

Abstract
Dopaminergic hyperactivity within frontostriatal brain systems is a key feature of schizophrenia, and an objective neural correlate of positive schizophrenia symptoms. N-methyl-D-aspartate (NMDA) receptors are known to play a prominent role in regulation of frontostriatal dopamine release. Furthermore, disturbances in glutamatergic function are increasingly being linked to pathophysiology of both positive and negative symptoms of schizophrenia. Prior studies have demonstrated that subchronic continuous administration of the NMDA antagonist phencyclidine (PCP) induces schizophrenia-like hyper-reactivity of frontostriatal dopamine release to amphetamine (AMPH) in rodents, and that effects were reversed by glycine and the prototypic glycine transport inhibitor (GTI) NFPS. The present study investigates effectiveness of the novel, high affinity and well tolerated GTIs, R231857, R231860 and Org29335, to reverse schizophrenia-like enhancement of AMPH-induced DA release, along with effects of the partial glycine-site agonist D-cycloserine. As previously, PCP had no significant effect on basal DA levels, but significantly enhanced AMPH-induced DA release in prefrontal cortex. All GTIs tested, as well as D-cycloserine, significantly reduced PCP-induced enhancement of DA release in prefrontal cortex. Neither PCP nor GTIs significantly affected striatal DA release. Overall, these findings suggest that treatments which target the glycine modulatory site of the NMDA receptor may significantly reverse NMDA receptor antagonist-induced dysregulation of frontal DA systems, consistent with potential beneficial effects on positive-, in addition to negative-, symptoms of schizophrenia.
Keywords: NMDA, Dopamine, Glycine Transport Inhibitor, Prefrontal Cortex, Striatum, Schizophrenia
INTRODUCTION
Schizophrenia is a severe neuropsychiatric disorder characterized by positive and negative symptoms and cognitive impairments. Traditional models of schizophrenia focus on dysfunction of brain dopaminergic systems, with all current medications blocking neurotransmission at brain dopamine (D2) receptors (rev. in Howes and Kapur, 2009). More recent models posit dysfunction in brain glutamatergic systems as well, and especially of transmission mediated at N-methyl-D-aspartate (NMDAR) type glutamate receptors (Coyle, 1996; D’Souza et al., 1995; Javitt and Zukin, 1991; Moghaddam and Javitt, 2012). Glutamatergic medications for schizophrenia, however, remain in preclinical or early clinical development.
NMDARs are modulated by glycine, levels of which in turn are regulated by glycine transporters (GLYT1) that are co-localize with NMDARs. It has thus been proposed that high affinity GLYT1 inhibitors (GTIs) might represent a novel class of compound for treatment of persistent symptoms of schizophrenia (rev. in Javitt, 2009). The present study evaluates effectiveness of the prototype high affinity glycine transport inhibitors, R231857/60 and Org25935, in preclinical model relevant to DA dysfunction in schizophrenia.
Original conceptualizations of DA dysfunction in schizophrenia were supported both by the ability of DA-releasing agents, such as AMPH, to induce symptoms resembling schizophrenia, in addition to the clinical efficacy of antipsychotic medications, which function by blocking D2-type DA receptors (Howes and Kapur, 2009). DA dysfunction has also been confirmed by PET and SPECT radioreceptor binding studies that show increased in vivo dopamine release to AMPH challenge during acute clinical decompensation, although not during more chronic stages of the disease(Laruelle et al., 1999). Neurochemical mechanisms underlying this well-documented DA dysfunction in schizophrenia, however, remain poorly understood.
Glutamatergic models of schizophrenia are supported by the observations that NMDAR antagonists, such as phencyclidine (PCP) or ketamine, also produce psychotic symptoms in normal volunteers, with psychosis incorporating negative and cognitive, in addition to positive symptoms (Coyle, 1996; Javitt and Zukin, 1991; Krystal et al., 2002; Linn et al., 2007). Moreover, NMDAR antagonists induce schizophrenia-like deficits in DA regulation in both rodents (Balla et al., 2001b; Balla et al., 2003) and humans (Kegeles et al., 2000), suggesting that NMDA dysfunction may contribute to the dopaminergic instability observed in schizophrenia. As such, NMDAR-based interventions may provide an alternative mechanism for new treatment development in schizophrenia.
To date, NMDAR-based treatments have targeted primarily the glycine/D-serine positive allosteric modulatory site of the NMDAR complex. Several small-scale studies have demonstrated efficacy for both glycine and D-serine (rev. in Kantrowitz and Javitt, 2010). Although negative studies have also been reported (e.g. Buchanan et al., 2007), recent meta-analyses suggest moderate-size beneficial effects of NMDAR/glycine site agonists on persistent negative symptoms when added to typical or atypical antipsychotics, with more modest effects on positive symptoms (Singh and Singh, 2011; Tsai and Lin, 2010).
The use of GTIs in treatment of schizophrenia in place of NMDAR/glycine-site agonists was first proposed over a decade ago, based on actions of the glycine derivative glycyldodecylamine (Javitt and Frusciante, 1997; Javitt et al., 1997), and was subsequently supported by preclinical studies with prototype GLYT1 inhibitors such as [3-(4″-fluorophenyl)-3-(4″-phenylphenoxy) propyl]sarcosine (NFPS, ALX5407) (Bowie et al., 2008). These compounds increased brain glycine levels in vivo (e.g. Umbricht et al., 2010) and potentiate NMDA receptor-mediated neurotransmission both in vitro(Bergeron et al., 1998) and in vivo (Chen et al., 2003), consistent with underlying glutamatergic theories (rev. in Javitt, 2009).
