selective inhibition of catalysis
Development of selective small molecule inhibitors is a basic component of drug design. The active sites in 12 subunit mini-ferritin (Dps protein), differ in structure (number of ligands, location in the protein cage)(Table 1), oxidant (H2O2 vs. O2) and the order of addition of the two substrates (binding of the second Fe2+ requires oxidant binding) 15, 20, 26. Currently, mini-ferritins of human pathogens have been targeted as antigens in vaccines. However, the idea of weakening bacteria during infection by inhibiting the catalytic functions of their mini-ferrityns has been little considered, even though the active protein is associated with bacterial virulence.
b. Ferritin protein cages as targets for chelators in transfusional iron overload during treatment of genetic anemias
Ferritin is the site of most of the excess iron in the genetic diseases of iron overload. In Hereditary Hemochromatosis, excess iron accumulates because of a genetic defect in a protein, HFE, which interferes with normal signaling of adequate iron absoprtion. In the genetic anemias, Thalassemia and Sickle Cell Disease, the treatment itself, transfusion of normal red blood cells, causes iron overload. A major physiological response to excess iron is increased ferritin protein biosynthesis mainly through the effects of Fe2+-IRE-RNA binding on interactions with regulatory protein binding (IRP or eIF-4F) described in part 4. Under the best of conditions, however, the amounts of protein synthesized are too small for the amounts of accumulated iron and, during iron overload, the average size of the iron mineral in ferritin protein cage increases 88, 89. The homeostatic responses that have evolved are clearly overwhelmed by contemporary medical therapies.
In Hereditary Hemochromatosis the excess iron is removed by regular phlebotomy and the blood is used by blood banks 90, since iron excretion is very low in mammals67. However, the excess iron from hypertransfusions in genetic anemia cannot be removed by phlebotomy, because the genetic defects are in red cell production.
To remove the excess iron in hypertransfusion iron overload, a variety of iron chelators have been developed over the last 40 years. Some chelators are from bacterial siderophore models. The three iron chelators in common use today are deferral, desferriox, and deferiprone 91. Combinations of iron chelators are particularly effective92, due in part to differences in the subcellular location of the different chelators 93. The iron chelator desferrioxamine (Desferal) folds without iron, and thus, enters cells by endocytosis into the lysosomal compartment; desferrioxamine (Desferal). By contrast, deferriprone and desferasirox require iron to fold, and the linear molecules enter cells by transport through the plasma membrane. Desferal is administered intravenously and deferriprone and desferasirox are administered orally. The major problem with current iron chelators is that they target low molecular iron (the labile iron pool”) 16, 71, which is only a small fraction of the excess iron. Most of the excess iron in the solid protein coated, ferritin mineral. The rationale used for such chelators is that all the iron in living cells, including iron in the ferritin mineral, is in equilibrium. As a result, current chelation regimens are very slow and long, sometimes as long as 40 hours/week. The idea of targeting chelators to ferritin protein pores, using specific binding partners, e.g. peptide (see Fig 2), that readily enters cells, (HepG2cell cultures, E.C. Theil and H. Wang, unpublished observations) remains underdeveloped.
c) Ferritin mRNA riboregulators as small molecule targets to increase ferritin protein synthesis, and minimize toxic hemosiderin (damaged ferritin)
The compensatory responses to iron overload are incompletely matched. For example, even though ferritin protein biosynthesis increases to manage the cellular iron, the iron content of each ferritin protein cage increases 25. Denatured ferritin (toxic hemosiderin) also increases. Little iron is excreted in the urine during iron overload, because, unlike other metal ions, ion excretion is minimal, even when the tissue iron content is abnormally high.
