

Abstract
High-efficiency genetic modification of human embryonic stem (hES) cells would enable manipulation of gene activity, routine gene targeting, and development of new human disease models and treatments. Chemical transfection, nucleofection, and electroporation of hES cells result in low transfection efficiencies. Viral transduction is efficient but has significant drawbacks. Here we describe techniques to transiently and stably express transgenes in hES cells with high efficiency using a widely available vector system. The technique combines nucleofection of single hES cells with improved methods to select hES cells at clonal density. As validation, we reduced Oct4 and Nanog expression using siRNAs and shRNA vectors in hES cells. Furthermore, we derived many hES cell clones with either stably reduced alkaline phosphatase activity or stably overexpressed green fluorescent protein. These clones retained stem cell characteristics (normal karyotype, stem cell marker expression, self-renewal, and pluripotency). These studies will accelerate efforts to interrogate gene function and define the parameters that control growth and differentiation of hES cells.
Keywords: Human embryonic stem cells, Nucleofection, Transfection, Transgene expression, RNA interference, Green fluorescent protein, Oct4, Nanog
Introduction
Human embryonic stem (hES) cells are potentially important tools for studying human development and for identifying genetic and environmental factors that affect developmental processes [1–4]. In addition, they are a potential resource for developing models of disease and cell-based treatments. This potential stems from their dual abilities to self-renew and differentiate. A powerful method to address questions about regulation of these dual abilities is to genetically manipulate hES cells in culture. The ability to transfect these cells with high efficiency would allow analysis of gene regulation using RNA interference (RNAi), expression vectors, and targeting constructs and also enable large-scale, genome-wide screens to be performed.
Two impediments to progress are the relatively poor growth of hES cells in culture and the difficulty in transfecting hES cells [4 – 6]. Recently, we demonstrated that addition of neurotrophins (NTs) to hES cell cultures increases survival of single cells and could consequently improve transfection techniques and clonal selection [7]. Several methods have been used to transfect hES cells, including chemical transfection, electroporation, nucleofection, and viral transduction, with various efficiencies (reviewed in [6]). Chemical and electrical methods of transfection result in transient efficiencies from <1% to 40% and very low, stable efficiencies from one in 105–107 cells [8 –15]. Expression of genes introduced into hES cells by retroviruses and lentiviruses is more successful (~85%) [16–18]. However, there are significant drawbacks associated with viral transduction [6, 19, 20].
One promising technique for introducing DNA into cells is nucleofection, which delivers DNA directly into the nucleus with high efficiency [21–23]. Methods for nucleofecting hES cells have been reported, with various transient transfection efficiencies of 20%– 65% [10, 11]. The reported stable transfection rate was low (1 in 106 cells), and little characterization of stem cell markers, karyotype, and pluripotency was reported [11]. Here we describe improved methods for obtaining stably transfected hES cells via nucleofection of individual hES cells mediated by increased clonal survival in the presence of NTs. We report a maximum transient transfection efficiency of 85% (similar to viral transduction) and a maximum stable efficiency of 1.2 in 104 cells (100 times better than current nucleofection rates). The method described here provides an opportunity to generate genetically modified diploid hES cell lines, which retain full pluripotency with very high efficiency.
Materials and Methods
hES Cell Culture
hES cell lines (H1, H9) were cultured as previously described [7, 24] on PMEFs (Chemicon, Temecula, CA, http://www.chemicon.com) and passaged using collagenase IV (Invitrogen, Carlsbad, CA, http://www.invitrogen.com). For nucleofection, cells were harvested using 0.05% trypsin-EDTA (Invitrogen), dissociated into single cells, and, following addition of trypsin inhibitor (Invitrogen), passed through a 0.4-mm cell strainer (Becton Dickinson, Franklin Lakes, NJ, http://www.bd.com; Falcon).
Vector and siRNA Design
Three cytomegalovirus (CMV)-immediate early (IE) promoter-based vectors were used: pmaxGFP (Amaxa Biosystems, Gaithersburg, MD, http://www.amaxa.com; 589-base pair [bp] PCMV IE, referred to as green fluorescent protein [GFP] vector) for transient transfections, phrGFP II-1 (catalog no. 240143; Stratagene, La Jolla, CA, http://www.stratagene.com; 665-bp CMV promoter, referred to as GFP-neo vector) for stable transfections, and pRNATin-H1.2/Neo (catalog no. SD1223; GenScript, Piscataway, NJ, http://www.genscript.com; 587-bp CMV promoter, referred to as shRNA vector) for RNAi. shRNAs corresponding to target genes were designed using GenScript’s Target Finder with the following sequences: alkaline phosphatase (AP; accession number: AB011406; alkaline phosphatase, liver/bone/kidney [ALPL], also known as the tissue-nonspecific alkaline phosphatase [TNAP]), 5′-AAGAGCT-TCAAACCGAGATA-3′ and 5′-GGACTATGCTCACAACAATA-3′. The Oct4 target was 5′-AGCAGCTTGGGCTCGAGAA-3′ [25], the Nanog sequence was 5′-AAGGGTTAAGCTGTAACATAC-3′ [17], and the β2M target sequence was 5′-GATTCAGGTTTACT-CACGT-3′ [25] Stem-loop structures for these targets were built into pRNATin-H1.2/Neo. For RNAi by cotransfection, synthesized siRNAs (Qiagen, Gaithersburg, MD, http://www1.qiagen.com) were transfected with the pmaxGFP vector (5:1).
