

Summary
Background
Tks5 regulates invadopodium formation, but the precise timing during invadopodium lifetime (initiation, stabilization, maturation) when Tks5 plays a role is not known.
Results
We report new findings based on high-resolution spatiotemporal live-cell imaging of invadopodium precursor assembly. Cortactin, N-WASP, cofilin, and actin arrive together to form the invadopodium precursor, followed by Tks5 recruitment. Tks5 is not required for precursor initiation but is needed for precursor stabilization, which requires the interaction of the phox homology (PX) domain of Tks5 with PI(3,4)P2. During precursor formation, PI(3,4)P2 is uniformly distributed but subsequently starts accumulating at the precursor core 3–4 min after core initiation, and conversely, PI(3,4,5)P3 gets enriched in a ring around the precursor core. SHIP2, a 5′-inositol phosphatase, localizes at the invadopodium core and regulates PI(3,4)P2 levels locally at the invadopodium. The timing of SHIP2 arrival at the invadopodium precursor coincides with the onset of PI(3,4)P2 accumulation. Consistent with its late arrival, we found that SHIP2 inhibition does not affect precursor formation but does cause decreases in mature invadopodia and matrix degradation, whereas SHIP2 overexpression increases matrix degradation.
Conclusions
Together, these findings lead us to propose a new sequential model that provides novel insights into molecular mechanisms underlying invadopodium precursor initiation, stabilization, and maturation into a functional invadopodium.
Introduction
Invadopodia are actin-rich protrusive structures of cancer cells, 0.05–1 μm in diameter and 0.5–10 μm long [1–3]. The primary function of these structures is to degrade extracellular matrix (ECM), creating a passage that cancer cells can utilize during metastasis [4–6], the leading cause of death among cancer patients. Invadopodia are characterized by a core containing several proteins, including cortactin, Tks5, cofilin, N-WASP, Nck1, Arp2/3, p190RhoGAP, F-actin, and MT1-MMP [2, 7–9], that is surrounded by a ring of other proteins: integrin α5 [10], Hic-5, paxillin, vinculin [6, 11], p190RhoGEF, and RhoC activity [2]. One of the invadopodial core proteins, Tks5, is essential for invadopodia and podosome formation in many different cell types [12–16]. Tks5 directly binds to the ADAM family proteases [12], N-WASP, dynamin-2, and Grb2 [14, 15]. In vivo, decreased Tks5 expression correlates with reductions in tumor growth, metastasis, and angiogenesis [17].
Tks5 contains a phox homology (PX) domain that binds the membrane phosphoinositides PI(3)P and PI(3,4)P2 [12]. In Src-transformed NIH 3T3 cells, Tks5 and PI(3,4)P2 localize to podosomes via a Grb2-dependent mechanism [14]. However, podosomes and invadopodia are distinct matrix-degrading structures with differences in molecular composition, dynamics, and structure [18–20]. In particular, invadopodia in carcinoma cells contain the Nck1 adaptor protein but lack Grb2 [20]. Furthermore, both localization of PI(3,4)P2 at invadopodia and the precise timing of Tks5 arrival during invadopodium assembly are unknown, and their involvement in invadopodium assembly and maturation is not well described. Furthermore, integrin-associated podosomes do not move laterally in the cell membrane [14], whereas invadopodia in carcinoma cells show rapid lateral movement there [9]. These differences suggest differences between the molecular regulation of invadopodium and podosome assembly and membrane linkage as well as differences in the regulation of the interaction between the PX domain of Tks5 and PI(3,4)P2.
So far, ten mammalian 5′-phosphatases have been identified [21], and one of these 5′-phosphatases, synaptojanin-2, was shown to regulate invadopodium formation in glioblastoma cells [22]. SHIP2 overexpression was recently shown to increase invadopodia number and matrix degradation in head and neck cancer cells [23]. Therefore, studying the spatiotemporal localization and dynamics of these 5′-phosphatases and how they regulate PI(3,4)P2 levels would yield important insights into the molecular mechanisms of invadopodium formation.
Previous studies have demonstrated that invadopodium assembly is a sequential process [7, 8], starting with the formation of an invadopodium precursor that matures into a degradation-competent invadopodium, but relatively little is known about the early and intermediate stages of maturation during which the invadopodium precursor is anchored to the membrane and stabilized. In this study, we investigate the order of arrival of different invadopodium core proteins (Tks5, cortactin, N-WASP, cofilin, actin, and SHIP2) and PI(3,4)P2 during invadopodium precursor assembly and maturation using high-resolution live-cell imaging. Our results yield novel insights into how invadopodial core proteins and phospholipids cooperate to stabilize the invadopodium precursor, a key step for carcinoma invasion and metastasis.
