

Abstract
The mechanisms underlying the selective targeting of specific brain regions by different neurodegenerative diseases is one of the most intriguing mysteries in medicine. For example, it is known that Alzheimer’s disease primarily affects parts of the brain that play a role in memory, whereas Parkinson’s disease predominantly affects parts of the brain that are involved in body movement. However, the reasons that other brain regions remain unaffected in these diseases are unknown. A better understanding of the phenomenon of selective vulnerability is required for the development of targeted therapeutic approaches that specifically protect affected neurons, thereby altering the disease course and preventing its progression. Prion diseases are a fascinating group of neurodegenerative diseases because they exhibit a wide phenotypic spectrum caused by different sequence perturbations in a single protein. The possible ways that mutations affecting this protein can cause several distinct neurodegenerative diseases are explored in this Review to highlight the complexity underlying selective vulnerability. The premise of this article is that selective vulnerability is determined by the interaction of specific protein conformers and region-specific microenvironments harboring unique combinations of subcellular components such as metals, chaperones and protein translation machinery. Given the abundance of potential contributory factors in the neurodegenerative process, a better understanding of how these factors interact will provide invaluable insight into disease mechanisms to guide therapeutic discovery.
KEY WORDS: Huntington’s disease, Neurodegeneration, Spinocerebellar ataxia, Knock-in mice, Neuropathology, Prion diseases
Introduction
Neurodegenerative diseases affect tens of millions of people worldwide every year. Trying to limit this devastating assault on human health is one of the most pressing challenges in current medicine. Neurodegenerative diseases, which are generally lethal, are typified by the physical decay and eventually loss of neurons. Like cancer, neurodegenerative disorders are a phenotypically heterogeneous group of diseases, with each having unique characteristics; however, several key features are shared. All neurodegenerative diseases are thought to be caused by the misfolding of specific proteins and the eventual clumping of misfolded proteins into aggregates, as the disease progresses. Although the aggregates found in histological sections of brains affected by different neurodegenerative diseases have different shapes and tinctorial properties, they seem to develop from a common pathway, as demonstrated by their universal reactivity with a pair of antibodies generated against an Alzheimer’s disease (AD)-related peptide (Kayed et al., 2003). The antibodies react with either small oligomeric forms or higher-order aggregates from several neurodegenerative disease-related proteins, including prion diseases (PrDs) (Aidt et al., 2013), indicating that the misfolded proteins share common conformational transition states (Glabe, 2006; Kayed et al., 2003). Inherited mutations in the genes encoding many of these proteins are causally linked to familial forms of neurodegeneration, further highlighting the importance of the malformed proteins for disease development. Neurodegenerative diseases generally present clinical signs at mid-late life, which is intriguing in the case of familial neurodegenerative diseases, in which the disease-causing mutant protein is present throughout life. Pathological changes commonly observed in neurodegenerative diseases include reactive astrocytosis and microgliosis (Aguzzi et al., 2013; Sofroniew and Vinters, 2010). With so many similarities between the different neurodegenerative diseases, the knowledge gained from studying one can often be applied to others. Nonetheless, there are many differences between the diseases, and understanding the mechanisms involved will be crucial for designing therapies. However, there are many pieces to the puzzle, making this an extremely challenging problem.
One reason this is an enormous challenge is because the brain is extraordinarily complex. It is built of highly interconnected networks of numerous neural nuclei. These nuclei consist of many types of neurons and, in most neurodegenerative diseases, only a subset of neurons in specific nuclei are initially targeted. For example, cholinergic neurons of the cerebral cortex as well as hippocampal neurons are targeted in AD (Francis et al., 1999), whereas dopaminergic neurons of the substantia nigra are targeted in Parkinson’s disease (PD) (Sulzer and Surmeier, 2013). Spinocerebellar ataxia-1 (Sca1) and Huntington’s disease (HD), two members of a broad class of diseases linked to long CAG (encoding glutamine)-repeat mutations, both affect GABAergic neurons [utilizing γ-aminobutyric acid (GABA)]. The cell populations affected, however, differ: Sca1 targets giant Purkinje cells of the cerebellum (Zoghbi, 2000), whereas HD targets medium-sized spiny neurons of the striatum (Graveland et al., 1985). Interestingly, the genes causing Sca1 and HD are expressed in both regions, implying that a single mutation, the long CAG repeat, can cause a number of diseases depending on which gene carries it. The reasons that both regions are not affected in both diseases are poorly understood.
Selective vulnerability is also a hallmark feature of the PrDs. In this group of disorders, GABAergic neurons are also affected, yet the diseases have some important features that distinguish them from the CAG-repeat diseases. Aside from the frightening names of some, such as chronic wasting disease, ‘mad cow’ disease and fatal familial insomnia (FFI), PrDs are infamous for the cases where disease has spread between individuals, sometimes across species (Prusiner, 1998). The causative agent is believed to be a prion: an infectious agent formed of abnormal, misfolded protein. Practices that facilitate the spread of PrDs between individuals include medical procedures involving contaminated tools or tissues, consumption of contaminated food and human cannibalism (Prusiner, 1998). A less well-known, but arguably more interesting, characteristic is that PrDs can have quite disparate clinical features (Kovács et al., 2002). Familial PrDs, which are all caused by dominant mutations in the protein-coding sequence of the same gene, PRNP, encoding the prion protein (PrP), are emblematic of this clinical pleiotropy. These diseases, caused by >30 different mutations, are classified into three types based on a combination of clinical and neuropathological changes (Kovács et al., 2002; Rossetti et al., 2011). Creutzfeldt-Jakob disease (CJD) generally presents as a cognitive disease characterized by severe neuronal loss, spongiform degeneration (spongiosis) and amorphous PrP aggregates, and primarily targets the cortex (Fig. 1) (Gambetti et al., 2003; Gambetti et al., 1995). Gerstmann-Straussler-Scheinker syndrome (GSS) is a motor system disease in which the cerebellum (Fig. 1) is affected by amyloid PrP aggregates (stainable with special dyes such as thioflavin T or Congo red); however, these brains generally show little neuronal loss or spongiosis (Ghetti et al., 1994; Piccardo et al., 1998). Finally, FFI, which is characterized by disruptions in sleep homeostasis and autonomic nervous system functions, primarily targets the thalamus (Fig. 1), with severe neuronal loss and reactive gliosis but relatively little PrP aggregation or spongiosis (Gambetti et al., 2003; Gambetti et al., 1995). Although individual mutations cause some additional, unique features that can distinguish their associated phenotypes from others assigned to the same disease type and, furthermore, the different disease types have some overlap (for example, CJD and GSS can both affect the cortex and cerebellum, but to a different extent), mutations in PRNP cause diseases that are consistently assigned to one of these three disease types (Kovács et al., 2002). Thus, familial PrDs contrast with CAG-repeat diseases because a single gene can cause multiple diseases depending on which mutation it carries. It is important to note that once a disease has progressed for many months or years, it spreads into new areas (Braak and Braak, 1991) and the selectivity technically diminishes. However, the diseases start in a specific area (Braak and Braak, 1991; Graveland et al., 1985; Hyman et al., 1984) and understanding what factors determine this selectivity is not only scientifically fascinating, but also medically important, because early interventions are likely to be the most efficacious.