Increasing ferritin protein synthesis with small molecules targeted to the ferritin mRNA riboregulator, will decrease the iron content of ferritin protein cages and of hemosiderin during iron overload. Synthesizing more ferritin protein during iron overload will decrease ferritin cages that are by the mineralization of excess caged iron. Moreover, additional sites for safe iron storage will be produced. Three observations support the rationale: First, the ferritin riboregulator (IRE-RNA) selectively binds small molecules and two regulatory proteins, one of which blocks ribosome binding to mRNA and the other of which enhances ribosome binding to mRNAs 3, 68; conformational change in IRE-RNA induced by RNA-Fe2+ binding3 is the trigger that decreases IRP binding and increases eIF-4F binding. The result is a change in ferritin protein biosynthesis rates in vitro whether the IRE-RNA is changed by binding a small, organic molecule 94 or by binding Fe2+3. Second, during iron overload, a significant fraction of ferritin mRNA molecules remain repressed in vivo 95. Third, increasing ferritin protein concentration by activating the pool of ferritin mRNA still blocked during iron overload 95 is benign to human physiology: overproduction of ferritin subunits and assembled cages, caused by mutations in the IRE riboregulator (hereditary hyperferrtinemia cataract syndrome), for example has only minor effects on human physiology96.
d) Ferritin protein cages as nanovessels for drug or sensor delivery
Ferritin protein cages are emerging as novel component of disease therapies 97. The use of human ferritin protein cages has the added benefit of minimizing immune reactions. Intact ferritin enters living cells by receptor mediated endocytosis 98, 99. Ferritin protein cages have been used to deliver sensors of uniform sizes for imaging 100, 101, for detecting vascular inflammation and angiogenesis in carotid and aortic disease 62 and compounds that are potential antitumor agents. A potential limit in using ferritin to deliver drugs or agents is fitting the desired cargo inside the cage. In some natural ferritins, crosslinks between pairs of subunits along the two fold symmetry axes of the protein cage, change the size of the caged ferritin mineral 81. Recently an engineered ferritin cage was produced with changed interactions along the cage dimer interface so that cage disassembly/assembly was controlled with the Cu2+ 8. The modified ferritin protein cage was disassembled and reassembled around a large molecular cargo. Such an approach allows large molecules close to the size of the ferritin mineral cavity to be captured inside of ferritin. How cells will react to such complexes remains to be explored. However, when an animal cell absorbs a foreign (plant) ferritin by endocytosis, the plant ferritin protein cage is degraded and the iron mineral content is made available to the cell for metabolism 98, 102. With such observations in hand to place desired compounds inside the ferritin cages, and modifications of ferritin surfaces for selective delivery, the ability to deliver concentrated doses of drugs to particular cell types is imminent.
6. Perspective
Ferritin protein chemistry protects cells from Fenton chemistry substrates Fe2+, O2 or H2O2, which are released by from their normal locations by oxidative damage. Ferritin converts Fe2+ and O2 to the caged, Fe/O mineral. The mineral also preserves the iron in a concentrated from for use in cofactor synthesis during cell recovery. Fe2+ binding to ferritin ion entry/exit channels and protein catalytic sites, as well as the binding of Fe3+O(H) multimeric products near active sites in protein nucleation channels and around the 4-fold symmetry axes inside the cage, are only partly understood; they also appear to depend on which member of the ferritin superfamily is being studied. The variations in ferritin amino acid sequence (up to 80%), active site ligands, location of the active sites (Table 1), protein cage size (12 or 24 subunit cages), numbers of catalytically active subunits/cage (4–24), and the interrelated variations in protein mineral crystallinity (Fig. 3), indicate that Nature has already matched ferritin structures to multiple physiological and environmental conditions. As long as chemists avoid the trap of blurring the variations with overly rigid reductionism, the natural, biological variety among ferritin proteins provides a rich platform to develop a ferritin nanocage for many technical and medical need.