Results
Arrival Kinetics of Invadopodium Core Proteins during Precursor Assembly
Given the key roles of invadopodia during carcinoma invasion and metastasis, it is critical to understand the order of arrival of core invadopodial proteins during invadopodium precursor assembly in carcinoma cells. We chose highly metastatic rat mammary adenocarcinoma MTLn3 cells as a model because these cells use epidermal growth factor (EGF)-dependent invadopodia [9] to invade and disseminate metastatic tumors in vivo [5]. Furthermore, they make robust invadopodia when plated on gelatin and/or fibronectin substrates in vitro, and EGF stimulation of these cells induces the formation of invadopodium precursors that mature and go on to degrade the underlying matrix [2, 8]. To study relative arrival of core invadopodial proteins in newly forming invadopodium precursors, we stimulated MTLn3 cells with EGF and performed high-resolution live-cell fluorescence imaging, with frames every 3 s. First, we transfected cells with TagRFP-cortactin and GFP-Tks5 and plated them on thin gelatin matrix [8, 24]. Within 1 min of EGF stimulation, these cells formed many invadopodium precursors identified as cortactin- and Tks5-rich puncta lacking protease activity (Figure 1A; see also Movie S1 available online). We analyzed the dynamics of fluorescence signal at the site of newly forming invadopodium precursors and found that cortactin localization preceded Tks5 localization (Figure 1B). We tracked newly forming invadopodium precursors in cortactin and Tks5 channels using the “Invadopodia Tracker” ImageJ plugin [25] (Figure S1A; Movie S2) and quantified the relative arrival of Tks5 with respect to cortactin; we found that Tks5 appears approximately 20 s after cortactin arrival (Figure 1G). To determine the relative arrival of N-WASP, cofilin, and actin with respect to cortactin, we transfected cells with their plasmids and repeated time-lapse imaging as described above (Figures 1C and 1E; Movies S3 and S4), and we found that N-WASP, cofilin, and actin appear at the same time as cortactin (Figures 1D, 1F, and S1B). Tracking of invadopodium precursors and quantification confirmed that there is no delay in N-WASP, cofilin, or actin arrival at the precursor compared to cortactin (Figure 1G).
Figure 1. Arrival Kinetics of Invadopodium Precursor Core Proteins during Precursor Assembly.

(A, C, and E) MTLn3 cells showing cortactin- and Tks5- (A), cortactin- and N-WASP- (C), and cortactin- and cofilin- (E) positive invadopodia before (top panels) and after (bottom panels) EGF stimulation (see Movies S1, S3, and S4). White arrows in the merge images point to examples of newly assembled invadopodium precursors. Scale bars represent 10 μm.
(B, D, and F) Time-lapse montage of cortactin and Tks5 (B), cortactin and N-WASP (D), and cortactin and cofilin (F) fluorescence channels for newly assembled invadopodium precursors. Red circles indicate the first appearance of precursor puncta in each channel. Time 0:00 corresponds to the EGF addition.
(G) Tks5, N-WASP, cofilin, and actin arrival delays with respect to cortactin during invadopodium precursor assembly. Red lines indicate mean with 95% confidence interval. n = 17 (Tks5), 46 (N-WASP), 45 (cofilin), and 26 (actin) invadopodium precursors.
See also Figure S1.
Next, we investigated the site where invadopodium precursor assembly takes place. We found that some invadopodium precursors initiate at the proximal tip of focal adhesions (Figures S1C and S1D; Movie S5), consistent with observations made for podosome initiation [14, 26]. However, some invadopodium precursors form with no nearby focal adhesion (Figure S1E). In addition, unlike podosomes, which remain stationary, some invadopodia formed at focal adhesions are motile (Figure S1F; [9]).