Fig. 1.

Different brain regions affected by different familial PrDs. (A) A powerful technique employed by neurologists to diagnose disease is to non-invasively look at a patient’s brain using magnetic resonance imaging (MRI). Two images in the sagittal plane from a healthy individual facing right are used as maps to indicate the regions affected in various neurodegenerative diseases and to highlight that the regions are widely distributed. The image on the left is at the mid-line, the other image is parallel but towards one side. (B) A line chart showing the spatial distribution of mRNAs that cause specific neurodegenerative diseases when mutated. These data were measured by in situ hybridization of the adult mouse brain. Expression levels are not appreciably higher in the areas that are targeted. Data were acquired from the Allen Brain Atlas website (www.brain-map.org). Brain region abbreviations: OLF, olfactory bulb; HPF, hippocampus; CTXsp, subregion of the cortex (bottom of the brain); STR, striatum; PAL, pallidum; CB, cerebellum; TH, thalamus; HY, hypothalamus; MB, midbrain; P, pons; MY, medulla; CTX, cortex (main region on top of the brain).
One potential explanation for selective vulnerability is that the gene that triggers protein misfolding is expressed at higher levels in areas that are affected the most. However, this hypothesis is unlikely to be vindicated because many neurodegenerative disease-related genes have similar levels of expression in both affected and unaffected areas (Fig. 1). Moreover, this cannot be the case for PrDs because, as mentioned, mutations in a single gene give rise to all the disease variants. Indeed, this makes the study of familial PrD pathology particularly important because they are all caused by mutations in PRNP, and comparisons between different diseases are not confounded by expression pattern differences. Here, I discuss several factors that might influence the selective targeting of brain regions in different diseases, using familial PrDs as a model.
The role of PRNP mutations in selective vulnerability
PrP is initially translated as a 254 amino acid polypeptide that is translocated into the endoplasmic reticulum and passages through the Golgi apparatus during synthesis (Taraboulos et al., 1992). Eventually, it is retained with a glycosyl-phosphatidylinositol anchor in an extracellular lipid raft environment of the cell membrane (Stahl et al., 1987). The mature form is a 208 amino acid globular glycoprotein with an unstructured N-terminus (Riek et al., 1996; Riek et al., 1997), two glycan chains (Haraguchi et al., 1989) and a disulfide bridge (Turk et al., 1988). [See Riesner (Riesner, 2003) for an intelligently designed schematic.] Many roles have been proposed for PrP, including the maintenance of neural stem cells (Steele et al., 2006) and myelin sheath (Bremer et al., 2010), metal homeostasis (Brown et al., 1997; Kralovicova et al., 2009; Pushie et al., 2011), modulation of NMDA (N-methyl-D-aspartate) receptors (Khosravani et al., 2008), and protection from ischemia-induced degeneration (Weise et al., 2006). It was recently proposed to be a receptor for a toxic protein fragment in AD (Laurén et al., 2009; Resenberger et al., 2011), but this hypothesis remains controversial (Calella et al., 2010; Larson et al., 2012). All of these important aspects of prion biology could potentially drive the selective vulnerability of familial PrDs; however, at present there is no overriding theory.
A logical strategy to understand how PRNP mutations target specific brain regions is to search for underlying patterns that link individual mutations with features of the diseases they cause (Capellari et al., 2011). One possibility is that mutations destabilize PrP to various extents, and the more severely PrP is misfolded, the more rapidly the disease progresses and the more widely distributed the neuropathological lesions, assuming that there would be a higher proportion of cells that cannot tolerate the misfolded protein. However, there is strong evidence to refute this hypothesis (Capellari et al., 2011). A mutant that strongly destabilizes PrP (F198S, Table 1) results in a late-onset, rather slowly developing PrD, whereas mutants causing moderate (D178N) or no (E200K) destabilization cause earlier and faster-progressing diseases (Apetri et al., 2004; Capellari et al., 2011; Kovács et al., 2002). Mutations also occur in the unstructured N-terminal region, including substitutions of prolines for leucines and increases or decreases in the size of an octapeptide repeat (Piccardo et al., 1998). Interestingly, the clinical phenotypes associated with the N-terminal mutations include GSS, CJD and even an HD-like disease. Because these mutations should have no influence on the stability or three-dimensional structure, and yet they target different brain regions, factors other than destabilizing changes to PrP must be able to direct selective vulnerability.
Table 1.
Effects of mutations in PRNP on protein structure
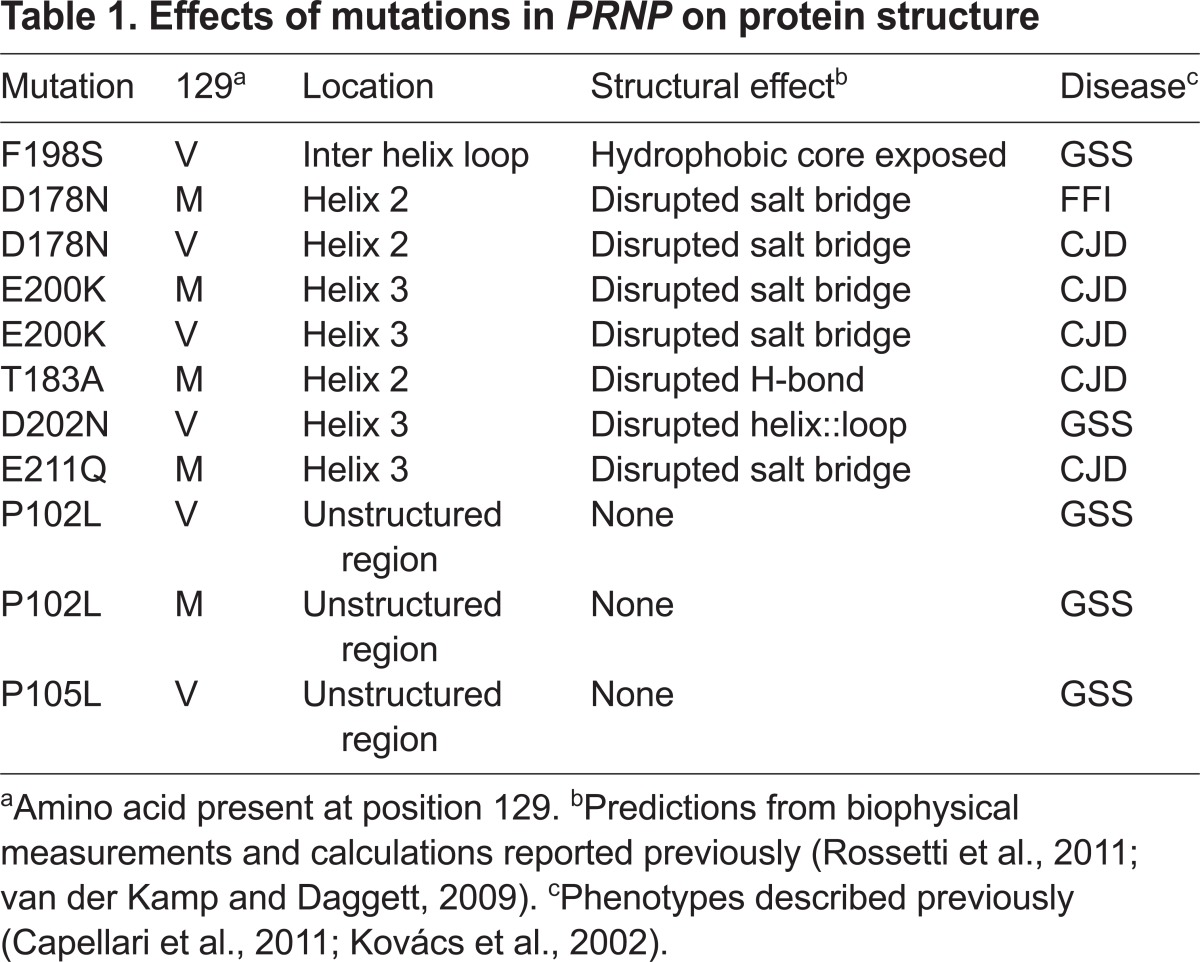
Changes in the overall protein charge could also drive selective vulnerability, by favoring aggregation pathways associated with a specific PrD subtype. Indeed, this would explain why over half of the mutations identified for PrDs involve changes in charge (Rossetti et al., 2011; Shen and Ji, 2011). However, mutants resulting in the same charge change can cause different diseases. For example, D178N causes FFI, D202N causes GSS and E211Q causes CJD, yet all of these mutations involve the substitution of a negatively charged amino acid with a polar uncharged amino acid (Capellari et al., 2011; Kovács et al., 2002). Moreover, mutations without charge changes (e.g. T183A and F198S) can also cause different diseases (CJD and GSS, respectively).