The amount of iron needed for health is well defined. In humans who are not growing or reproducing, only 1–2 mg/day absorbed is needed to maintain normal health. (Absorption from food iron is ~ 10–20% efficient among all the different chemical forms of iron in food so ~ 10 – 20 mg/day need to be eaten). Nevertheless, iron deficiency is the major nutritional deficiency in the world today, ~500 years after the original diagnoses and development of treatments 103. Successful organisms are programmed to synthesize ferritin in times of “plenty”, in anticipation of future use. Thus, ferritin is most abundant in stationery cultures of bacteria awaiting triggers for rapid growth 5, 104, in legume nodules preparing for nitrogen fixation 104 where nitrogenases (32 Fe/protein) and leghemoglobin are synthesized in abundance, in immature leaves preparing for photosynthesis and ferredoxin synthesis 105, in embryonic erythrocytes before the rapid cell replacement associated with hemoglobin “switching” 106, and in fetal liver107 in anticipation of the developmental, neonatal iron deficit. Ferritin iron mineral dissolution occurs when cellular demands inside the organisms change, such as cell division, or rapid growth (childhood, puberty). The signals that regulate cellular developmental programs to “plan ahead” for iron use are still obscure, although a recent study indicates a study role for the heme binding proteins Bach1 as a mitosis regulator 108.
Gaps in the chemical and molecular biological information about normal iron homeostasis and normal development negatively impact understanding of the medical problems associated with massive iron accumulations above normal homeostatic mechanisms. Toxic iron accumulations occur as a result of hypertransfusion treatment in hereditary hemoglobin defects (Thalassemia, Sickle Cell Disease), transfusions after chemotherapy or before transplants (e.g., CNS tumors, leukemia, sarcomas, lymphomas, renal carcinomas), and excess iron absorption from food in hereditary hemochromatosis. Ferritin and hemosiderin (damaged ferritin) are the major sites of the abnormal iron deposits in humans. Small molecule iron chelators can access only a small fraction of the iron needed to be removed because solid ferritin minerals are protected from the chelators and cellular reductants by the folded protein cage. New strategies such as binding peptides which modulate folding of ferritin proteins pores 20, 23, (Figs 4,5) to increase chelation are in very early stages and require extensive “translation” and development before they are ready for removing iron from humans with toxic iron overload. Other uses of ferritins being developed for use in disease are as nanovessels for drug delivery and templates for sensors, e.g. 61, 62, 107. Finally, ferritin mRNA riboregulators are both models for mRNA exploitation in general and potential aids during iron excess, in particular. During the billions of years ferritins have existed, differences in the protein cages have evolved to match particular environmental niches (aerobic, anaerobic, single cells, multiple cells and tissues) and for different physiological purposes (DNA protection, recovery from oxidant stress, providing metabolic iron concentrates, sequestering iron from invading pathogens). The attractive, natural array of ferritin variants requires further exploration for full understanding and novel applications to human health and disease.
HIGHLIGHTS/Synopsis.
Ferritin protein cages and iron biominerals are described. Possible targets in ferritin protein : protein and protein – iron interactions for therapeutic development in disease modulate disease are indicated, such as the catalytic sites (pathogen-specific), folded ferritin pores (improved iron chelation), the ferritin mRNA riboregulator (stabilizing excess iron accumulations in hypertransfusion therapies, and protein cage assembly (nanodrug/nanosensor delivery).
Acknowledgments
FUNDING SOURCE: The work of the author cited here was supported by the CHORI Partners and National Institutes of Health Grant DK20251.
The author thanks all her group members and colleagues for their enthusiastic intellectual and technical contributions to the work described here. Support from the NIH (R01-DK20251), the CHORI Foundation and CHORI Partners has been crucial.