Early in vivo studies with NFPS, while encouraging (Harsing, 2003; Javitt et al., 2004), were limited by the poor pharmacodynamic properties of the compound. Other compounds, such as SSR103800 (Vanneste and De Ridder, 2011), while also showing encouraging preclinical effects, have yet to be entered into human clinical trials (rev. in Javitt, 2009). The present study reports on effects of two compounds, both of which have been entered into early stage clinical development. R231857 is a high affinity GLYT1 antagonist, and recently evaluated in a scopolamine challenge model in humans (Liem-Moolenaar et al., 2010). Although no significant beneficial effects were observed, it was nevertheless considered safe for human use. Org25935 is GlyT1 inhibitor that has shown to be safe in human use, and to reverse acute psychotomimetic effects of ketamine in healthy volunteers (D’Souza et al., 2012).
The goal of this study was two-fold: First, to further investigate the role of NMDA receptors in DA dysregulation relevant to schizophrenia using available high-affinity compounds; second, to validate PCP-induced augmentation of AMPH-induced DA release as a translational model for new treatment development in schizophrenia. We hypothesized that, as with glycine and NFPS (Javitt et al., 2004), significant reversal of AMPH-induced DA release would be observed during subchronic treatment with NMDAR antagonists. As compared to earlier studies with NFPS, both R231857 and Org25935 are well-tolerated in rodents, permitting evaluation of sustained efficacy during subchronic administration. Because D-serine is nephrotoxic in rats, D-cycloserine (DCS) was used along with glycine as an active control compound, at doses at which it has been previously shown to be effective in animal models of schizophrenia (Carlsson et al., 1994).
EXPERIMENTAL PROCEDURES
Animals
Studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. Male Sprague-Dawley rats (160–200 g and 280–320 g) bred in our animal colony were used. The rats were maintained under a 10h/14 h dark/light cycle, and were allowed food and water ad libitum during the microdialysis procedure.
Drug Administration
Phencyclidine hydrochloride (obtained from the National Institute of Drug Abuse) was dissolved in sterile physiological saline and was given through an osmotic pump model 2ML4 (ALZA Corporation) implanted under the skin. The pumps were filled based on the animal weight at the start of the experiment to deliver 15 mg/kg/day. Osmotic pumps filled with saline were used in control animals. The implantation was carried under anesthesia with ketamine hydrochloride and acepromazine maleate 1:1 mixture (1 μL/g i.m.). D-Cycloserine (30 mg/kg/ip per day for 2 weeks), or Org25935 (5 – 10 mg/kg/day by gavage) were administered for 2 weeks (prepared in a concentration of 1 mg/ml saline- gavage ~ 1.25 – 2.5 ml depending on body weight). R231857 and R231860 were dissolved in β-cyclodextrin (1 mg/ml) and administered i.p. daily for 2 weeks at a dose of 5 mg/kg. R231860 was used in higher 9.2mg/kg/daily dose also, since the lower dose proved less effective. Glycine (GLY) was administered for 2 weeks in the form of glycine-enriched food (16% by weight) made by Diets Inc. This formulation has previously been shown to produce plasma GLY levels similar to those observed during effective clinical trials (Javitt et al., 2004).
In Vivo Microdialysis
Microdialysis was performed 16–21 days after pump implantation in control, PCP, PCP plus D-cycloserine, or ORG 25935G treatment groups. Animals were anesthetized with chloral hydrate (400 mg/kg i.p.) and mounted in a stereotaxic frame (David Kopf Instruments). CMA 12 guide cannulas were implanted relative to bregma into medial prefrontal cortex (PFC) (AP: +4.1, L: +1.0, V:−1.2 at 15° angle) and into striatum (AP: +1.0, L:+2.5, V: −4.0) according to the rat brain atlas of Paxinos and Watson 1998, 48 hours before microdialysis measurement. The cannulas and two stainless-steel screws embedded in the skull were cemented with dental acrylic. Buprenorphine (0.5 mg/kg) was given post-op.
CMA 12 probes (0.5 mm × 2.0 mm or 4.0 mm membrane length with a molecular cut-off 20 000 Dalton) were used for the striatum and PFC respectively. The dialysis probes were perfused continuously at a flow rate of 1.0 μl/min using a syringe pump CMA/100 (Carnegie Medicine) with Mg2+-free Ringer solution containing (NaCl 147 mM; KCl 4 mM; CaCl2 1.2 mM; degassed). After a 2 hour wash-out period, 3 baseline samples were collected and then animals were challenged with AMPH (1 mg/kg sc) and samples collected for 210 min with a fraction collector (Bioanalytical Systems). The treatment groups were randomized over the course of the study.
Determination of the Probe Placement
The following day after the microdialysis study, the rats were anesthetized with ketamine hydrochloride and acepromazine maleate 1:1 mixture (1 μl/g i.m.). A blood sample was taken from a heart-puncture and plasma obtained after centrifugation for DCS and PCP analysis. The rat brain was fixed with 4% formaldehyde solution perfused through the heart; the brain was stored in 30% glucose solution. The placement of the probes was determined histologically.
High-Pressure Liquid Chromatography
The dialysate samples (30 μl) – collected in 15 mM NaEDTA and 10% ethyl alcohol mixture – were injected with an autosampler. For a determination of catecholamines, a reverse-phase high-pressure liquid chromatography (BAS-480 system) with electrochemical detection was used. A microbore C18 100 × 2 mm millibore column, glassy carbon electrodes vs. Ag/AgCl reference electrode at 0.60 V and 0.75 V was used (BAS).
A filtered, degassed mobile phase (sodium phosphate monobasic 25 mM; sodium citrate 50 mM; disodium-EDTA 27 μM; diethylamine-HCl 10 mM; 1-octanesulfonic acid, sodium salt; methanol 3% v/v; dimethylacetamide 2.1% v/v; pH = 3.5) with a flow rate 0.4 ml/min gave the following retention times in minutes: DOPAC 3.8; DA 4.9; HIAA 6.7; HVA 7.7.