Nucleofection
All nucleofections were performed using the Nucleofector II (Amaxa Biosystems). For optimization, six Nucleofector settings (A-06, A-12, A-13, A-23, A-27, and B-16), two nucleofection solutions (solution V and mouse embryonic stem [mES] solution), and two cell harvesting methods (collagenase and trypsin) were analyzed for effects on hES cell survival and transfection. GFP-positive colonies were visualized using a Nikon Eclipse T100 microscope (Nikon, Tokyo, http://www.nikon.com) and counted manually. For all subsequent transfections, hES cells were harvested by trypsinization and transfected using mES solution and program A-23. A total of 2 × 106 trypsinized hES cells were resuspended in 100 μl of mES cell solution and incubated (37°C) for 5 minutes. Two to three micrograms of DNA (or siRNA) was added to the cell solution. The mixture was transferred to a cuvette, nucleofected, and quickly transferred into 500 μl of prewarmed (37°C) RPMI medium (Invitrogen). Nucleofected cells were incubated (37°C) for 5 minutes and plated at 1 × 106 cells per well in a PMEF-Neor-coated six-well plate containing hES medium supplemented with NTs (10 ng/ml of brain-derived neurotrophic factor, NT-3, NT-4). To maximize viability and efficiency, the entire procedure was completed within 20 minutes. To select for stably transfected hES cell colonies, G418 sulfate (25 μg/ml; Invitrogen) was added 96 hours post-transfection. G418 concentration was increased to 50 μg/ml at 1 week post-transfection and finally to 100 μg/ml at 10 days post-transfection.
Stem Cell Characterization
Flow cytometric analysis of GFP and SSEA-4 (Developmental Studies Hybridoma Bank, Iowa City, IA, http://dshb.biology.uiowa.edu) expression was performed using a FACSCalibur flow cytometer (Becton Dickinson). Immunocytochemistry and AP staining was performed as previously described [7] Antibodies to Oct4, SSEA-3, and SSEA-4 were obtained from R&D Systems Inc. (Minneapolis, http://www.rndsystems.com), TRA1–60 and TRA1–81 from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, http://www.scbt.com), and anti-GFP IgG-Alexa Fluor 488 and secondary antibodies from Invitrogen.
Polymerase Chain Reaction Analyses
For reverse transcription (RT)-polymerase chain reaction (PCR), RNA was prepared using the RNeasy Kit (Qiagen) and performed using the Superscript One-Step Kit (Invitrogen) for 25–35 cycles. For PCR analyses, DNA was prepared using the DNeasy Blood and Tissue Kit (Qiagen) and performed using the Taq PCR Core Kit (Qiagen) for 35 cycles. Supplemental online Table 1 has primers and annealing temperature for each gene.
Karyotype Analysis
Analyses were performed by Johns Hopkins Cytogenetics Facility (Baltimore, MD) or Genzyme Genetics (Orange, CA, http://www.genzymegenetics.com).
Differentiation Analyses
Embryoid body and teratoma formation assays were carried out as previously described [7, 24, 26]. Animal work was performed under the Johns Hopkins University and University of California Irvine Institutional Animal Care and Use Committee protocols. Tumors were processed at the Johns Hopkins Comparative Medicine Department (Baltimore, MD) and analyzed at University of California Irvine Medical Center (Orange, CA).
Results
To optimize conditions for high efficiency nucleofection of hES cells, a 3.5-kilobase (kb) plasmid expressing GFP from the CMV promoter (GFP vector) was transfected into H9 hES cells, and the percentage of GFP-positive colonies was determined visually 96 hours after nucleofection. Six nucleofection programs and two solutions were compared, and nucleofected collagenase-derived clumps or trypsin-derived single hES cells gave colonies with cells that expressed GFP (18%–35% or 11%–54%, respectively; Fig. 1A, 1B). Nucleofection of trypsin-derived single cells using program A-23 and mES solution resulted in the most GFP-positive colonies (61%). In some transfections, NTs were omitted. Approximately 30% fewer colonies were observed in the absence of NTs (data not shown), suggesting that NTs enhance survival of clones after nucleofection. Notably, 70% of trypsin-derived colonies were entirely GFP-positive, whereas <10% of collagenase-derived colonies were uniformly GFP-positive (Fig. 1C, 1D). In fact, >90% of the collagenase-derived colonies contained <50% GFP-positive cells, suggesting that nucleofection does not effectively transfect all cells in collagenase-derived hES cell clumps.