ABBREVIATIONS
- DFP
diferric peroxo
- HH
Hereditary Hemochormatosis
- WT
wild type
References
- 1.Hsiao C, Chou IC, Okafor CD, Bowman JC, O'Neill EB, Athavale SS, Petrov AS, Hud NV, Wartell RM, Harvey SC, Williams LD. Nat Chem. 2013;5:525–8. doi: 10.1038/nchem.1649. [DOI] [PubMed] [Google Scholar]
- 2.Khan MA, Walden WE, Goss DJ, Theil EC. J Biol Chem. 2009;284:30122–8. doi: 10.1074/jbc.M109.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma J, Haldar S, Khan MA, Sharma SD, Merrick WC, Theil EC, Goss DJ. Proc Natl Acad Sci U S A. 2012;109:8417–22. doi: 10.1073/pnas.1120045109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haldar S, Bevers LE, Tosha T, Theil EC. J Biol Chem. 2011;286:25620–25627. doi: 10.1074/jbc.M110.205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nandal A, Huggins CC, Woodhall MR, McHugh J, Rodriguez-Quinones F, Quail MA, Guest JR, Andrews SC. Mol Microbiol. 2010;75:637–57. doi: 10.1111/j.1365-2958.2009.06977.x. [DOI] [PubMed] [Google Scholar]
- 6.St Pierre T, Tran KC, Webb J, Macey DJ, Heywood BR, Sparks NH, Wade VJ, Mann S, Pootrakul P. Biol Met. 1991;4:162–165. doi: 10.1007/BF01141308. [DOI] [PubMed] [Google Scholar]
- 7.Theil EC. Curr Opin Chem Biol. 2011;15:304–11. doi: 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huard DJ, Kane KM, Tezcan FA. Nat Chem Biol. 2013 doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P, Woolfson DN. Science. 2013;340:595–9. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. J Am Chem Soc. 2008;130:9441–9450. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tosha T, Behera RK, Ng HL, Bhattasali O, Alber T, Theil EC. J Biol Chem. 2012;287:13016–13025. doi: 10.1074/jbc.M111.332734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tosha T, Behera RK, Theil EC. Inorg Chem. 2012;51:11406–11. doi: 10.1021/ic3010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ragland M, Briat JF, Gagnon J, Laulhere JP, Massenet O, Theil EC. J Biol Chem. 1990;265:18339–18344. [PubMed] [Google Scholar]
- 14.Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. Nat Struct Biol. 1998;5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- 15.Chiancone E, Ceci P. Biochim Biophys Acta. 2010;1800:798–805. doi: 10.1016/j.bbagen.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Lal A, Porter J, Sweeters N, Ng V, Evans P, Neumayr L, Kurio G, Harmatz P, Vichinsky E. Blood Cells Mol Dis. 2013;50:99–104. doi: 10.1016/j.bcmd.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rucker P, Torti FM, Torti SV. Protein Eng. 1997;10:967–73. doi: 10.1093/protein/10.8.967. [DOI] [PubMed] [Google Scholar]
- 18.Matzapetakis M, Turano P, Theil EC, Bertini I. J Biomol NMR. 2007;38:237–242. doi: 10.1007/s10858-007-9163-9. [DOI] [PubMed] [Google Scholar]
- 19.Turano P, Lalli D, Felli IC, Theil EC, Bertini I. Proc Natl Acad Sci U S A. 2010;107:545–550. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu XS, Patterson LD, Miller MJ, Theil EC. J Biol Chem. 2007;282:31821–5. doi: 10.1074/jbc.C700153200. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Pielak GJ. J Am Chem Soc. 2009;131:1368–9. doi: 10.1021/ja808428d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bermel W, Bertini I, Felli IC, Matzapetakis M, Pierattelli R, Theil EC, Turano P. J Mag Reson. 