The working standard solutions were stored at −80°C and 10 μl of the standard solution was injected between biological samples.
Data analysis
Data were analyzed using change in dopamine concentration from pre-AMPH baseline (-90 to 0 min), designated ΔDA. Neither PCP (F1,49=.2, p=.6) nor R231857/R231860 (F3,49=.5, p=.7) treatment significantly altered basal DA levels (Table 1). Effects of treatment on ΔDA levels were assessed using repeated measures multivariate analysis of variance (rmANOVA, equivalent to Wilks-Lambda) across indicated time points after AMPH administration (0–210 min). PCP and GTI treatment were included as separate factors. Follow-up comparisons were conducted by post-hoc least significant difference (LSD) analysis. Data in text represent mean ± SD unless otherwise indicated.
Table 1.
Mean (± std dev, pg/10 μl) basal dopamine levels (DA) levels in PFC.
| Condition | Control | PCP |
|---|---|---|
| R231857/60 | ||
| Control | 1.93 ± .69 (8) | 2.16 ± .61 (8) |
| R231857, 5 mg | 1.92 ± .77 (8) | 1.94 ± .57 (8) |
| R231860, 5 mg | 1.65 ± .60 (8) | 2.20 ± .56 (8) |
| R231860, 10 mg | 2.66 ± .09 (8) | 2.32 ± 1.99 (8) |
| Glycine (16% by diet) | --- | 4.41 ± 3.02 (8) |
| Org25935 | ||
| Control (vehicle) | .96 ± .70 (10) | 1.03 ± .80 (16) |
| Org25935, 5 mg1 | 1.86 ± 1.54 (10) | 1.71 ± 1.31 (11) |
| Org25935, 10 mg | .47 ± .27 (9) | .54 ± .35 (11) |
| DCS (30 mg) | 1.20 ± 1.55 (9) | 1.05 ± .63 (4) |
p=.007 vs. control, post-hoc LSD
RESULTS
The effects of NMDAR modulation on AMPH-induced DA were assessed using high affinity GTIs R231857, R231860, and ORG 25935, along with glycine and the partial glycine-site agonist D-cycloserine (DCS). In all experiments, test compounds were administered daily for 14 days either orally (GlyT1 inhibitors) or i.p. (DCS), while PCP or saline were administered by osmotic minipump at a dose of 15 mg/kg/d. This regimen has been shown previously to be sensitive to effects of orally administered high dose glycine (Javitt et al., 2004), with maximal effect over the 120 -210 time period following AMPH administration. Similar metrics were used in the present study.
Prefrontal Cortex
R231857/R231860
Initial studies were performed with both compounds at a daily dose of 5 mg/kg. A subsequent series of experiments investigated the effect of R231860 administered at 10 mg/kg. Across all compounds and doses, PCP significantly increased AMPH-induced DA release throughout the 0–210 min period (F1,49=43.4, p<.0001) (Figure 1). There was also a significant main effect of R231857/R231860 treatment (F3,49=4.24, p=.003) and a significant PCP X drug interaction (F3,49=5.02, p=.004), reflecting greater effect in the presence (F1,26=8.15, p=.001) than absence (F3,23=.3, p=.98) of PCP.
Figure 1.

Effect of R231857/R231860 and glycine (A,B) and Org25935 and DCS (C,D) on amphetamine (AMPH)-stimulated dopamine (DA) release in prefrontal cortex (PFC) in animals treated subchronically with either saline (Control) (A,C) or phencyclidine (PCP) 15 mg/kg/d × 14d (B,D). Values are mean of 4–16 experiments each as indicated in Table 1.
Finally, there was a significant PCP X drug X time interaction (F18,129=2.63, p=.001), reflecting greater PCP X GTI treatment effect during the 60–210 min post-AMPH treatment interval (F3,48=4.76, p=.006) than in the 0–60 min (F3,47=1.24, p=.31). When data were summed across the 60 – 210 min interval, significant main effects were observed for PCP (F1,49=47.7, p<.0001) and drug (F3,49=3.75, p=.017) along with a significant PCP X drug interaction (F 3,49=5.43, p=.003). Effect of PCP on AMPH-induced DA release was highly significant (LSD p<.0001). In the presence of PCP, R231857 (LSD p<.0001), R231860 - 5 mg (LSD p=.007) and R231860 - 10 mg (LSD p<.0001) all significantly inhibited AMPH-induced DA release (Figure 2A). By contrast, in the absence of PCP no significant effects were observed for any of the compounds (R231857, p=.7; R231860 - 5 mg, p=.94; R231860 - 10 mg, p=.7). Glycine was included as a positive control, and also significantly reduced DA release in the presence of PCP (LSD p=.001).
Figure 2.
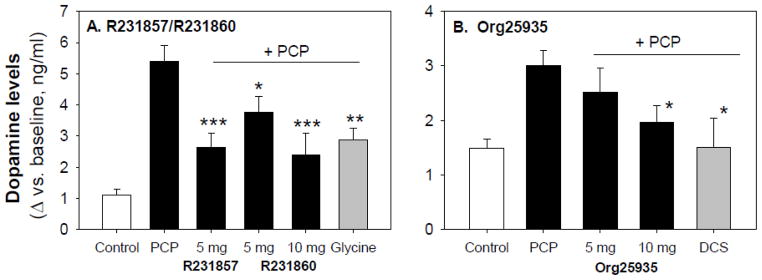
Summed activity in PFC over the 60–210 min range (mean ± sem) in animals treated with either R21857/R231860 and glycine (A) or Org25935 and DCS (B).
*p<.05, ** p<.01, ***p<.001 vs. PCP alone.