Interestingly, a very common polymorphism affecting amino acid position 129 in the PrP polypeptide chain, which does not itself cause disease, can modify the effects of disease-causing mutations. Presence of the D178N mutation together with a methionine at amino acid 129 (129M) causes FFI and targets the thalamus, but, when the same mutation is in cis with valine at amino acid 129 (129V), the thalamus is relatively unscathed and instead the cortex is targeted, resulting in CJD. In contrast, compared with the 129M variant, 129V in cis with the E200K mutation steers pathology towards the thalamus, but still causes CJD. The only trend between sequence changes and disease phenotype is that most cases of GSS include 129V on the mutant allele, but the presence of this polymorphism in some familial CJD cases diminishes the correlation. The fact that this polymorphism at amino acid 129 without a charge change partially determines which brain regions are targeted indicates that factors other than charge differences are driving selective vulnerability. Therefore, no clear pattern exists, such as extent of destabilization of the native structure, position in the linear sequence or charge change, which explains how each mutation causes a specific disease. It seems that different mutations lead PrP to form distinct misfolded conformers or aggregates, and each of these unique structures somehow targets specific regions.
Interest in studying the familial PrDs has led to the development of several mouse models. Many carry randomly integrated transgenes expressing the mutant protein, an approach that generally leads to expression levels that are much higher than endogenous levels (Dossena et al., 2008; Friedman-Levi et al., 2011; Hsiao et al., 1990; Yang et al., 2009). However, overexpression of wild-type PrP is toxic to mice (Chiesa et al., 2008; Watts et al., 2012; Westaway et al., 1994), making it difficult to distinguish the effects of the mutation from any confounding effects associated with overexpression in mouse models designed this way. Another problem is that the spatial expression patterns are highly dependent on the genomic integration site, vary from line to line and do not accurately mimic the endogenous expression pattern (Borchelt et al., 1996; Faas et al., 2010; Karapetyan et al., 2009).
In recent years we have been developing an allelic series of knock-in mice to model six of the most interesting familial PrDs. The knock-in approach introduces mutations to the endogenous gene at the site that it naturally exists in the genome, thereby facilitating endogenous levels of expression. Two lines, D178N-129M and E200K-129M, which model FFI or CJD, respectively, show selective vulnerability, with the former targeting the thalamus and the latter targeting the hippocampus (Jackson et al., 2009; Jackson et al., 2013). These two structures are in very close proximity in the brain: the hippocampus, which resembles a partial shell, virtually cups the thalamus, which is shaped roughly like a disc (Fig. 2). However, they have few neuroanatomical connections (Somogyi and Klausberger, 2005). How these two mutations, located just 60 base pairs away in the context of the 3-billion base pairs of mouse genome, encode such specific targeting in the brain is currently under intensive investigation in our group.
Fig. 2.

The thalamus and hippocampus are intertwined in the mouse brain. (A) A computer rendering of the mouse brain oriented so that, in an intact mouse, it would be facing slightly to the right of the observer. (B) The same model as in A but with the external regions made translucent so that the space occupied by the thalamus can be observed. (C) The same model as in B but with the hippocampus highlighted instead. (D) The hippocampus and thalamus shown together. These images were created on the Allen Brain Atlas website (www.brain-map.org).
Tau is a microtubule protein that has been implicated in the pathogenesis of neurodegenerative diseases, including PrDs. Like PrP, mutations affecting Tau are associated with clinical pleiotropy (Wolfe, 2009). An allelic series of Tau knock-in mice carrying mutations linked to familial human diseases with different neuropathological changes could also be an invaluable tool for neurodegenerative disease research.
Brain factors determining selective vulnerability
Because mutant proteins cause different problems in different brain regions, brain-region-specific factors must play a role in determining selective vulnerability. One possible factor is that intrinsic firing properties or metabolic activity of specific cell types might make specific regions more vulnerable to degeneration. For example, GABA neurons, mentioned earlier as being affected in CAG-repeat diseases and PrDs, are inherently highly active cells. The constant firing is energetically expensive because of the frequent need to reestablish ion gradients. The resulting high metabolic rate could prime these cells for damage due to the high production of free radicals. The overworked cellular components would become prone to damage, requiring more degradation and re-synthesis of cellular components and further increased metabolic demand, setting a vicious cycle into motion. As the brain ages, it would likely struggle to cope with the heavy burden of degrading damaged cellular components and making repairs, particularly in the presence of a misfolded protein. A similar argument was made for the subset of dopaminergic neurons that have an intrinsically high firing rate and are selectively targeted in PD (Chan et al., 2007). Data from mouse models, however, suggest that there is more to the story. Knock-in mutations in mice modeling HD, Sca1 and FFI primarily affect the striatum, cerebellum and thalamus, respectively (Jackson et al., 2009; Lin et al., 2001; Watase et al., 2002), just as in humans, suggesting that the mouse lines model the corresponding diseases relatively well. However, the mouse thalamus has a far lower proportion of GABAergic neurons compared with the human thalamus (Arcelli et al., 1997). This leads to an alternative hypothesis, at least for FFI, that their location in the brain, rather than simply their neurochemical or intrinsic firing properties, can influence a neuron’s vulnerability. In addition, non-neuronal cells (e.g. glial and vascular cells) are also involved in the disease process and could cause a mixture of positive and negative effects, both of which would influence selective vulnerability. There are many unanswered questions in this regard, highlighting the need for further research in this area.