2007;188:301–310. doi: 10.1016/j.jmr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Jin W, Theil EC. Proc Natl Acad Sci U S A. 2003;100:3653–8. doi: 10.1073/pnas.0636928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasan MR, Tosha T, Theil EC. J Biol Chem. 2008;283:31394–400. doi: 10.1074/jbc.M806025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arosio P, Levi S. Biochim Biophys Acta. 2010;1800:783–92. doi: 10.1016/j.bbagen.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Le Brun NE, Crow A, Murphy ME, Mauk AG, Moore GR. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbagen.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Honarmand Ebrahimi K, Bill E, Hagedoorn PL, Hagen WR. Nat Chem Biol. 2012;8:941–8. doi: 10.1038/nchembio.1071. [DOI] [PubMed] [Google Scholar]
- 28.Hempstead PD, Yewdall SJ, Fernie AR, Lawson DM, Artymiuk PJ, Rice DW, Ford GC, Harrison PM. J Mol Biol. 1997;268:424–448. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Theil EC. Proc Natl Acad Sci USA. 2004;101:8557–8562. doi: 10.1073/pnas.0401146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP. J Mol Biol. 2007;365:440–52. doi: 10.1016/j.jmb.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Tosha T, Hasan MR, Theil EC. Proc Natl Acad Sci U S A. 2008;105:18182–7. doi: 10.1073/pnas.0805083105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda T, Goto F, Yoshihara T, Mikami B. J Biol Chem. 2009 doi: 10.1074/jbc.M109.059790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tosha T, Ng HL, Bhattasali O, Alber T, Theil EC. J Am Chem Soc. 2010;132:14562–9. doi: 10.1021/ja105583d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stillman TJ, Connolly PP, Latimer CL, Morland AF, Quail MA, Andrews SC, Treffry A, Guest JR, Artymiuk PJ, Harrison PM. J Biol Chem. 2003;278:26275–86. doi: 10.1074/jbc.M207354200. [DOI] [PubMed] [Google Scholar]
- 35.Crow A, Lawson TL, Lewin A, Moore GR, Le Brun NE. J Am Chem Soc. 2009;131:6808–13. doi: 10.1021/ja8093444. [DOI] [PubMed] [Google Scholar]
- 36.Tatur J, Hagen WR, Matias PM. J Biol Inorg Chem. 2007;12:615–30. doi: 10.1007/s00775-007-0212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Kim K, Leighton T, Theil EC. J Biol Chem. 2006;28:27827–27835. doi: 10.1074/jbc.M601398200. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz JK, Liu XS, Tosha T, Diebold A, Theil EC, Solomon EI. Biochemistry. 2010;49:10516–25. doi: 10.1021/bi101346c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papinutto E, Dundon WG, Pitulis N, Battistutta R, Montecucco C, Zanotti G. J Biol Chem. 2002;277:15093–8. doi: 10.1074/jbc.M112378200. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Yun S, Yang X, Zhao G. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbagen.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Waldo GS, Theil EC. Biochemistry. 1993;32:13262–13269. doi: 10.1021/bi00211a039. [DOI] [PubMed] [Google Scholar]
- 42.Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM. Biochemistry. 1997;36:432–441. doi: 10.1021/bi961830l. [DOI] [PubMed] [Google Scholar]