ORG25935
In experiments involving Org25935, PCP treatment again had no significant effect on basal DA levels (F1,61=0, p=.98) (Table 1), but significantly increased AMPH-induced DA release across the 0 – 210 min interval (F1,54=20.7, p<.0001) (Figure 1). Org259355 mg/kg treatment led to a significant increase in basal DA levels across control and PCP treatment conditions (LSD p=.007), while the 10 mg/kg dose produced a non-significant decrease (LSD p=.09)(Table 1).
Across the entire 0–210 min interval, Org25935 significantly inhibited AMPH-induced DA release (F1,54=3.73, p=.031) (Figure 1C,D), with the effect again being most pronounced during the 60–210 min post-AMPH treatment interval (F2,55=5.06, p=.010) than in the 0–60 min interval (F2,57=.56, p=.95) (Figure 1C,D). During the 60–210 min period, PCP treatment significantly increased AMPH-induced DA release vs. control (LSD p<.0001). In the presence of PCP, the 5 mg dose of Org25935 did not significantly reduced AMPH-induced DA release vs. PCP alone (LSD p=.2). In contrast, the 10 mg dose was effective (post-hoc p=.008) (Figure 2B). In the absence of PCP, Org25935 had no significant effect on AMPH-induced DA release at either the 5 mg (LSD p=.3) or 10 mg (LSD p=.13) dose.
D-cycloserine (DCS)
DCS studies were intermixed with Org25935 and utilized the same control groups. Plasma concentrations obtained during treatment with DCS were approx. 10 μg/ml. There were no metabolic interactions between DCS and PCP (Table 2). DCS treatment had no significant effect on basal DA levels (F1,26=2.13, p=.2), but significantly reduced AMPH-induced DA release during the 60–210 min time period (F1,26=10.1, p=.004) (Figure 2B). As with Org25935, DCS had no significant effect on AMPH-induced DA release in the absence of PCP (LSD p=.17).
Table 2.
Levels of plasma and brain D-cycloserine (DCS) and phencyclidine (PCP) after 2-week administration
| Treatment | D-Cylcoserine (DCS)
|
Phencyclidine (PCP)
|
||
|---|---|---|---|---|
| Plasma (μg/ml) | Brain (μg/g) | Plasma (ng/ml) | Brain (ng/g) | |
| DCS 30 mg/kg/day | 10.2 ± 1.6 | 6.1 ± 0.9 | ||
| DCS 30 mg/kg/day + PCP | 10.7 ± 1.2 | 5.5 ± 0.6 | 36.4 ± 7.9 | 126.3 ± 33.8 |
Plasma and brain tissue levels of DCS and PCP (mean ± std dev) after indicated dose (per day for 2 weeks) and in PCP-treated rats (n= 5 per group). PCP was administered via Alzet minipump based on delivering 15 mg/kg per day.
Striatum
In experiments involving R231857/R231860, PCP significantly potentiated AMPH-induced DA release across all time points (F1,31=18.6, p<.001). There was no significant main effect of R231857/231860 treatment (F3,31=2.08, p=.12) or PCP X drug treatment interaction (F3,31=1.52, p=.23) (Fig 3A,B). In experiments involving Org25935 and DCS, no significant effects were observed for either PCP (F1,51=2.0, p=.16), drug treatment (F1,2.20, p=.1) or PCP by treatment interaction (F3,51=.8, p=.5) (Fig 3C,D).
Figure 3.

Effect of R231857/R231860 (A,B) and Org25935, DCS (C,D) on amphetamine (AMPH)-stimulated dopamine (DA) release in striatum in animals treated subchronically with either saline (Control) (A,C) or phencyclidine (PCP) 15 mg/kg/d × 14d (B,D)
DISCUSSION
In schizophrenia, increased AMPH-stimulated DA release (Laruelle et al., 1999), along with increased dopamine metabolism (Howes et al., 2009; Kegeles et al., 2010) has been associated with increased symptoms during acute psychotic relapse. The present study supports prior research in humans (Kegeles et al., 2000) and rodents (Balla et al., 2001b; Balla et al., 2003) showing that schizophrenia-like dysregulation of subcortical dopamine release may be induced by ongoing, subchronic treatment with PCP or other NMDAR antagonists. In addition, it supports prior studies conducted with high-dose glycine and the prototypic GLYT1 antagonist NFPS showing significant reversal of AMPH-induced DA release by high affinity GLYT1 antagonists (Javitt et al., 2004). To the extent that DA dysfunction within PFC has been linked to cognitive deficits (Kellendonk et al., 2009) along with positive symptoms, the present studies support potential utility of GlyT1 antagonists against both positive symptoms and PFC-type cognitive deficits, along with negative symptoms, in schizophrenia.
Current theories of schizophrenia have attributed positive symptoms of schizophrenia to the hyperactivity of brain DA systems, particularly in frontostriatal brain regions (Fusar-Poli et al., 2010; Kegeles et al., 2010). Although etiology of DA hyperactivity remains unknown, deficits may be attributable to underlying NMDAR dysfunction based upon studies in both humans and rodents. Thus, in humans, administration of ketamine increases dopamine release in PFC (Aalto et al., 2005) and induces schizophrenia-like potentiation of AMPH-induced DA release (Laruelle et al., 1999). Similarly, in rodents, NMDAR antagonists induce significant acute increases in DA release during acute treatment, associated with acute impairments in PFC function (Kargieman et al., 2007; Lorrain et al., 2003; Verma and Moghaddam, 1996). Although these acute changes resolve following chronic treatment with NMDAR antagonists (Tsukada et al., 2005), nevertheless, DA systems show continued increased sensitivity to AMPH effects even during subchronic treatment (Balla et al., 2001b; Sershen et al., 2008), suggesting that this may serve as a pharmacological model for persistent dopaminergic hypersensitivity in schizophrenia.