Metal ions might also play an important role in making certain brain regions more vulnerable to neurodegenerative diseases. Maintenance of the appropriate stoichiometry of metals and their biomolecular partners is critical because metals are required for numerous synthesis, degradation and defensive processes, and can be toxic in free forms. Like the non-uniform distribution of neuron types, metals also have variable concentrations across brain regions and, interestingly, the levels of three common metal ions, Cu2+, Fe2+ and Zn2+, seem to be regulated by PrP in normal brains (Brown et al., 1997; Kralovicova et al., 2009; Pushie et al., 2011; Watt et al., 2012). Moreover, Cu2+ and Mn2+ levels change in PrDs (Hesketh et al., 2008; Thackray et al., 2002). In addition to PrP-mediated modification of metal concentrations, the reciprocal is also true, i.e. metals can modify the activities of PrP. For example, the inhibitory effect of PrP on NMDA-receptor activity occurs via a Cu2+-dependent mechanism (You et al., 2012). Interestingly, changes in metal levels change PrP levels (Kralovicova et al., 2009) as well as redirecting its trafficking through the secretory pathway (Brown and Harris, 2003). In support of a role for metal ions in modulating PrP function in the context of disease, excess Cu2+ aggravates disease in a mouse model of familial CJD (Canello et al., 2012). Furthermore, Zn2+ interacts with PrP to varying degrees depending on the mutation present, with weaker interactions being associated with mutations conferring charge changes (Spevacek et al., 2013). Therefore, differences in metal concentrations are likely to modify regional sensitivity to different PrP variants, but understanding how and to what extent this happens awaits further research.
Another factor potentially controlling selective vulnerability is that ubiquitous molecular machines that reduce or restrict toxicity, for example components of the protein-folding and quality-control systems, might vary between brain regions. Indeed, different strains of mouse prions causing different morphologies of aggregates and targeting different brain regions activate different combinations of chaperones (Asuni et al., 2013). Moreover, in the context of cell culture experiments, where a small number of cell lines is typically employed, PrP mutants associated with different diseases can have similar trafficking abnormalities, suggesting that trafficking defects cannot contribute to selective vulnerability (Ashok and Hegde, 2009; Capellari et al., 2000a; Capellari et al., 2000b; Ivanova et al., 2001; Petersen et al., 1996; Zou et al., 2011). However, the massive diversity of cell types in the brain provides an exponentially greater number of microenvironments for mutant PrPs to interact with. Thus, if the protein quality-control system has distinct combinations of components expressed in specific cell types, then some cells might be less capable of dealing with a specific misfolded conformer than others, thereby being more vulnerable to disease. Inconsistent with this hypothesis, mRNAs encoding protein chaperones are expressed at approximately equal levels across the adult mouse brain (Tebbenkamp and Borchelt, 2010), suggesting that there is little cell-type specificity for this critical system, and all cells should be equally capable of detoxifying a misfolded protein. However, the relative expression levels of ribosomal protein mRNAs vary across the body of the developing mouse, suggesting that levels of translation could vary (Kondrashov et al., 2011). Examination of ribosomal protein mRNAs in the adult mouse brain using data from the Allen Brain Atlas (www.brain-map.org) corroborates this, revealing a startling variability in expression between regions (Fig. 3). Some ribosomal protein mRNAs are approximately uniformly distributed across the brain (Fig. 3, Rpl17 and Rplpo), whereas others are highly variable (Fig. 3, Rps3 and RPS15). Moreover, of the 62 ribosomal genes examined, at least seven are apparently not expressed in the adult mouse brain (Rpl12, Rpl27, Rpl37, Rpl41, Rps13, Rps17, Rps20). The non-stoichiometric distribution of ribosomal protein mRNAs suggests that there is a large diversity of ribosomal configurations, distributed across specific cell types, which could impact the regulation of translation. Therefore, the chaperone repertoire of specific cell types in healthy brains is likely to be much more specialized than is reflected by Tebbenkamp and Borchelt’s mRNA localization study, and the same is probably true for components of other processes, including degradation and synthesis. Because the stressful conditions of acquired PrD cause protein translation to be sharply altered (Moreno et al., 2012), the molecular composition of each cell is likely to be drastically reshaped during disease.
Fig. 3.

A surprising distribution of ribosomal protein mRNAs in the brain. Small and large ribosomal subunit proteins are encoded by Rps and Rpl mRNAs, respectively. A small subunit binds to mRNAs first and recruits the large subunit for translation. A variable expression pattern of components of both subunits in the mouse brain suggests the existence of extra layers of gene regulation that were previously unrecognized. Data were acquired from the Allen Brain Atlas website (www.brain-map.org). The data were systematically normalized and therefore comparable to the mRNA distributions in Fig. 1. Abbreviations: OLF, olfactory bulb; HPF, hippocampus; CTXsp, subregion of the cortex (bottom of the brain); STR, striatum; PAL, pallidum; CB, cerebellum; TH, thalamus; HY, hypothalamus; MB, midbrain; P, pons; MY, medulla; CTX, cortex (main region on top of the brain).
A better understanding of how the multitude of various components of cells (e.g. protein quality-control machinery) are distributed across the brain, in the presence and absence of disease, is required to fully understand how these factors make certain regions most vulnerable.
Hitting a moving target
In recent years, there has been a reawakening of interest into the mechanism of spreading of neurodegenerative disease from one region to another. This concept was initially proposed following a systematic study of brains of humans who died after having various clinical stages of AD (Braak and Braak, 1991). Many investigators currently hypothesize that spreading involves the movement of misfolded protein conformers, possibly as lower-order oligomers or small bits of mature aggregates (Fig. 4A, ‘Moving aggregates’). Important events that led to the recent revitalized interest in neurodegenerative disease spreading were the observation of PD neuropathology in human transplanted tissue lying adjacent to endogenous, diseased tissue (Li et al., 2008) and the experimental transmission of AD-related aggregates in mouse models (Meyer-Luehmann et al., 2006; Meyer-Luehmann et al., 2003). Combined with reports of the spreading of additional neurodegenerative-disease-related proteins (Bolmont et al., 2007; Grad et al., 2011; Harris et al., 2010; Kane et al., 2000; Luk et al., 2012), these findings have resulted in an exponential growth of interest in this topic and have fuelled discussions on whether proteins other than PrP can spread via a prion-like mechanism (Brundin et al., 2010; Cushman et al., 2010; Frost and Diamond, 2010; Guest et al., 2011; Jucker and Walker, 2011). In light of the growing number of ‘infectious’ proteins identified, the term ‘prionoid’ was coined to distinguish protein aggregates that are likely to not be naturally transmissible between individuals from those that are (prions) (Aguzzi and Rajendran, 2009). Proposed mechanisms for the spread of protein aggregates include cellular release and free diffusion through the extracellular space, transport via secreted vesicles known as exosomes (Danzer et al., 2012; Emmanouilidou et al., 2010; Lee et al., 2012; Saman et al., 2012) or movement through intercellular tunnels known as nano-tubes (Costanzo et al., 2013; Marzo et al., 2012). Mammalian prions, considered to be ‘super spreaders’ because of their unique ability to spread between individuals, have been posited to transit the brain via each of the above mechanisms (Alais et al., 2008; Gousset et al., 2009; Gousset and Zurzolo, 2009; Porto-Carreiro et al., 2005; Vella et al., 2007). It has recently been shown that cytosolic proteins can, in addition to extracellular proteins such as PrP, spread to neighboring cells via a prion-like mechanism (Hofmann et al., 2013).