Figure 1.

Optimization of nucleofection in hES cells. (A): The mean percentage of colonies (±SEM; n = 3) that were GFP-positive after nucleofection of collagenase-dissociated hES cells in V solution (blue bars) or mES solution (green bars). (B): The percentage of colonies (±SEM; n = 3) that were GFP-positive after nucleofection of trypsin-dissociated hES cells in V solution (yellow bars) or mES solution (red bars). (C): The mean percentage of GFP-positive cells (n = 5) in each colony after nucleofection of collagenase-derived (blue and green bars) and trypsin-derived (yellow and red bars) hES cells using program A-23 in either V solution (blue and yellow bars) or mES solution (green and red bars). (D): BF (top) and GFP expression (bottom) of transfected collagenase-dissociated hES cells and trypsin-dissociated hES cells 4 days after nucleofection. Abbreviations: BF, bright-field; GFP, green fluorescent protein; hES, human embryonic stem; mES, mouse embryonic stem.
To determine the efficiency of nucleofection, we analyzed the number of cells transfected with either the GFP vector or a 4.9-kb vector containing hrGFP expressed from the CMV promoter (GFP-neo vector). H1 or H9 hES cells were trypsin-dissociated, nucleofected using program A-23 and mES solution, and cultured in hES cell medium supplemented with NTs for 96 hours. Nucleofected hES cells were analyzed by flow cytometry using SSEA-4, to distinguish hES cells from mouse embryonic fibroblasts and differentiated cells. When either H1 or H9 hES cells were nucleofected, ~66% of SSEA-4-positive hES cells expressed GFP (Fig. 2A). In seven nucleofections using the GFP vector in H9 hES cells, transfection efficiencies ranged from 60% to 85%, with a mean of 76%. In 20 nucleofections using the GFP-neo vector, transfection efficiencies ranged from 47% to 81%, with a mean of 67% (data not shown). An increase in transfection efficiency was observed over time as the ability to handle and maintain single hES cells during nucleofection improved. The vast majority of control hES cells (nucleofected without DNA) continued to express SSEA-4, indicating that nucleofection does not affect stem cell marker expression (Fig. 2A).
Figure 2.

Nucleofection of hES cells with DNA, shRNA vectors, or siRNAs alters gene expression. (A): Flow cytometric analysis of GFP and SSEA-4 expression in H1 and H9 hES cells 4 days after transfection with the GFP vector (middle and right) or no DNA (left). (B): BF (top) and GFP expression (bottom) of hES cells 4 days after co-transfection of the GFP vector and either β2M siRNA or Oct4 siRNA. GFP expression was used to identify transfected cells. (C): BF (top) and SSEA-4 (bottom) immunostaining of β2M KD and Oct4 KD hES cells. (D): AP staining of β2M KD and Oct4 KD hES cells. (E): Reverse transcription-polymerase chain reaction analysis of hES cells transfected with either shRNA vectors or siRNAs. β-Actin was used as a template loading control. Abbreviations: AP, alkaline phosphatase; BF, bright-field; DAPI, 4,6-diamidino-2-phenylindole; GFP, green fluorescent protein; hES, human embryonic stem; KD, knockdown.
Next we carried out a proof-of-principle experiment to demonstrate the utility of nucleofection in studies of gene function. Previous studies demonstrated that Oct4 and Nanog are required to maintain the stem cell state [17, 25, 27–30]. To test the utility of nucleofection in reducing Oct4 or Nanog expression, shRNAs expressed from a CMV-based vector system (shRNA vector) or siRNAs were introduced into either H1 or H9 hES cells. The β2M gene, which is expressed in hES cells but not required for self-renewal [25], was used as a control (β2M knockdown [KD]). shRNA vectors and siRNAs reduced expression of specific targets, as determined by RT-PCR (Fig. 2E). Because siRNAs were cotransfected with the GFP vector and the shRNA vector contains a GFP reporter gene, transfected cells could be identified visually by fluorescence microscopy. Ninety-six hours after nucleofection, all GFP-positive colonies transfected with Oct4-specific siRNA (Oct4 KD; >500 colonies per well) were markedly flattened and morphologically distinct from typical or β2M KD hES cell colonies (Fig. 2B, 2C). The stem cell markers AP, SSEA-3, SSEA-4, TRA1–60, and TRA1–81 were reduced in the flattened, GFP-positive Oct4 KD colonies, in contrast with β2M KD cells (Fig. 2C, 2D; data not shown). Similar results were obtained when shRNA vectors were used to reduce Oct4 or Nanog expression (Nanog KD; data not shown). Interestingly, reduction of Oct4 or Nanog expression caused reciprocal downregulation of these genes, consistent with our understanding of the stem cell regulatory network (Fig. 2E) [31, 32]. To determine the identity of the differentiated cells, expression of trophectodermal genes GCM1 and hCG was assayed by RT-PCR. In both Oct4 KD and Nanog KD cells, expression of GCM1 and hCG increased, in comparison with β2M KD cells (Fig. 2E). These data strongly support the idea that nucleofection of siRNAs/shRNA vectors effectively alters gene expression in hES cells.