- 43.Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, Stemmler TL, Philpott CC. J Biol Chem. 2013;288:17791–17802. doi: 10.1074/jbc.M113.460253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871–6. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 45.Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM. Biochemistry. 1999;38:5290–5. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 46.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 47.Zhao G, Arosio P, Chasteen ND. Biochemistry. 2006;45:3429–36. doi: 10.1021/bi052443r. [DOI] [PubMed] [Google Scholar]
- 48.Bellapadrona G, Stefanini S, Zamparelli C, Theil EC, Chiancone E. J Biol Chem. 2009;284:19101–9. doi: 10.1074/jbc.M109.014670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawson DM, Treffry A, Artymiuk PJ, Harrison PM, Yewdall SJ, Luzzago A, Cesareni G, Levi S, Arosio P. FEBS Lett. 1989;254:207–210. doi: 10.1016/0014-5793(89)81040-3. [DOI] [PubMed] [Google Scholar]
- 50.Ha Y, Shi D, Small GW, Theil EC, Allewell NM. J Biol Inorg Chem. 1999;4:243–256. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 51.Bertini I, Lalli D, Mangani S, Pozzi C, Rosa C, Theil EC, Turano P. J Am Chem Soc. 2012;134:6169–6176. doi: 10.1021/ja210084n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ceci P, Di Cecca G, Falconi M, Oteri F, Zamparelli C, Chiancone E. J Biol Inorg Chem. 2011;16:869–80. doi: 10.1007/s00775-011-0784-9. [DOI] [PubMed] [Google Scholar]
- 53.Calhoun LN, Kwon YM. J Appl Microbiol. 2011;110:375–86. doi: 10.1111/j.1365-2672.2010.04890.x. [DOI] [PubMed] [Google Scholar]
- 54.Gauss GH, Benas P, Wiedenheft B, Young M, Douglas T, Lawrence CM. Biochemistry. 2006;45:10815–27. doi: 10.1021/bi060782u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cairo G, Rappocciolo E, Tacchini L, Schiaffonati L. Biochem J. 1991;275(Pt 3):813–6. doi: 10.1042/bj2750813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levi S, Santambrogio P, Cozzi A, Rovida E, Corsi B, Tanborini E, Spada S, Albertini A, Arosio P. J Mol Biol. 1994;238:649–654. doi: 10.1006/jmbi.1994.1325. [DOI] [PubMed] [Google Scholar]
- 57.Theil EC, Small GW, He L, Tipton A, Danger D. Inorg Chim Acta. 2000;297:242–251. [Google Scholar]
- 58.Bou-Abdallah F, Biasiotto G, Arosio P, Chasteen ND. Biochemistry. 2004;43:4332–4337. doi: 10.1021/bi0498813. [DOI] [PubMed] [Google Scholar]
- 59.Mann S. Nat Mater. 2009;8:781–92. doi: 10.1038/nmat2496. [DOI] [PubMed] [Google Scholar]
- 60.Yamashita I, Iwahori K, Kumagai S. Biochim Biophys Acta. 2010;1800:846–57. doi: 10.1016/j.bbagen.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 61.Terashima M, Uchida M, Kosuge H, Tsao PS, Young MJ, Conolly SM, Douglas T, McConnell MV. Biomaterials. 2011;32:1430–7. doi: 10.1016/j.biomaterials.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitagawa T, Kosuge H, Uchida M, Dua MM, Iida Y, Dalman RL, Douglas T, McConnell MV. Mol Imaging Biol. 2012;14:315–24. doi: 10.1007/s11307-011-0495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Theil EC, Behera RKB. The Chemistry of Nature's Iron Biominerals in Ferritin Protein Cages. Vol. 1 John Wiley & Sons; 2013. [Google Scholar]
- 64.Jameson GNL, Jin W, Krebs C, Perreira AS, Tavares P, Liu X, Theil EC, Huynh BH. Biochemistry. 2002;41:13435–13443. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 65.Funk F, Lenders JP, Crichton RR, Schneider W. Eur J Biochem. 1985;152:167–72. doi: 10.1111/j.1432-1033.1985.tb09177.x. [DOI] [PubMed] [Google Scholar]