Fig. 4.

Models of protein aggregate spread. Three models of how the localization of aggregates in the brain might change as neurodegenerative disease progresses. (A) Moving aggregates. Aggregates generated in one region physically move to another, covering a large distance. (B) Serial dysfunction. Due to network dysfunction, conditions inducing de novo synthesis of protein aggregates move across the brain. (C) Falling dominoes. Similar to serial dysfunction, where conditions permissive to aggregate formation move, but with the distinction that misfolded protein aggregates seed the formation of new aggregates in close proximity, creating a chain reaction. The lightning bolts represent induction of conditions in different brain regions that accommodate the appearance of protein aggregates. Conditions might include aberrant network activity or impairment of protein quality-control machinery. The arrows in A show that an aggregate formed in the left end of the brain moves towards the right end. The dots in C represent small oligomeric misfolded protein seeds, which spread and can form full size aggregates once permissive conditions are present (lightning bolt) but not in areas without permissive conditions (above and below the aggregates).
A caveat of the hypothesis of disease spread being caused by the movement of aggregates is that the role of aggregates in disease remains quite contentious. It seems that some aggregates are toxic, some are benign and some might even be helpful (Kayed et al., 2003; Piccardo et al., 2007; Resenberger et al., 2011). Another problem is that, if aggregates are toxic, why is this the case? Is the mere presence of a mature aggregate toxic, or is toxicity a byproduct of the process of generating the aggregate? Do identical aggregates cause different problems in different brain regions? These are all outstanding questions that need to be resolved. Nonetheless, the presence of aggregates indicates that a non-physiological process is active in that area and a discussion of neurodegenerative disease spreading, and the corresponding aggregate pathology, is warranted even without a complete understanding of the mechanism(s) of toxicity.
Despite being well poised to flow through the brain, PrDs display somewhat restricted spreading rather than affecting the entire brain to give rise to what would be classified as a single disease. Instead, specific patterns of neural targeting are maintained. The spreading potential is partially controlled by the incorporated mutation, shown by the finding that some mutants are experimentally transmissible [for example, D178N and E200K (Tateishi et al., 1995; Tateishi and Kitamoto, 1995)], whereas others seem not to be [for example, A117V and F198S (Bugiani et al., 2000; Tateishi et al., 1996)]. Intracellular factors might also restrict spreading, such as restriction of PrP synthesis, interactions of PrP with intracellular molecules, modifications to PrP such as glycosylation or cleavage (Goh et al., 2007; Haigh et al., 2009; Kuczius and Kelsch, 2013; Moleres and Velayos, 2007), reuptake of misfolded PrP from the cell surface, or restricted production of exosomes or nano-tubes. Extracellular factors that might restrict spreading include physical barriers created by the dense mass of various cell types or perineuronal nets, extracellular chaperones or proteases, or engulfment by native phagocytic cells (micro- or astroglia). A crucial element required for significant spreading might be a breakdown of the barriers that restrict it. Therefore, if protein aggregates do indeed move through the brain, there are many variables that could influence the route and thereby the final pathological changes that accompany this movement.
An alternative explanation is that disease is caused by neurons that are adversely affected by receiving improper signaling from an earlier affected and misfiring neuron in its network (Sperling et al., 2009; Verret et al., 2012). The sum result would be the spreading of pathology. This mode of spreading is distinct from that described above in that a misfolded protein conformer, and its associated toxicity, is not physically moving. Importantly, however, in this scenario aggregates would still appear to have moved because the distribution of neurons succumbing to the stressful effects of dysfunctioning network members would expand, eventually causing degeneration and the production of aggregates (Fig. 4B, ‘Serial dysfunction’). In the case of familial neurodegenerative disease (and PrD) the mutant protein would be expressed nearly everywhere, making local de novo production of aggregates even more feasible because it is prone to misfolding. A fusion of these disparate hypotheses is that misfolded protein oligomers induce the conversion of neighboring proteins to misfold and form more oligomers, causing a chain reaction resembling the way a line of standing dominoes appears to move as they fall. The oligomers themselves are difficult to detect and highly transient, but they would seed the formation of aggregates and thereby cause the appearance of the spreading of aggregates. The spread would be most successful in stressed regions that are not able to degrade the oligomers (Fig. 4C, ‘Falling dominoes’). These three models have subtle but important distinctions. The ‘moving aggregate’ model posits that aggregates form in a diseased area and later move large distances into unaffected areas, inducing degenerative processes upon arrival. In the ‘serial dysfunction’ model, the aggregates never physically move but form de novo once an area becomes susceptible. The ‘falling dominoes’ model also requires that an area is first primed by network-dysfunction-induced stress, then oligomers act as scaffolds to seed the formation of neighboring aggregates; nonetheless, the proteins incorporated into the aggregates remain essentially in their original location. The previously cited PD-related aggregates observed in transplanted tissue (Li et al., 2008) could have developed through any of these mechanisms. Of course, additional models can be easily imagined. All of these models bring to light the following questions: how do neuronal and non-neuronal (glial, vascular, etc.) cells limit or facilitate spread? And do different misfolded proteins spread via a common mechanism or does each have its own unique strategy? PrP is emblematic of this latter question because different mutations induce different structures of aggregates in addition to targeting different regions. Therapeutic strategies designed to interfere with the toxic events related to protein misfolding will likely benefit from an understanding of how pathology spreads.
Conclusions and future outlook
Understanding how mutant proteins target specific brain regions is clearly not a simple issue. Ultimately, the mutations carry the disease-encoding information that determines the protein conformation, but it is the interaction of mutant proteins with the combination of cell-type-specific microenvironments and brain-region-specific neural features that ultimately determines selective vulnerability. The spreading of pathology is likely controlled by a combination of opposing factors that facilitate or restrict spread. The Prnp knock-in lines modeling FFI and CJD mentioned above (Jackson et al., 2009; Jackson et al., 2013) will likely be a valuable tool for this issue. Each line develops pathologies in specific regions that spread into neighboring regions but not into the region primarily targeted by the opposite mutation. The separation of pathologies in the two mouse models is quite remarkable given PrP’s inherent ability to spread, and further emphasizes the point that the spreading phenomenon is likely to be more complicated than currently envisioned. An understanding of the factors that impede or facilitate spreading will make important contributions to the development of therapeutics, particularly those targeting extracellular molecules that might be more easily engaged than intracellular molecules.