For many applications, stable long-term alteration in gene expression is necessary. To determine whether nucleofection can produce hES cells with stable modifications in gene expression, the GFP-neo vector was introduced into hES cells, and stable colonies were isolated by gradually increasing the G418 concentration. To isolate stable clones, G418 was added at a concentration of 25 μg/ml for 3 days, then 50 μg/ml for 3 days, and finally 100 μg/ml for at least 2 weeks. The GFP vector was used as a control in these experiments. GFP expression in control cells was rapidly lost in the absence of G418, and all cells grown in G418 (at only 25 μg/ml) died after 3 days. After 2 weeks in the presence of G418, an average of 60 colonies per well (n = 12) and a maximum of 120 colonies per well were observed in cultures transfected with the GFP-neo vector. GFP was expressed in most of the cells of G418-resistant colonies (Fig. 3A). G418-resistant, GFP-positive colonies were manually passaged 3 weeks after nucleofection (>2 weeks in selection) and maintained for 17 months in culture (>75 passages). Flow cytometry of the stably transfected hES cells showed that ~87% of the cells expressed GFP (data not shown), suggesting that some G418-resistant hES cells lose GFP expression. To monitor the stability of transgene expression in prolonged culture, we derived clonal cell lines from GFP-positive hES cell lines. A total of 144 clones were isolated by dilution cloning, half in G418 and half in the absence of G418. In both growth conditions, approximately 85% of the derived clones expressed GFP (Fig. 3B), consistent with the fluorescence-activated cell sorting analysis of the nonclonal G418-resistant, GFP-positive cells from which the clones were isolated. Genomic DNA from 14 clones was analyzed by PCR and demonstrated that the CMV promoter and the hrGFP gene are present contiguously in all clones, even those that do not express GFP (Fig. 3C), suggesting that silencing of the GFP transgene occurs in some cells of the stably transfected hES cell colonies [16, 18, 33].
Figure 3.

GFP expression in stable GFP-neo cell lines. (A): BF (left) and GFP (right) expression of hES cells stably transfected with the GFP-neo vector (GFP-neo cells). (B): Clonal cell lines derived from GFP-neo cells grown with or without G418. (C): Polymerase chain reaction analysis of genomic DNA isolated from clonal cell lines. Both GFP-positive and GFP-negative clones contain the CMV promoter and hrGFP transgene contiguously in their genomes. Untransfected hES cells served as a control, and β-actin was used a template loading control. Abbreviations: BF, bright-field; CMV, cytomegalovirus; GFP, green fluorescent protein; hES, human embryonic stem; NT, neurotrophin.
Cell lines and clones derived by nucleofection that stably express GFP retained an hES cell morphology and expressed GFP in long-term culture. All stable cell lines expressed the stem cell markers Oct4, SSEA-4, SSEA-3, TRA1–60, TRA1–81, and AP (Fig. 4A, 4B; data not shown). Nucleofected hES cells maintained a normal karyotype for >8 months of continuous culture (Fig. 4B; data not shown), suggesting that nucleofection does not induce major genomic instability. Stable cell lines retained their ability to differentiate, as demonstrated by formation of embryoid bodies (EBs) expressing GFP (Fig. 4C) and markers of all three germ layers (Fig. 5C). GFP-positive hES cells were also injected into NOD/SCID mice, producing teratomas that, by comparison with tumors from GFP-negative clones and nontransfected cells, expressed GFP broadly throughout the tumor (data not shown). Histological examination of those tumors revealed that they contained derivatives of all three germ layers, indicating that the cells were indeed pluripotent (Fig. 4D). Fluorescence microscopy of sections indicated that the GFP transgene continued to be expressed in most of the tumor cells (Fig. 4D). Clonal cell lines also readily formed teratomas expressing GFP (Fig. 4E), indicating long-term, stable expression of the transgene in vivo.
Figure 4.

Stably transfected clonal hES cell lines retain stem cell characteristics, including expression of stem cell markers, normal karyotype, and the ability to differentiate in vitro and in vivo. (A): DAPI (top), GFP (middle), Oct4 (bottom left), and SSEA-4 (bottom right) staining of GFP-neo hES cells. (B): AP staining (left) and karyotype (right) of GFP-neo hES cells after 25 passages. (C): BF (left) and GFP expression (right) of EBs derived from GFP-neo cells. (D): Hematoxylin and eosin (H&E; top panels), GFP, and DAPI staining (bottom panels) of sectioned GFP-neo teratomas. (E): Gross image of teratomas formed from GFP-neo hES cell clones. Abbreviations: AP, alkaline phosphatase; BF, bright-field; DAPI, 4,6-diamidino-2-phenylindole; GFP, green fluorescent protein.