Hyperfunction of brain dopaminergic and serotonergic systems is observed as well in NMDAR knock-out mice (Miyamoto et al., 2001), providing further evidence of regulation of dopaminergic function by the NMDAR system. Finally, genetic variations of the GlyT1 transporter have shown genetic association with methamphetamine-use disorder (Morita et al., 2008), suggesting that the GlyT1-mediated regulation of frontal dopamine release may be of functional significance in disorders of dopaminergic regulation.
Initial studies investigating effects of chronic NMDAR stimulation on in vivo prefrontal and striatal DA release were conducted with glycine and the prototype GlyT1 inhibitor NFPS (Javitt et al., 2004). Since that time, high affinity GlyT1 inhibitors have been developed by several pharmaceutical companies (Javitt, 2009; Pinard et al., 2010) and shown to be free of toxic side effects associated with earlier compounds such as NFPS. In addition, these compounds show reliable effects on extracellular brain glycine levels and NMDAR activity in a number of schizophrenia assays, including inhibition of NMDAR-antagonist induced rodent hyperactivity (Alberati et al., 2011; Javitt, 2009; Singer et al., 2009a), modulation of hippocampal LTP in vitro (Martina et al., 2004) and reversal of NMDA-antagonist induced abnormalities in latent inhibition (Gaisler-Salomon et al., 2008; Zaehle et al., 2011). The high affinity GlyT1 inhibitor Org24461 was also shown to reduce extracellular levels of dopamine when administered in combination with risperidone (Nagy et al., 2010). In addition to modulating dopamine, these compounds may also normalize serotonergic function in brain (Papp et al., 2008). The majority of studies to date, however, have utilized only acute dosing, while effects during chronic treatment remain relatively understudied.
The present study evaluates effects of two newer GlyT1 inhibitors, R231587/60 and Org25935 on DA alterations observed during subchronic PCP administration. An advantage of these compounds vs. previous agents is their tolerability during subchronic systemic administration. As in earlier studies with NMDAR antagonists, significant enhancement in prefrontal DA release was observed following subchronic PCP treatment at concentrations relevant to PCP-induced psychosis in humans (Balla et al., 2001a; Balla et al., 2001b; Balla et al., 2003). Furthermore, as in subchronic studies with the NMDAR modulator glycine or an acute study with the prototype GlyT1 inhibitor NFPS (Javitt et al., 2004), significant effects were observed on AMPH-induced DA release in PFC. Similar effects were observed as well with the partial glycine antagonist DCS that has also shown effectiveness in some, but not all, studies in schizophrenia (Heresco-Levy and Javitt, 2004; Singh and Singh, 2011). For all compounds, dissociation was observed with these compounds significantly altering AMPH-induced DA release in PFC, but not in striatum. Overall, these studies provide continued support for ability of high affinity GlyT1 inhibitors to reverse effects of NMDAR antagonists and potentiate NMDAR-mediated neurotransmission in vivo.
The present rodent findings support recent clinical research with NMDAR glycine-site agonists and GlyT1 inhibitors. Clinical studies have been conducted with the glycine-site agonists, glycine, D-serine and D-cycloserine, as well as with the naturally occurring GlyT1 inhibitor sarcosine, and, most recently, with the high affinity GlyT1 inhibitor RG1678 (Pinard et al., 2010; Umbricht et al., 2010). In most studies, the primary outcome measure has been persistent negative symptoms, with meta-analyses showing medium effect-size improvement across agents. Nevertheless, significant improvements have been noted as well on positive symptoms (Singh and Singh, 2011; Tsai and Lin, 2010), suggesting that these agents may also significantly normalize disrupted DA transmission.
Similarly, in an acute challenge study, Org259335 reversed not only total symptoms induced by ketamine, but also tended (p=.067) to reverse ketamine-induced positive symptoms (D’Souza et al., 2012). A more recently developed compound, RG1678, has been found to have significant results not only in animal models but also in a recently completed phase II clinical trial (Pinard et al., 2010; Umbricht et al., 2010). The present study thus provides a preclinical analog for the preclinical findings and a model for further investigating underlying mechanisms. Furthermore, to date these compounds have been used primarily in combination with antipsychotics, although efficacy was reported as well in one small monotherapy study (Lane et al., 2008). The present results argue for further development of NMDAR agonists as monotherapy treatment for schizophrenia.
In the present study, although significant PCP-induced enhancement of AMPH-induced DA release was observed in PFC, no significant enhancement was observed in striatum. In a prior study using a dual probe placement similar to that used in the present study, which is intended to target dorsal striatum along with PFC, significant (p<.04) enhancement was observed in striatum, but was substantially less robust than that observed in PFC (p=.001) (Javitt et al., 2004). By contrast, no enhancement was observed in a study in which probes were placed in ventral striatum (Balla et al., 2003). Increasingly, dopaminergic disturbances in schizophrenia are being localized to associative striatum, which projects to dorsolateral prefrontal cortex, rather than limbic or sensorimotor regions (Howes et al., 2009; Kegeles et al., 2010), consistent with regional changes in NMDA receptor-related transcripts in dorsolateral PFC (Kristiansen et al., 2007). Findings of selective PCP effects on PFC dopamine changes are thus consistent with the pattern of deficit observed in schizophrenia.