These rare diseases provide a plethora of clues about the tools our brains employ to limit degenerative processes. It is up to us to decipher their meanings. As the fellow with the original Curious Case – Benjamin Button – noted, “Our lives are defined by opportunities, even the ones we miss”. Is the secret to selective vulnerability and a cure for neurodegenerative diseases right in front of us?
Acknowledgments
I am grateful to Drs Ina Vorberg and Gabor Petzold for helpful discussions.
Footnotes
This article is part of a review series on protein-folding diseases. See related articles at http://dmm.biologists.org/site/protein-folding-disease.xhtml.
Funding
This work was funded by the German Center for Neurodegenerative Diseases in the Helmholtz Gemeinschaft.
Competing interests
The author declares no competing financial interests.
References
- Aguzzi A., Rajendran L. (2009). The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790 [DOI] [PubMed] [Google Scholar]
- Aguzzi A., Barres B. A., Bennett M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aidt F. H., Hasholt L. F., Christiansen M., Laursen H. (2013). Localization of A11-reactive oligomeric species in prion diseases. Histopathology 62, 994–1001 [DOI] [PubMed] [Google Scholar]
- Alais S., Simoes S., Baas D., Lehmann S., Raposo G., Darlix J. L., Leblanc P. (2008). Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 100, 603–618 [DOI] [PubMed] [Google Scholar]
- Apetri A. C., Surewicz K., Surewicz W. K. (2004). The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 279, 18008–18014 [DOI] [PubMed] [Google Scholar]
- Arcelli P., Frassoni C., Regondi M. C., De Biasi S., Spreafico R. (1997). GABAergic neurons in mammalian thalamus: a marker of thalamic complexity? Brain Res. Bull. 42, 27–37 [DOI] [PubMed] [Google Scholar]
- Ashok A., Hegde R. S. (2009). Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 5, e1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni A. A., Pankiewicz J. E., Sadowski M. J. (2013). Differential molecular chaperone response associated with various mouse adapted scrapie strains. Neurosci. Lett. 538, 26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T., Clavaguera F., Meyer-Luehmann M., Herzig M. C., Radde R., Staufenbiel M., Lewis J., Hutton M., Tolnay M., Jucker M. (2007). Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am. J. Pathol. 171, 2012–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt D. R., Davis J., Fischer M., Lee M. K., Slunt H. H., Ratovitsky T., Regard J., Copeland N. G., Jenkins N. A., Sisodia S. S., et al. (1996). A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 13, 159–163 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A. D., Toyka K. V., Nave K. A., Weis J., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318 [DOI] [PubMed] [Google Scholar]
- Brown L. R., Harris D. A. (2003). Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J. Neurochem. 87, 353–363 [DOI] [PubMed] [Google Scholar]
- Brown D. R., Qin K., Herms J. W., Madlung A., Manson J., Strome R., Fraser P. E., Kruck T., von Bohlen A., Schulz-Schaeffer W., et al. (1997). The cellular prion protein binds copper in vivo. Nature 390, 684–687 [DOI] [PubMed] [Google Scholar]
- Brundin P., Melki R., Kopito R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani O., Giaccone G., Piccardo P., Morbin M., Tagliavini F., Ghetti B. (2000). Neuropathology of Gerstmann-Sträussler-Scheinker disease. Microsc. Res. Tech. 50, 10–15 [DOI] [PubMed] [Google Scholar]
- Calella A. M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I. M., Aguzzi A. (2010). Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canello T., Friedman-Levi Y., Mizrahi M., Binyamin O., Cohen E., Frid K., Gabizon R. (2012). Copper is toxic to PrP-ablated mice and exacerbates disease in a mouse model of E200K genetic prion disease. Neurobiol. Dis. 45, 1010–1017 [DOI] [PubMed] [Google Scholar]
- Capellari S., Parchi P., Russo C. M., Sanford J., Sy M. S., Gambetti P., Petersen R. B. (2000a). Effect of the E200K mutation on prion protein metabolism. Comparative study of a cell model and human brain. Am. J. Pathol. 157, 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellari S., Zaidi S. I., Long A. C., Kwon E. E., Petersen R. B. (2000b). The Thr183Ala Mutation, not the loss of the first glycosylation site, alters the physical properties of the prion protein. J. Alzheimers Dis. 2, 27–35 [DOI] [PubMed] [Google Scholar]
- Capellari S., Strammiello R., Saverioni D., Kretzschmar H., Parchi P. (2011). Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 121, 21–37 [DOI] [PubMed] [Google Scholar]
- Chan C. S., Guzman J. N., Ilijic E., Mercer J. N., Rick C., Tkatch T., Meredith G. E., Surmeier D. J. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086 [DOI] [PubMed] [Google Scholar]
- Chiesa R., Piccardo P., Biasini E., Ghetti B., Harris D. A. (2008). Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J. Neurosci. 28, 13258–13267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Abounit S., Marzo L., Danckaert A., Chamoun Z., Roux P., Zurzolo C. (2013). Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 126, 3678–3685 [DOI] [PubMed] [Google Scholar]
- Cushman M., Johnson B. S., King O. D., Gitler A. D., Shorter J. (2010). Prion-like disorders: blurring the divide between transmissibility and infectivity. J. Cell Sci. 123, 1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer K. M., Kranich L. R., Ruf W. P., Cagsal-Getkin O., Winslow A. R., Zhu L., Vanderburg C. R., McLean P. J. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossena S., Imeri L., Mangieri M., Garofoli A., Ferrari L., Senatore A., Restelli E., Balducci C., Fiordaliso F., Salio M., et al. (2008). Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 60, 598–609 [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S. D., Ntzouni M., Margaritis L. H., Stefanis L., Vekrellis K. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faas H., Jackson W. S., Borkowski A. W., Wang X., Ma J., Lindquist S., Jasanoff A. (2010). Context-dependent perturbation of neural systems in transgenic mice expressing a cytosolic prion protein. Neuroimage 49, 2607–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis P. T., Palmer A. M., Snape M., Wilcock G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry 66, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman-Levi Y., Meiner Z., Canello T., Frid K., Kovacs G. G., Budka H., Avrahami D., Gabizon R. (2011). Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease. PLoS Pathog. 7, e1002350. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Frost B., Diamond M. I. (2010). Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P., Parchi P., Petersen R. B., Chen S. G., Lugaresi E. (1995). Fatal familial insomnia and familial Creutzfeldt-Jakob disease: clinical, pathological and molecular features. Brain Pathol. 5, 43–51 [DOI] [PubMed] [Google Scholar]
- Gambetti P., Kong Q., Zou W., Parchi P., Chen S. G. (2003). Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66, 213–239 [DOI] [PubMed] [Google Scholar]
- Ghetti B., Tagliavini F., Giaccone G., Bugiani O., Frangione B., Farlow M. R., Dlouhy S. R. (1994). Familial Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Mol. Neurobiol. 8, 41–48 [DOI] [PubMed] [Google Scholar]