Figure 5.
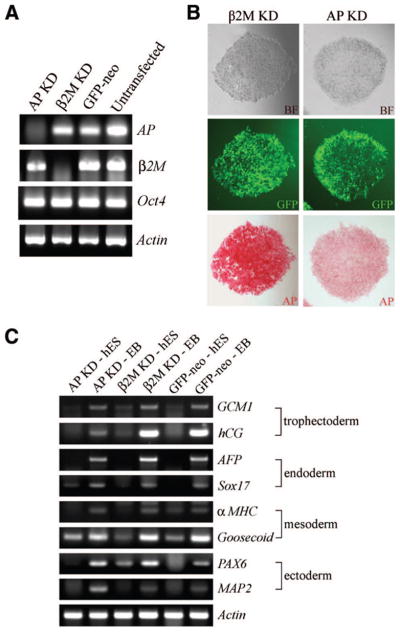
shRNA-mediated knockdown of AP does not affect the expression of other stem cell genes. (A): Reverse transcription (RT)-polymerase chain reaction (PCR) analysis of AP KD and β2M KD hES cells. GFP-neo and untransfected hES cells were used as controls. β-Actin was used a template loading control. (B): BF (top), GFP (middle), and AP (bottom) staining of AP KD and β2M KD hES cells. (C): RT-PCR analysis of AP KD and β2M KD hES cells and EBs. GFP-neo cells served as a control, and β-actin was used a template loading control. Abbreviations: αMHC, alpha myosin heavy chain; AFP, alpha fetoprotein; AP, alkaline phosphatase; BF, bright-field; GCM1, glial cells missing-1; GFP, green fluorescent protein; hES, human embryonic stem; KD, knockdown.
The function of the stem cell marker ALPL (GenBank accession no. AB011406) in hES cell growth and pluripotency is not known. In mice, loss of function of TNAP (AP homolog) has no effect on early development in vivo [34, 35]. We investigated the long-term effects of AP reduction in hES cells by creating stable cell lines transfected with a vector encoding an AP-specific shRNA (AP KD). As a control, we also created β2M-specific shRNA stable cell lines and maintained the cell lines for more than 8 months of continuous culture. shRNA-mediated reduction of AP in hES cells produced a marked decrease in, but not complete loss of, AP activity, as judged by RT-PCR and histochemical staining (Fig. 5A, 5B). The morphology of AP KD cells was indistinguishable from untreated and β2M KD hES cells (Fig. 5B). AP KD hES cells also expressed Oct4 (Fig. 5A), SSEA-3, SSEA-4, TRA1–60, and TRA1–81 (data not shown). AP KD hES cells were still pluripotent, since the cells differentiated into EBs containing cells derived from all three germ layers and formed well-differentiated teratomas (data not shown). Stem cell marker expression and EB differentiation were performed approximately every 2 months and teratoma analyses were performed 7 months after derivation. These data suggest that nucleofection of an AP-specific shRNA reduces expression of the AP gene without affecting hES cell characteristics.
Discussion
In mice, ES cells are used to analyze many aspects of development because of the relative ease with which they can be genetically modified. This ability facilitates studies to delineate gene function, define lineage development, and identify genes that function in self-renewal and pluripotency [36]. hES cells provide the opportunity to carry out similar studies with human cells. Differentiated human cells could have enormous potential utility for disease treatment, drug development, and analysis of development [1–3]. Although a few examples of genetic manipulation of hES cells have been reported, major improvements in the transfection efficiency will be necessary if hES cells are to reach their full potential. Here we describe a method for reproducible, high-efficiency production of genetically modified, clonally derived hES cell lines. Using nucleofection combined with culture conditions that improve the survival of hES cells as single cells, we observed a transient transfection efficiency of up to 85% and a stable transfection efficiency of up to 1.2 in 104 cells.
Previously, results using chemical methods of transfection in hES cells were reported, with transient transfection efficiencies from <1% to 30%, consistent with our own observations ([8, 11, 13, 15]; data not shown). Addition of NTs following chemical transfection (i.e., lipofection) had no effect on transfection efficiency. The efficiency of deriving stably modified hES cell lines appeared to be low, possibly because clumps of hES cells were transfected [8, 9]. In addition to the low transfection efficiency, chemical transfection may not be an effective method for carrying out homologous recombination [37, 38], a widely used technique in mouse genetics [36]. The low efficiency of chemical transfection, in general, has hindered studies of gene function in hES cells and made high-throughput screens to identify genetic pathways all but impossible.