In the present study, no behavioral measures were obtained because of the dialysis probe setup. However, in other studies, GTIs have been shown to reverse PCP- or MK-801 induced hyperactivity (Boulay et al., 2010 ; Depoortere et al., 2005; Harsing, 2003; Javitt et al., 1999; Javitt et al., 1997; Singer et al., 2009b), as well as hyperactivity associated with NR1 knockdown mice (Boulay et al., 2010), and to reverse PCP- or MK-801 induced impairments in prepulse (Lipina et al., 2005) and latent (Black et al., 2009; Gaisler-Salomon et al., 2008) inhibition, and ketamine-induced working memory deficits in monkeys (Roberts et al., 2010). In contrast, GTIs do not reverse hyperactivity induced by AMPH or other dopaminergic manipulations (Boulay et al., 2010; Depoortere et al., 2005; Harsing, 2003; Javitt et al., 1997; Singer et al., 2009b), suggesting relative specificity for NMDA-mediated behavioral disruption. In animals receiving combined PCP and AMPH, the time course of behavioral effect is characterized by prolonged hyperactivity with significant increase in activity persisting beyond 210 min following AMPH administration (Balla et al., 2001b), similar to the time course of DA change observed in PFC but not striatum in the present study. Thus, as in other model systems, locomotor hyperactivity is temporally dissociated from corticolimbic dopaminergic activity (Adams and Moghaddam, 1998), and follows more closely the pattern observed for prefrontal dopaminergic activity.
In summary, all current treatments for schizophrenia function by antagonism of dopamine D2-mediated neurotransmission. However, there has been increasing interest in alternative treatments based upon glutamatergic mechanisms, and in particular in treatments that reverse putative deficits NMDAR mediated neurotransmission (Javitt et al., 2011; Moghaddam and Javitt, 2012). The present study demonstrates effectiveness of several novel GlyT1 inhibitors in modulating dopaminergic neurotransmission during subchronic treatment and supports continued development of these compounds for treatment of persistent positive, as well as negative, symptoms of schizophrenia.
Acknowledgments
Data were presented in part at the 2011 meeting of the European College for Neuropsychopharmacology annual meeting, Paris, France, Sept 3–8, 2011. We wish to acknowledge the technical contributions of John Aspromonte and Sarah Burch, who contributed to data collection and entry.
Footnotes
Contributors:
AB collected and performed primary analysis of the data presented in the paper; SS contributed significantly to data analysis and manuscript preparation; HS contributed significantly to study design, implementation and oversight and to data analysis and manuscript preparation. DCJ oversaw all aspects of study design and implementation and contributed significantly to data interpretation and manuscript preparation. We would like to acknowledge the technical contributions of John Aspromonte and Sarah Burch who assisted with data collection and entry.
Conflict of Interest
Dr. Javitt’s work has been funded by the NIDA and NIMH. He holds intellectual property rights for use of GTIs in schizophrenia. He holds equity interest in Glytech, Inc. Over the past 3 years, he has consulted for Sanofi, Lundbeck, Schering-Plough, Takeda, NPS, Solvay, Sepracor, AstraZeneca, Pfizer, Cypress, Merck, Sunovion, Lilly, and Bristol-Myers-Squibb. He has received research support from Pfizer, Roche, and Jazz. He serves on the advisory board of Promentis, Inc. Other authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aalto S, Ihalainen J, Hirvonen J, Kajander J, Scheinin H, Tanila H, Nagren K, Vilkman H, Gustafsson LL, Syvalahti E, Hietala J. Cortical glutamate-dopamine interaction and ketamine-induced psychotic symptoms in man. Psychopharmacology (Berl) 2005;182:375–383. doi: 10.1007/s00213-005-0092-6. [DOI] [PubMed] [Google Scholar]
- Adams B, Moghaddam B. Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci. 1998;18:5545–5554. doi: 10.1523/JNEUROSCI.18-14-05545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberati D, Borroni E, Moreau J, Hainzl D, Pinnard E, Wettstein JG. Partial occupancy of the glycine transporter type 1 in rat by RG1678 leads to efficacy in models relevant to schizophrenia. Schizophr Bull. 2011;37 (Suppl 1):286. [Google Scholar]
- Balla A, Hashim A, Burch S, Javitt DC, Lajtha A, Sershen H. Phencyclidine-induced dysregulation of dopamine response to amphetamine in prefrontal cortex and striatum. Neurochem Res. 2001a;26:1001–1006. doi: 10.1023/a:1012396820510. [DOI] [PubMed] [Google Scholar]
- Balla A, Koneru R, Smiley J, Sershen H, Javitt DC. Continuous phencyclidine treatment induces schizophrenia-like hyperreactivity of striatal dopamine release. Neuropsychopharmacology. 2001b;25:157–164. doi: 10.1016/S0893-133X(01)00230-5. [DOI] [PubMed] [Google Scholar]
- Balla A, Sershen H, Serra M, Koneru R, Javitt DC. Subchronic continuous phencyclidine administration potentiates amphetamine-induced frontal cortex dopamine release. Neuropsychopharmacology. 2003;28:34–44. doi: 10.1038/sj.npp.1300019. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci U S A. 1998;95:15730–15734. doi: 10.1073/pnas.95.26.15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MD, Varty GB, Arad M, Barak S, De Levie A, Boulay D, Pichat P, Griebel G, Weiner I. Procognitive and antipsychotic efficacy of glycine transport 1 inhibitors (GlyT1) in acute and neurodevelopmental models of schizophrenia: latent inhibition studies in the rat. Psychopharmacology (Berl) 2009;202:385–396. doi: 10.1007/s00213-008-1289-2. [DOI] [PubMed] [Google Scholar]