- Glabe C. G. (2006). Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 [DOI] [PubMed] [Google Scholar]
- Goh A. X., Li C., Sy M. S., Wong B. S. (2007). Altered prion protein glycosylation in the aging mouse brain. J. Neurochem. 100, 841–854 [DOI] [PubMed] [Google Scholar]
- Gousset K., Zurzolo C. (2009). Tunnelling nanotubes: a highway for prion spreading? Prion 3, 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gousset K., Schiff E., Langevin C., Marijanovic Z., Caputo A., Browman D. T., Chenouard N., de Chaumont F., Martino A., Enninga J., et al. (2009). Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336 [DOI] [PubMed] [Google Scholar]
- Grad L. I., Guest W. C., Yanai A., Pokrishevsky E., O’Neill M. A., Gibbs E., Semenchenko V., Yousefi M., Wishart D. S., Plotkin S. S., et al. (2011). Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 108, 16398–16403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveland G. A., Williams R. S., DiFiglia M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773 [DOI] [PubMed] [Google Scholar]
- Guest W. C., Silverman J. M., Pokrishevsky E., O’Neill M. A., Grad L. I., Cashman N. R. (2011). Generalization of the prion hypothesis to other neurodegenerative diseases: an imperfect fit. J. Toxicol. Environ. Health A 74, 1433–1459 [DOI] [PubMed] [Google Scholar]
- Haigh C. L., Lewis V. A., Vella L. J., Masters C. L., Hill A. F., Lawson V. A., Collins S. J. (2009). PrPC-related signal transduction is influenced by copper, membrane integrity and the alpha cleavage site. Cell Res. 19, 1062–1078 [DOI] [PubMed] [Google Scholar]
- Haraguchi T., Fisher S., Olofsson S., Endo T., Groth D., Tarentino A., Borchelt D. R., Teplow D., Hood L., Burlingame A., et al. (1989). Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 274, 1–13 [DOI] [PubMed] [Google Scholar]
- Harris J. A., Devidze N., Verret L., Ho K., Halabisky B., Thwin M. T., Kim D., Hamto P., Lo I., Yu G. Q., et al. (2010). Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 68, 428–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh S., Sassoon J., Knight R., Brown D. R. (2008). Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell. Neurosci. 37, 590–598 [DOI] [PubMed] [Google Scholar]
- Hofmann J. P., Denner P., Nussbaum-Krammer C., Kuhn P. H., Suhre M. H., Scheibel T., Lichtenthaler S. F., Schätzl H. M., Bano D., Vorberg I. M. (2013). Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc. Natl. Acad. Sci. USA 110, 5951–5956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K. K., Scott M., Foster D., Groth D. F., DeArmond S. J., Prusiner S. B. (1990). Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 250, 1587–1590 [DOI] [PubMed] [Google Scholar]
- Hyman B. T., Van Hoesen G. W., Damasio A. R., Barnes C. L. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170 [DOI] [PubMed] [Google Scholar]
- Ivanova L., Barmada S., Kummer T., Harris D. A. (2001). Mutant prion proteins are partially retained in the endoplasmic reticulum. J. Biol. Chem. 276, 42409–42421 [DOI] [PubMed] [Google Scholar]
- Jackson W. S., Borkowski A. W., Faas H., Steele A. D., King O. D., Watson N., Jasanoff A., Lindquist S. (2009). Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63, 438–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson W. S., Borkowski A. W., Watson N. E., King O. D., Faas H., Jasanoff A., Lindquist S. (2013). Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proc. Natl. Acad. Sci. USA 110, 14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M., Walker L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M. D., Lipinski W. J., Callahan M. J., Bian F., Durham R. A., Schwarz R. D., Roher A. E., Walker L. C. (2000). Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J. Neurosci. 20, 3606–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetyan Y. E., Saá P., Mahal S. P., Sferrazza G. F., Sherman A., Salès N., Weissmann C., Lasmézas C. I. (2009). Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS ONE 4, e5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- Khosravani H., Zhang Y., Tsutsui S., Hameed S., Altier C., Hamid J., Chen L., Villemaire M., Ali Z., Jirik F. R., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov N., Pusic A., Stumpf C. R., Shimizu K., Hsieh A. C., Xue S., Ishijima J., Shiroishi T., Barna M. (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács G. G., Trabattoni G., Hainfellner J. A., Ironside J. W., Knight R. S., Budka H. (2002). Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 249, 1567–1582 [DOI] [PubMed] [Google Scholar]
- Kralovicova S., Fontaine S. N., Alderton A., Alderman J., Ragnarsdottir K. V., Collins S. J., Brown D. R. (2009). The effects of prion protein expression on metal metabolism. Mol. Cell. Neurosci. 41, 135–147 [DOI] [PubMed] [Google Scholar]
- Kuczius T., Kelsch R. (2013). The effect of copper and zinc binding on the solubility and resistance to proteolysis of physiological prion protein PrP depends on the tissue source and the PrP glycotypes. J. Cell. Biochem. 114, 2690–2698 [DOI] [PubMed] [Google Scholar]
- Larson M., Sherman M. A., Amar F., Nuvolone M., Schneider J. A., Bennett D. A., Aguzzi A., Lesné S. E. (2012). The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Kim W., Li Z., Hall G. F. (2012). Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int. J. Alzheimers Dis. 2012, 172837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Y., Englund E., Holton J. L., Soulet D., Hagell P., Lees A. J., Lashley T., Quinn N. P., Rehncrona S., Björklund A., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 [DOI] [PubMed] [Google Scholar]
- Lin C. H., Tallaksen-Greene S., Chien W. M., Cearley J. A., Jackson W. S., Crouse A. B., Ren S., Li X. J., Albin R. L., Detloff P. J. (2001). Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 10, 137–144 [DOI] [PubMed] [Google Scholar]
- Luk K. C., Kehm V. M., Zhang B., O’Brien P., Trojanowski J. Q., Lee V. M. (2012). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo L., Gousset K., Zurzolo C. (2012). Multifaceted roles of tunneling nanotubes in intercellular communication. Front Physiol 3, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Stalder M., Herzig M. C., Kaeser S. A., Kohler E., Pfeifer M., Boncristiano S., Mathews P. M., Mercken M., Abramowski D., et al. (2003). Extracellular amyloid formation and associated pathology in neural grafts. Nat. Neurosci. 6, 370–377 [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A. L., et al. (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 [DOI] [PubMed] [Google Scholar]
- Moleres F. J., Velayos J. L. (2007). The neurochemical nature of PrP(c)-containing cells in the rat brain. Brain Res. 1174, 143–151 [DOI] [PubMed] [Google Scholar]
- Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., Martin M. G., Halliday M., Morgan J., Dinsdale D., Ortori C. A., et al. (2012). Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen R. B., Parchi P., Richardson S. L., Urig C. B., Gambetti P. (1996). Effect of the D178N mutation and the codon 129 polymorphism on the metabolism of the prion protein. J. Biol. Chem. 271, 12661–12668 [DOI] [PubMed] [Google Scholar]