Viruses were also used successfully to introduce genes into hES cells. Lentiviruses produce a very high rate of transient transfection (70%–85%) [16–18]. Here we report similar transient transfection efficiencies using nucleofection (59%–85%). Lentiviral transduction also results in stable transgene expression during prolonged culture. Flow cytometric analyses of stable cell lines demonstrated that 70%–85% of hES cells expressed eGFP from the EF1α promoter after 7–38 weeks in culture [16, 18, 33]. Here we demonstrated similar stable expression of hrGFP from the CMV promoter using nucleofection (87%). Importantly, the use of lentiviruses involves a significant investment in vector development, and important safety concerns must be considered [6]. Also, the DNA insert size in viral vectors is limited, and host immune and inflammatory responses to cells after transduction may occur [19]. The use of CMV-based vectors eliminates many of these problems.
Electroporation is an attractive method for transfecting hES cells because of the utility of this technique in mES cells, relative ease of the method, and its utility for generating homologous recombination events [37, 38]. Electroporation of hES cells resulted in slightly higher transient transfection efficiencies than chemical methods (up to 40%) [8, 10, 11, 13]. However, these reports involved electroporation of clumps of hES cells, likely reducing the gene transfer efficiency and making clonal selection difficult. In addition, such methods make it difficult to determine the gene transfer efficiency because it is difficult to determine the number of cells per clump. Indeed, the reported rates of deriving stable clones were low. For example, only one genetically modified hES cell line was produced using mES cell electroporation conditions [12]. Recently, two stably modified clones were derived following electroporation of 1.5–3.0 × 107 cells, giving an efficiency of one clone in 107 cells [14]. Here we report a stable transfection efficiency of up to one clone in 104 transfected cells, a significant improvement over electroporation methods. Our data suggest that nucleofection of single hES cells followed by growth in NTs is as efficient as lentivirus-mediated transfection for transient modification of hES cells and more efficient than other methods published to date for generating stably modified hES cell lines.
The utility of genetic modification of hES cells using nucleofection was demonstrated in two sets of experiments. First, we performed siRNA- and shRNA-mediated knockdown of two genes involved in maintenance of the pluripotent state, Oct4 and Nanog. In both cases, reduced gene expression resulted in the differentiation of hES cells into trophectoderm-like cells. Isolation of stable cell lines with constitutive reduction in Oct4 and Nanog was not possible, consistent with the idea that these genes are required for hES cell maintenance [17, 25, 27–30]. In the second experiment, the expression of the stem cell marker gene AP was constitutively reduced in hES cells, showing that stable downregulation of AP does not appear to affect hES cell growth or developmental potency, consistent with the conclusions from the mouse studies [34, 35].
An important feature of the work described here is the use of a CMV IE-based vector system to drive gene expression. CMV forms the basis of many widely available vector systems and would enable use of many reagents that already exist (e.g., cDNA/shRNA libraries). Our studies suggest that CMV IE-based systems are expressed effectively in hES cells and can drive long-term gene expression in vitro and in vivo. The ability to manipulate gene function with a commonly used vector system is a major advantage in studies of gene function in hES cells.
A central problem for all methods of genetic modification of hES cells is the difficulty of clonal selection. In most studies, transfection of hES cell clumps makes the process of clonal selection difficult because transfected cells confer resistance to nontransfected cells by communication through gap junctions, and nontransfected hES cells may overgrow recovering, genetically modified cells [25, 39, 40]. Nucleofection of a population of single cells allows the development of true clonal cell lines despite the difficulties normally seen when they are passaged as single cells. The hES cell lines used in this study are routinely passaged by enzymatic dissociation into clumps but gently dissociated by trypsinization to yield a population of single cells for nucleofection. We expect that hES cell lines passaged in a similar manner will yield results equivalent to those reported here. hES cell lines that are passaged exclusively by mechanical dissociation may be more sensitive to gentle trypsinization and may have a higher mortality rate [41, 42]. However, the actions of NTs may ameliorate this effect. The development of true clonal cell lines is important both for determining the effect of regulating gene expression on cell physiology and for development of robust methods for carrying out successful gene targeting. Increasing the efficiency of hES cell transfection methods may also improve efficiency of deriving stable cell lines. Recently, introduction of DNA into Accutase-dissociated cells by nucleofection resulted in the derivation of four clones from 2–4 × 106 cells, representing a stable clone derivation efficiency of one to two clones in 106 cells [11]. In conclusion, we report the derivation of clonally derived stable cells lines with an efficiency of up to one clone in 104 cells. The improved efficiency should greatly facilitate some of the techniques of genetic modification of hES cells, including homologous recombination, gene trapping, and cDNA and siRNA/shRNA library screens. These studies should accelerate efforts to interrogate gene function in hES cells and to define the parameters that control the growth and differentiation of these cells.