- Boulay D, Bergis O, Avenet P, Griebel G. The glycine transporter-1 inhibitor SSR103800 displays a selective and specific antipsychotic-like profile in normal and transgenic mice. Neuropsychopharmacology. 2010;35:416–427. doi: 10.1038/npp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie CR, Reichenberg A, McClure MM, Leung WL, Harvey PD. Age-associated differences in cognitive performance in older community dwelling schizophrenia patients: differential sensitivity of clinical neuropsychological and experimental information processing tests. Schizophr Res. 2008;106:50–58. doi: 10.1016/j.schres.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, Heresco-Levy U, Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Carlsson ML, Engberg G, Carlsson A. Effects of D-cycloserine and (+)-HA-966 on the locomotor stimulation induced by NMDA antagonists and clonidine in monoamine-depleted mice. J Neural Transm Gen Sect. 1994;95:223–233. doi: 10.1007/BF01271568. [DOI] [PubMed] [Google Scholar]
- Chen L, Muhlhauser M, Yang CR. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol. 2003;89:691–703. doi: 10.1152/jn.00680.2002. [DOI] [PubMed] [Google Scholar]
- Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harvard review of psychiatry. 1996;3:241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Charney D, Krystal J. Glycine site agonists of the NMDA receptor: a review. CNS Drug Reviews. 1995;1:227–260. [Google Scholar]
- D’Souza DC, Singh N, Elander J, Carbuto M, Pittman B, de Haes JU, Sjogren M, Peeters P, Ranganathan M, Schipper J. Glycine transporter inhibitor attenuates the psychotomimetic effects of ketamine in healthy males: preliminary evidence. Neuropsychopharmacology. 2012;37:1036–1046. doi: 10.1038/npp.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C, Poncelet M, Heaulme M, Santucci V, Decobert M, Cudennec A, Voltz C, Boulay D, Terranova JP, Stemmelin J, Roger P, Marabout B, Sevrin M, Vige X, Biton B, Steinberg R, Francon D, Alonso R, Avenet P, Oury-Donat F, Perrault G, Griebel G, George P, Soubrie P, Scatton B. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. 2005;30:1963–1985. doi: 10.1038/sj.npp.1300772. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, Grasby PM, McGuire PK. Abnormal frontostriatal interactions in people with prodromal signs of psychosis: a multimodal imaging study. Arch Gen Psychiatry. 2010;67:683–691. doi: 10.1001/archgenpsychiatry.2010.77. [DOI] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Diamant L, Rubin C, Weiner I. Abnormally persistent latent inhibition induced by MK801 is reversed by risperidone and by positive modulators of NMDA receptor function: differential efficacy depending on the stage of the task at which they are administered. Psychopharmacology (Berl) 2008;196:255–267. doi: 10.1007/s00213-007-0960-3. [DOI] [PubMed] [Google Scholar]
- Harsing L. The glycine transporter-1 inhibitors NFPS and Org 24461: a pharmacological study. Phamacol Biochem Behav. 2003;74:811–825. doi: 10.1016/s0091-3057(02)01078-x. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC. Comparative effects of glycine and D-cycloserine on persistent negative symptoms in schizophrenia: a retrospective analysis. Schizophr Res. 2004;66:89–96. doi: 10.1016/S0920-9964(03)00129-4. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, Bramon-Bosch E, Valmaggia L, Johns L, Broome M, McGuire PK, Grasby PM. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Glycine transport inhibitors for the treatment of schizophrenia: symptom and disease modification. Current opinion in drug discovery & development. 2009;12:468–478. [PubMed] [Google Scholar]
- Javitt DC, Balla A, Burch S, Suckow R, Xie S, Sershen H. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29:300–307. doi: 10.1038/sj.npp.1300313. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Balla A, Sershen H, Lajtha A. A.E. Bennett Research Award. Reversal of phencyclidine-induced effects by glycine and glycine transport inhibitors. Biol Psychiatry. 1999;45:668–679. doi: 10.1016/s0006-3223(98)00237-6. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Frusciante M. Glycyldodecylamide, a phencyclidine behavioral antagonist, blocks cortical glycine uptake: implications for schizophrenia and substance abuse. Psychopharmacology (Berl) 1997;129:96–98. doi: 10.1007/s002130050168. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Schoepp D, Kalivas PW, Volkow ND, Zarate C, Merchant K, Bear MF, Umbricht D, Hajos M, Potter WZ, Lee CM. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3:102mr102. doi: 10.1126/scitranslmed.3002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Sershen H, Hashim A, Lajtha A. Reversal of phencyclidine-induced hyperactivity by glycine and the glycine uptake inhibitor glycyldodecylamide. Neuropsychopharmacology. 1997;17:202–204. doi: 10.1016/S0893-133X(97)00047-X. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83:108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargieman L, Santana N, Mengod G, Celada P, Artigas F. Antipsychotic drugs reverse the disruption in prefrontal cortex function produced by NMDA receptor blockade with phencyclidine. Proc Natl Acad Sci U S A. 2007;104:14843–14848. doi: 10.1073/pnas.0704848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, Cooper TB, Carlsson A, Laruelle M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48:627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Kandel ER. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci. 2009;32:347–358. doi: 10.1016/j.tins.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen LV, Huerta I, Beneyto M, Meador-Woodruff JH. NMDA receptors and schizophrenia. Curr Opin Pharmacol. 2007;7:48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Anand A, Moghaddam B. Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry. 2002;59:663–664. doi: 10.1001/archpsyc.59.7.663. [DOI] [PubMed] [Google Scholar]