- Piccardo P., Dlouhy S. R., Lievens P. M., Young K., Bird T. D., Nochlin D., Dickson D. W., Vinters H. V., Zimmerman T. R., Mackenzie I. R., et al. (1998). Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J. Neuropathol. Exp. Neurol. 57, 979–988 [DOI] [PubMed] [Google Scholar]
- Piccardo P., Manson J. C., King D., Ghetti B., Barron R. M. (2007). Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc. Natl. Acad. Sci. USA 104, 4712–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto-Carreiro I., Février B., Paquet S., Vilette D., Raposo G. (2005). Prions and exosomes: from PrPc trafficking to PrPsc propagation. Blood Cells Mol. Dis. 35, 143–148 [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. (1998). Prions. Proc. Natl. Acad. Sci. USA 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushie M. J., Pickering I. J., Martin G. R., Tsutsui S., Jirik F. R., George G. N. (2011). Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 3, 206–214 [DOI] [PubMed] [Google Scholar]
- Resenberger U. K., Harmeier A., Woerner A. C., Goodman J. L., Müller V., Krishnan R., Vabulas R. M., Kretzschmar H. A., Lindquist S., Hartl F. U., et al. (2011). The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wüthrich K. (1996). NMR structure of the mouse prion protein domain PrP (121–231). Nature 382, 180–182 [DOI] [PubMed] [Google Scholar]
- Riek R., Hornemann S., Wider G., Glockshuber R., Wüthrich K. (1997). NMR characterization of the full-length recombinant murine prion protein, mPrP (23–231). FEBS Lett. 413, 282–288 [DOI] [PubMed] [Google Scholar]
- Riesner D. (2003). Biochemistry and structure of PrP(C) and PrP(Sc). Br. Med. Bull. 66, 21–33 [DOI] [PubMed] [Google Scholar]
- Rossetti G., Cong X., Caliandro R., Legname G., Carloni P. (2011). Common structural traits across pathogenic mutants of the human prion protein and their implications for familial prion diseases. J. Mol. Biol. 411, 700–712 [DOI] [PubMed] [Google Scholar]
- Saman S., Kim W., Raya M., Visnick Y., Miro S., Saman S., Jackson B., McKee A. C., Alvarez V. E., Lee N. C., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Ji H. F. (2011). Mutation directional selection sheds light on prion pathogenesis. Biochem. Biophys. Res. Commun. 410, 159–163 [DOI] [PubMed] [Google Scholar]
- Sofroniew M. V., Vinters H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P., Klausberger T. (2005). Defined types of cortical interneurone structure space and spike timing in the hippocampus. J. Physiol. 562, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Laviolette P. S., O’Keefe K., O’Brien J., Rentz D. M., Pihlajamaki M., Marshall G., Hyman B. T., Selkoe D. J., Hedden T., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spevacek A. R., Evans E. G., Miller J. L., Meyer H. C., Pelton J. G., Millhauser G. L. (2013). Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 21, 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N., Borchelt D. R., Hsiao K., Prusiner S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240 [DOI] [PubMed] [Google Scholar]
- Steele A. D., Emsley J. G., Ozdinler P. H., Lindquist S., Macklis J. D. (2006). Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 103, 3416–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D., Surmeier D. J. (2013). Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 28, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A., Raeber A. J., Borchelt D. R., Serban D., Prusiner S. B. (1992). Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 3, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi J., Kitamoto T. (1995). Inherited prion diseases and transmission to rodents. Brain Pathol. 5, 53–59 [DOI] [PubMed] [Google Scholar]
- Tateishi J., Brown P., Kitamoto T., Hoque Z. M., Roos R., Wollman R., Cervenáková L., Gajdusek D. C. (1995). First experimental transmission of fatal familial insomnia. Nature 376, 434–435 [DOI] [PubMed] [Google Scholar]
- Tateishi J., Kitamoto T., Hoque M. Z., Furukawa H. (1996). Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 46, 532–537 [DOI] [PubMed] [Google Scholar]
- Tebbenkamp A. T., Borchelt D. R. (2010). Analysis of chaperone mRNA expression in the adult mouse brain by meta analysis of the Allen Brain Atlas. PLoS ONE 5, e13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray A. M., Knight R., Haswell S. J., Bujdoso R., Brown D. R. (2002). Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J. 362, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk E., Teplow D. B., Hood L. E., Prusiner S. B. (1988). Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176, 21–30 [DOI] [PubMed] [Google Scholar]
- van der Kamp M. W., Daggett V. (2009). The consequences of pathogenic mutations to the human prion protein. Protein Eng. Des. Sel. 22, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella L. J., Sharples R. A., Lawson V. A., Masters C. L., Cappai R., Hill A. F. (2007). Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590 [DOI] [PubMed] [Google Scholar]
- Verret L., Mann E. O., Hang G. B., Barth A. M., Cobos I., Ho K., Devidze N., Masliah E., Kreitzer A. C., Mody I., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K., Weeber E. J., Xu B., Antalffy B., Yuva-Paylor L., Hashimoto K., Kano M., Atkinson R., Sun Y., Armstrong D. L., et al. (2002). A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34, 905–919 [DOI] [PubMed] [Google Scholar]
- Watt N. T., Taylor D. R., Kerrigan T. L., Griffiths H. H., Rushworth J. V., Whitehouse I. J., Hooper N. M. (2012). Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun 3, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J. C., Giles K., Stöhr J., Oehler A., Bhardwaj S., Grillo S. K., Patel S., DeArmond S. J., Prusiner S. B. (2012). Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc. Natl. Acad. Sci. USA 109, 3498–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise J., Sandau R., Schwarting S., Crome O., Wrede A., Schulz-Schaeffer W., Zerr I., Bähr M. (2006). Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 37, 1296–1300 [DOI] [PubMed] [Google Scholar]
- Westaway D., DeArmond S. J., Cayetano-Canlas J., Groth D., Foster D., Yang S. L., Torchia M., Carlson G. A., Prusiner S. B. (1994). Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 76, 117–129 [DOI] [PubMed] [Google Scholar]
- Wolfe M. S. (2009). Tau mutations in neurodegenerative diseases. J. Biol. Chem. 284, 6021–6025 [DOI] [PubMed] [Google Scholar]
- Yang W., Cook J., Rassbach B., Lemus A., DeArmond S. J., Mastrianni J. A. (2009). A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J. Neurosci. 29, 10072–10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H., Tsutsui S., Hameed S., Kannanayakal T. J., Chen L., Xia P., Engbers J. D., Lipton S. A., Stys P. K., Zamponi G. W. (2012). Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 109, 1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi H. Y. (2000). Spinocerebellar ataxias. Neurobiol. Dis. 7, 523–527 [DOI] [PubMed] [Google Scholar]
- Zou R. S., Fujioka H., Guo J. P., Xiao X., Shimoji M., Kong C., Chen C., Tasnadi M., Voma C., Yuan J., et al. (2011). Characterization of spontaneously generated prion-like conformers in cultured cells. Aging (Albany, NY Online) 3, 968–984 [DOI] [PMC free article] [PubMed] [Google Scholar]