Supplementary Material
Acknowledgments
We thank Drs. John Gearhart, Mike Shamblott, Nicholas Christoforou, David Valle, Erika Matunis, Daniel Shain, and Grant MacGregor for helpful suggestions and comments; Scott D’Andrea and Vicki Pomodoro (Amaxa Biosystems) and Rick Marolt (Carl Zeiss Microimaging, Inc., Thornwood, NY, http://www.zeiss.com) for technical help; Jorge Martin and Holly Wellington for technical assistance; and Jeff Bonadio for teratoma analyses. We also thank members of the Donovan and Lock laboratories for assistance and encouragement. We are also grateful to Sean Donovan and Aaron Elliott for their patient support. L.F.L. and P.J.D. dedicate this paper to Dr. Camilynn Brannan (1963–2002). This work was supported in part by funding from NIH (HD49488 and HD46765) (to P.J.D.), funds from University of California Irvine (to P.J.D. and L.F.L.), and by the Human Genetics and Molecular Biology Training Program (NIH 2T32GM07814) at Johns Hopkins University School of Medicine (to K.A.H.). L.F.L. and P.J.D. are co-senior authors.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Pera MF, Reubinoff B, Trounson A. Human embryonic stem cells. J Cell Sci. 2000;113:5–10. doi: 10.1242/jcs.113.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Jones JM, Thomson JA. Human embryonic stem cell technology. Semin Reprod Med. 2000;18:219–223. doi: 10.1055/s-2000-12560. [DOI] [PubMed] [Google Scholar]
- 3.Donovan PJ, Gearhart J. The end of the beginning for pluripotent stem cells. Nature. 2001;414:92–97. doi: 10.1038/35102154. [DOI] [PubMed] [Google Scholar]
- 4.Lerou PH, Daley GQ. Therapeutic potential of embryonic stem cells. Blood Rev. 2005;19:321–331. doi: 10.1016/j.blre.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi N, Rivas-Carrillo JD, Soto-Gutierrez A, et al. Gene delivery to embryonic stem cells. Birth Defects Res C Embryo Today. 2005;75:10–18. doi: 10.1002/bdrc.20031. [DOI] [PubMed] [Google Scholar]
- 6.Yates F, Daley GQ. Progress and prospects: Gene transfer into embryonic stem cells. Gene Ther. 2006;13:1431–1439. doi: 10.1038/sj.gt.3302854. [DOI] [PubMed] [Google Scholar]
- 7.Pyle AD, Lock LF, Donovan PJ. Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol. 2006;24:344–350. doi: 10.1038/nbt1189. [DOI] [PubMed] [Google Scholar]
- 8.Eiges R, Schuldiner M, Drukker M, et al. Establishment of human embryonic stem cell-transfected clones carrying a marker for undifferentiated cells. Curr Biol. 2001;11:514–518. doi: 10.1016/s0960-9822(01)00144-0. [DOI] [PubMed] [Google Scholar]
- 9.Vallier L, Rugg-Gunn PJ, Bouhon IA, et al. Enhancing and diminishing gene function in human embryonic stem cells. Stem Cells. 2004;22:2–11. doi: 10.1634/stemcells.22-1-2. [DOI] [PubMed] [Google Scholar]
- 10.Lakshmipathy U, Pelacho B, Sudo K, et al. Efficient transfection of embryonic and adult stem cells. Stem Cells. 2004;22:531–543. doi: 10.1634/stemcells.22-4-531. [DOI] [PubMed] [Google Scholar]
- 11.Siemen H, Nix M, Endl E, et al. Nucleofection of human embryonic stem cells. Stem Cells Dev. 2005;14:378–383. doi: 10.1089/scd.2005.14.378. [DOI] [PubMed] [Google Scholar]
- 12.Costa M, Dottori M, Ng E, et al. The hESC line Envy expresses high levels of GFP in all differentiated progeny. Nat Methods. 2005;2:259–260. doi: 10.1038/nmeth748. [DOI] [PubMed] [Google Scholar]
- 13.Koch P, Siemen H, Biegler A, et al. Transduction of human embryonic stem cells by ecotropic retroviral vectors. Nucleic Acids Res. 2006;34:e120. doi: 10.1093/nar/gkl674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nolden L, Edenhofer F, Haupt S, et al. Site-specific recombination in human embryonic stem cells induced by cell-permeant Cre recombinase. Nat Methods. 2006;3:461–467. doi: 10.1038/nmeth884. [DOI] [PubMed] [Google Scholar]
- 15.Liew CG, Draper JS, Walsh J, et al. Transient and stable transgene expression in human embryonic stem cells. Stem Cells. 2007;25:1521–1528. doi: 10.1634/stemcells.2006-0634. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Ramezani A, Lewis R, et al. High-level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cells. 2003;21:111–117. doi: 10.1634/stemcells.21-1-111. [DOI] [PubMed] [Google Scholar]
- 17.Zaehres H, Lensch MW, Daheron L, et al. High-efficiency RNA interference in human embryonic stem cells. Stem Cells. 2005;23:299–305. doi: 10.1634/stemcells.2004-0252. [DOI] [PubMed] [Google Scholar]