- Lane HY, Liu YC, Huang CL, Chang YC, Liau CH, Perng CH, Tsai GE. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63:9–12. doi: 10.1016/j.biopsych.2007.04.038. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Liem-Moolenaar M, Zoethout RW, de Boer P, Schmidt M, de Kam ML, Cohen AF, Franson KL, van Gerven JM. The effects of a glycine reuptake inhibitor R231857 on the central nervous system and on scopolamine-induced impairments in cognitive and psychomotor function in healthy subjects. J Psychopharmacol. 2010;24:1681–1687. doi: 10.1177/0269881109105573. [DOI] [PubMed] [Google Scholar]
- Linn GS, O’Keeffe RT, Lifshitz K, Schroeder C, Javitt DC. Behavioral effects of orally administered glycine in socially housed monkeys chronically treated with phencyclidine. Psychopharmacology (Berl) 2007;192:27–38. doi: 10.1007/s00213-007-0771-6. [DOI] [PubMed] [Google Scholar]
- Lipina T, Labrie V, Weiner I, Roder J. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology (Berl) 2005;179:54–67. doi: 10.1007/s00213-005-2210-x. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, Bergeron R. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol. 2004;557:489–500. doi: 10.1113/jphysiol.2004.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Hyperfunction of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor epsilon1 subunit. J Neurosci. 2001;21:750–757. doi: 10.1523/JNEUROSCI.21-02-00750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Ujike H, Tanaka Y, Kishimoto M, Okahisa Y, Kotaka T, Harano M, Inada T, Komiyama T, Hori T, Yamada M, Sekine Y, Iwata N, Iyo M, Sora I, Ozaki N, Kuroda S. The glycine transporter 1 gene (GLYT1) is associated with methamphetamine-use disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:54–58. doi: 10.1002/ajmg.b.30565. [DOI] [PubMed] [Google Scholar]
- Nagy K, Marko B, Zsilla G, Matyus P, Pallagi K, Szabo G, Juranyi Z, Barkoczy J, Levay G, Harsing LG., Jr Alterations in brain extracellular dopamine and glycine levels following combined administration of the glycine transporter type-1 inhibitor Org-24461 and risperidone. Neurochem Res. 2010;35:2096–2106. doi: 10.1007/s11064-010-0241-0. [DOI] [PubMed] [Google Scholar]
- Papp A, Juranyi Z, Nagymajtenyi L, Matyus P, Harsing LG., Jr The synaptic and nonsynaptic glycine transporter type-1 inhibitors Org-24461 and NFPS alter single neuron firing rate in the rat dorsal raphe nucleus. Further evidence for a glutamatergic-serotonergic interaction and its role in antipsychotic action. Neurochem Int. 2008;52:130–134. doi: 10.1016/j.neuint.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Pinard E, Alanine A, Alberati D, Bender M, Borroni E, Bourdeaux P, Brom V, Burner S, Fischer H, Hainzl D, Halm R, Hauser N, Jolidon S, Lengyel J, Marty HP, Meyer T, Moreau JL, Mory R, Narquizian R, Nettekoven M, Norcross RD, Puellmann B, Schmid P, Schmitt S, Stalder H, Wermuth R, Wettstein JG, Zimmerli D. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfon yl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. Journal of medicinal chemistry. 2010;53:4603–4614. doi: 10.1021/jm100210p. [DOI] [PubMed] [Google Scholar]
- Roberts BM, Shaffer CL, Seymour PA, Schmidt CJ, Williams GV, Castner SA. Glycine transporter inhibition reverses ketamine-induced working memory deficits. Neuroreport. 2010;21:390–394. doi: 10.1097/WNR.0b013e3283381a4e. [DOI] [PubMed] [Google Scholar]
- Sershen H, Balla A, Aspromonte JM, Xie S, Cooper TB, Javitt DC. Characterization of interactions between phencyclidine and amphetamine in rodent prefrontal cortex and striatum: implications in NMDA/glycine-site-mediated dopaminergic dysregulation and dopamine transporter function. Neurochem Int. 2008;52:119–129. doi: 10.1016/j.neuint.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Singer P, Feldon J, Yee BK. The glycine transporter 1 inhibitor SSR504734 enhances working memory performance in a continuous delayed alternation task in C57BL/6 mice. Psychopharmacology (Berl) 2009a;202:371–384. doi: 10.1007/s00213-008-1286-5. [DOI] [PubMed] [Google Scholar]
- Singer P, Feldon J, Yee BK. Interactions between the glycine transporter 1(GlyT1) inhibitor SSR504734 and psychoactive drugs in mouse motor behaviour. Eur Neuropsychopharmacol. 2009b;19:571–580. doi: 10.1016/j.euroneuro.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Singh SP, Singh V. Meta-Analysis of the Efficacy of Adjunctive NMDA Receptor Modulators in Chronic Schizophrenia. CNS Drugs. 2011;25:859–885. doi: 10.2165/11586650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Current pharmaceutical design. 2010;16:522–537. doi: 10.2174/138161210790361452. [DOI] [PubMed] [Google Scholar]
- Tsukada H, Nishiyama S, Fukumoto D, Sato K, Kakiuchi T, Domino EF. Chronic NMDA antagonism impairs working memory, decreases extracellular dopamine, and increases D1 receptor binding in prefrontal cortex of conscious monkeys. Neuropsychopharmacology. 2005;30:1861–1869. doi: 10.1038/sj.npp.1300732. [DOI] [PubMed] [Google Scholar]
- Umbricht D, Yoo K, Youssef E, Dorflinger E, Martin-Facklam M, Bausch A, Arrowsmith R, Alberati D, Marder SR, Santarelli L. Glycine Transporter Type 1 (GLYT1) Inhibitor RG1678: Positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacol. 2010;35:s320–321. [Google Scholar]
- Vanneste S, De Ridder D. Bifrontal transcranial direct current stimulation modulates tinnitus intensity and tinnitus-distress-related brain activity. Eur J Neurosci. 2011;34:605–614. doi: 10.1111/j.1460-9568.2011.07778.x. [DOI] [PubMed] [Google Scholar]
- Verma A, Moghaddam B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 1996;16:373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaehle T, Beretta M, Jancke L, Herrmann CS, Sandmann P. Excitability changes induced in the human auditory cortex by transcranial direct current stimulation: direct electrophysiological evidence. Exp Brain Res. 2011;215:135–140. doi: 10.1007/s00221-011-2879-5. [DOI] [PubMed] [Google Scholar]