- 66.Takagi H, Shi D, Ha Y, Allewell NM, Theil EC. J Biol Chem. 1998;273:18685–8. doi: 10.1074/jbc.273.30.18685. [DOI] [PubMed] [Google Scholar]
- 67.Anderson CP, Shen M, Eisenstein RS, Leibold EA. Biochim Biophys Acta. 2012;1823:1468–83. doi: 10.1016/j.bbamcr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goss DJ, Theil EC. Acc Chem Res. 2011;44:1320–8. doi: 10.1021/ar2001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Mol Cell Biol. 2006;26:2845–56. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hintze KJ, Katoh Y, Igarashi K, Theil EC. J Biol Chem. 2007;282:34365–34371. doi: 10.1074/jbc.M700254200. [DOI] [PubMed] [Google Scholar]
- 71.Berdoukas V, Farmaki K, Carson S, Wood J, Coates T. J Blood Med. 2012;3:119–29. doi: 10.2147/JBM.S27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaisman B, Meyron-Holtz EG, Fibach E, Krichevsky AM, Konijn AM. Br J Haematol. 2000;110:394–401. doi: 10.1046/j.1365-2141.2000.02167.x. [DOI] [PubMed] [Google Scholar]
- 73.Davila-Hicks P, Theil EC, Lonnerdal B. Am J Clin Nutr. 2004;80:936–40. doi: 10.1093/ajcn/80.4.936. [DOI] [PubMed] [Google Scholar]
- 74.Leimberg MJ, Prus E, Konijn AM, Fibach E. J Cell Biochem. 2008;103:1211–8. doi: 10.1002/jcb.21499. [DOI] [PubMed] [Google Scholar]
- 75.Theil EC, Chen H, Miranda C, Janser H, Elsenhans B, Nunez MT, Pizarro F, Schumann K. J Nutr. 2012;142:478–83. doi: 10.3945/jn.111.145854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burton JW, Harlow C, Theil EC. J Plant Nutr. 1998;21:913–927. [Google Scholar]
- 77.Lynch SR, Beard JL, Dassenko SA, Cook JD. Am J Clin Nutr. 1984;40:42–7. doi: 10.1093/ajcn/40.1.42. [DOI] [PubMed] [Google Scholar]
- 78.Beard JL, Burton JW, Theil EC. J Nutr. 1996;126:154–160. doi: 10.1093/jn/126.1.154. [DOI] [PubMed] [Google Scholar]
- 79.Murray-Kolb LE, Welch R, Theil EC, Beard JL. Am J Clin Nutr. 2003;77:180–184. doi: 10.1093/ajcn/77.1.180. [DOI] [PubMed] [Google Scholar]
- 80.Martinez Meyer MR, Rojas A, Santanen A, Stoddard FL. Food Chem. 2013;136:87–93. doi: 10.1016/j.foodchem.2012.07.105. [DOI] [PubMed] [Google Scholar]
- 81.Mertz JR, Theil EC. J Biol Chem. 1983;258:11719–11726. [PubMed] [Google Scholar]
- 82.Treffry A, Harrison PM. Biochem J. 1984;220:357–859. doi: 10.1042/bj2200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. Embo J. 2006;25:5396–404. doi: 10.1038/sj.emboj.7601409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Theil EC, Goss DJ. Chem Rev. 2009;109:4568–79. doi: 10.1021/cr900052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Darbani B, Briat JF, Holm PB, Husted S, Noeparvar S, Borg S. Biotechnol Adv. 2013 doi: 10.1016/j.biotechadv.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Ferreira F, Bucchini D, Martin ME, Levi S, Arosio P, Grandchamp B, Beaumont C. J Biol Chem. 2000;275:3021–3024. doi: 10.1074/jbc.275.5.3021. [DOI] [PubMed] [Google Scholar]
- 87.Dehner C, Morales-Soto N, Behera RK, Shrout J, Theil ECM, Aurice PA, Dubois JL. J Biol Inorg Chem. 2013;18:371–81. doi: 10.1007/s00775-013-0981-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oshtrakh MI, Alenkina IV, Vinogradov AV, Konstantinova TS, Kuzmann E, Semionkin VA. Biometals. 2013;26:229–39. doi: 10.1007/s10534-012-9602-2. [DOI] [PubMed] [Google Scholar]