- 18.Xiong C, Tang DQ, Xie CQ, et al. Genetic engineering of human embryonic stem cells with lentiviral vectors. Stem Cells Dev. 2005;14:367–377. doi: 10.1089/scd.2005.14.367. [DOI] [PubMed] [Google Scholar]
- 19.Worgall S. A realistic chance for gene therapy in the near future. Pediatr Nephrol. 2005;20:118–124. doi: 10.1007/s00467-004-1680-0. [DOI] [PubMed] [Google Scholar]
- 20.Pfeifer A, Verma IM. Gene therapy: Promises and problems. Annu Rev Genomics Hum Genet. 2001;2:177–211. doi: 10.1146/annurev.genom.2.1.177. [DOI] [PubMed] [Google Scholar]
- 21.Hamm A, Krott N, Breibach I, et al. Efficient transfection method for primary cells. Tissue Eng. 2002;8:235–245. doi: 10.1089/107632702753725003. [DOI] [PubMed] [Google Scholar]
- 22.Dityateva G, Hammond M, Thiel C, et al. Rapid and efficient electroporation-based gene transfer into primary dissociated neurons. J Neurosci Methods. 2003;130:65–73. doi: 10.1016/s0165-0270(03)00202-4. [DOI] [PubMed] [Google Scholar]
- 23.Lenz P, Bacot SM, Frazier-Jessen MR, et al. Nucleoporation of dendritic cells: Efficient gene transfer by electroporation into human monocyte-derived dendritic cells. FEBS Lett. 2003;538:149–154. doi: 10.1016/s0014-5793(03)00169-8. [DOI] [PubMed] [Google Scholar]
- 24.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 25.Matin MM, Walsh JR, Gokhale PJ, et al. Specific knockdown of Oct4 and beta2-microglobulin expression by RNA interference in human embryonic stem cells and embryonic carcinoma cells. Stem Cells. 2004;22:659–668. doi: 10.1634/stemcells.22-5-659. [DOI] [PubMed] [Google Scholar]
- 26.Reubinoff BE, Pera MF, Fong CY, et al. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 27.Hay DC, Sutherland L, Clark J, et al. Oct-4 knockdown induces similar patterns of endoderm and trophoblast differentiation markers in human and mouse embryonic stem cells. Stem Cells. 2004;22:225–235. doi: 10.1634/stemcells.22-2-225. [DOI] [PubMed] [Google Scholar]
- 28.Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 29.Chambers I, Colby D, Robertson M, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 30.Mitsui K, Tokuzawa Y, Itoh H, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 31.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Rao S, Chu J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 33.Gropp M, Itsykson P, Singer O, et al. Stable genetic modification of human embryonic stem cells by lentiviral vectors. Mol Ther. 2003;7:281–287. doi: 10.1016/s1525-0016(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 34.MacGregor GR, Zambrowicz BP, Soriano P. Tissue non-specific alkaline phosphatase is expressed in both embryonic and extraembryonic lineages during mouse embryogenesis but is not required for migration of primordial germ cells. Development. 1995;121:1487–1496. doi: 10.1242/dev.121.5.1487. [DOI] [PubMed] [Google Scholar]
- 35.Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432–446. doi: 10.1002/(SICI)1097-0177(199703)208:3<432::AID-AJA13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 36.Brault V, Pereira P, Duchon A, et al. Modeling chromosomes in mouse to explore the function of genes, genomic disorders, and chromosomal organization. PLoS Genet. 2006;2:e86. doi: 10.1371/journal.pgen.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasquez KM, Marburger K, Intody Z, et al. Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci U S A. 2001;98:8403–8410. doi: 10.1073/pnas.111009698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- 39.Huettner JE, Lu A, Qu Y, et al. Gap junctions and connexon hemichannels in human embryonic stem cells. Stem Cells. 2006;24:1654–1667. doi: 10.1634/stemcells.2005-0003. [DOI] [PubMed] [Google Scholar]
- 40.Wong RC, Dottori M, Koh KL, et al. Gap junctions modulate apoptosis and colony growth of human embryonic stem cells maintained in a serum-free system. Biochem Biophys Res Commun. 2006;344:181–188. doi: 10.1016/j.bbrc.2006.03.127. [DOI] [PubMed] [Google Scholar]
- 41.Reubinoff BE, Pera MF, Fong C, et al. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–403. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 42.Inzunza J, Gertow K, Stromber MA, et al. Derivation of human embryonic stem cell lines in serum replacement medium using postnatal human fibroblasts as feeder cells. Stem Cells. 2005;23:544–549. doi: 10.1634/stemcells.2004-0201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.