- 89.Zuyderhoudt FM, Sindram JW, Marx JJ, Jorning GG, van Gool J. Hepatology. 1983;3:232–5. doi: 10.1002/hep.1840030216. [DOI] [PubMed] [Google Scholar]
- 90.Luten M, Roerdinkholder-Stoelwinder B, Rombout-Sestrienkova E, de Grip WJ, Bos HJ, Bosman GJ. Transfusion. 2008;48:436–41. doi: 10.1111/j.1537-2995.2007.01547.x. [DOI] [PubMed] [Google Scholar]
- 91.Musallam KM, Cappellini MD, Taher AT. Curr Opin Hematol. 2013;20:187–92. doi: 10.1097/MOH.0b013e32835f5a5c. [DOI] [PubMed] [Google Scholar]
- 92.Trachtenberg F, Vichinsky E, Haines D, Pakbaz Z, Mednick L, Sobota A, Kwiatkowski J, Thompson AA, Porter J, Coates T, Giardina PJ, Olivieri N, Yamashita R, Neufeld EJ. Am J Hematol. 2011;86:433–6. doi: 10.1002/ajh.21993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Domenico I, Ward DM, Kaplan J. Blood. 2009;114:4546–51. doi: 10.1182/blood-2009-05-224188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tibodeau JD, Fox PM, Ropp PA, Theil EC, Thorp HH. Proc Natl Acad Sci U S A. 2006;103:253–7. doi: 10.1073/pnas.0509744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Melefors O, Goossen B, Johansson HE, Stripecke R, Gray NK, Hentze MW. J Biol Chem. 1993;268:5974–8. [PubMed] [Google Scholar]
- 96.Bennett TM, Maraini G, Jin C, Sun W, Hejtmancik JF, Shiels A. Mol Vis. 2013;19:835–44. [PMC free article] [PubMed] [Google Scholar]
- 97.Uchida M, Kang S, Reichhardt C, Harlen K, Douglas T. Biochim Biophys Acta. 2010;1800:834–45. doi: 10.1016/j.bbagen.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.San Martin CD, Garri C, Pizarro F, Walter T, Theil EC, Nunez MT. J Nutr. 2008;138:659–66. doi: 10.1093/jn/138.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, Torti SV. PLoS One. 2011;6:e23800. doi: 10.1371/journal.pone.0023800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leung K. 2004 [Google Scholar]
- 101.Uchida M, Terashima M, Cunningham CH, Suzuki Y, Willits DA, Willis AF, Yang PC, Tsao PS, McConnell MV, Young MJ, Douglas T. Magn Reson Med. 2008;60:1073–81. doi: 10.1002/mrm.21761. [DOI] [PubMed] [Google Scholar]
- 102.Antileo E, Garri C, Tapia V, Munoz JP, Chiong M, Nualart F, Lavandero S, Fernandez J, Nunez MT. Am J Physiol Gastrointest Liver Physiol. 2013;304:G655–61. doi: 10.1152/ajpgi.00472.2012. [DOI] [PubMed] [Google Scholar]
- 103.Pasricha SR, Drakesmith H, Black J, Hipgrave D, Biggs BA. Blood. 2013 doi: 10.1182/blood-2012-09-453522. [DOI] [PubMed] [Google Scholar]
- 104.Ragland M, Theil EC. Plant Mol Biol. 1993;21:555–560. doi: 10.1007/BF00028813. [DOI] [PubMed] [Google Scholar]
- 105.Theil EC, Hase T. Iron Chelation in Plants and Soil Microorganisms. Academic Press; San Diego: 1993. Plant and microbial ferritins; pp. 133–156. [Google Scholar]
- 106.Theil EC. Red cell ferritin and iron storage during the early hemoglobin switch. In: Stamatoyannopoulos G, Nienhuis A, editors. Hemoglobins in Development and Differentiation. Alan R. Liss, Inc; New York: 1981. pp. 423–431. [Google Scholar]
- 107.Munro HN, Linder MC. Physiol Rev. 1978;58:318–396. doi: 10.1152/physrev.1978.58.2.317. [DOI] [PubMed] [Google Scholar]
- 108.Li J, Shiraki T, Igarashi K. Commun Integr Biol. 2013;5:477–9. doi: 10.4161/cib.21149. [DOI] [PMC free article] [PubMed] [Google Scholar]