Tks5 Is Not Required for Invadopodium Precursor Initiation
Because Tks5 arrives later at the invadopodium precursor than other core components do, we speculated that Tks5 might not be important for invadopodium precursor assembly. To test this hypothesis, first we knocked down Tks5 using siRNA, and we found an efficient Tks5 reduction (~85%–90%) (Figures S2A and S2B) with low cell-to-cell variability in Tks5 KD levels (Figure S2C). To check Tks5 KD effect on invadopodium precursor formation, we stimulated control and Tks5 siRNA-treated cells with EGF for 1 min (the earliest time that corresponds to a burst in precursor formation; [8]) and fixed and stained them with N-WASP and cortactin antibodies to identify invadopodium precursors. The number of invadopodium precursors in Tks5 KD cells compared to the control cells was unchanged (Figures 2A and 2B). We repeated this experiment with a different set of invadopodium markers (cortactin, cofilin, and Arp2) and again found that the number of invadopodium precursors is unaffected by Tks5 KD (Figure S2D). These results indicate that Tks5 is not required for initial assembly of invadopodium precursors in carcinoma cells.
Figure 2. Tks5 Is Dispensable for Invadopodium Precursor Initiation but Is Required for Precursor Stability.

(A) Control or Tks5 siRNA-treated cells were stimulated with 5 nM EGF for 1 min and stained with N-WASP and cortactin antibodies to identify invadopodium precursors (shown by white arrows).
(B) Quantification of invadopodium precursors (means ± SEM) in control and Tks5 siRNA-treated cells. n = 19 (control siRNA) and 22 (Tks5 siRNA) cells.
(C) Stills from Movie S6 showing invadopodia stability. White arrows in control siRNA panel point to a stable invadopodium, whereas white arrows in Tks5 siRNA panels point to examples of unstable invadopodium precursors.
(D) Invadopodia lifetime histograms for control and Tks5 siRNA-transfected cells. Note the complete absence of stable invadopodia (lifetime > 60 min) in Tks5 siRNA cells. n = 49 (control siRNA) and 60 (Tks5 siRNA) invadopodia.
(E and F) Representative invadopodium trajectories in control (E) and Tks5 siRNA-treated (F) cells. Numbers beside each trajectory indicate lifetime (min).
(G) Quantification of invadopodium motility as net distance per lifetime (means ± SEM). n = 39 (control siRNA) and 49 (Tks5 siRNA) invadopodia.
See also Figure S2.
Tks5 Is Required for Invadopodium Precursor Stability
Although Tks5 is not required for invadopodium precursor initiation, previous studies indicated that Tks5 KD reduced the number of invadopodia/podosomes and degradation area in many cell types [13–15]. Therefore, we hypothesized that Tks5 might be important for invadopodium precursor stability and required for their maturation into functional invadopodia [8]. We therefore performed long-duration time-lapse imaging (one frame every 2 min for 10 hr) of control and Tks5 siRNA-treated cells, with TagRFP-cortactin as an invadopodium marker. We found that compared to many stable invadopodia in control cells, most of the invadopodia in Tks5 siRNA cells were short lived (Figure 2C; Movie S6). Lifetime quantification showed that control siRNA-treated cells had invadopodia with a variety of lifetimes, ranging from very short lived (<10 min), some staying tens of minutes, to a very stable population (~40%) staying for more than 1 hr (Figure 2D). In contrast, most of the invadopodia in Tks5 siRNA-treated cells were unstable, with more than 50% of the invadopodia exhibiting a lifetime of less than 10 min and none remaining longer than 1 hr (Figure 2D). To examine individual invadopodia motility, we plotted their x-y trajectories (Figures 2E and 2F) and quantified net distance traveled by each invadopodium divided by its lifetime [9]. This ratio was higher for invadopodia in Tks5 siRNA cells compared to those in control cells (Figure 2G), indicating that invadopodia in Tks5 siRNA-treated cells are more motile than those in control cells. Together, these results indicate that Tks5 is necessary for stabilizing invadopodium precursors and helping to localize their positions.
To check whether ECM degradation and protrusion into the matrix might be required for invadopodium precursor stability, we treated cells with GM6001, a pan-MMP inhibitor, to block matrix degradation (Figure S2E) and examined invadopodia motility. We found that GM6001-treated cells show invadopodia lifetime distributions and motility similar to DMSO control (Figures S2F–S2I), suggesting that ECM degradation is not a requirement for precursor stability. Recently, reactive oxygen species (ROS) were implicated in podosome/invadopodia formation through a Tks5-dependent pathway [27]. To examine this, we treated cells with a widely used ROS inhibitor, diphenyleneiodonium chloride (DPI), and assayed for invadopodia formation and matrix degradation. We found that cells treated with DPI concentration as high as 20 μM made robust invadopodia and degraded matrix, similar to DMSO control (Figure S2J), suggesting that ROS does not play a role in invadopodium precursor formation and stability in MTLn3 cells.
Interaction of the PX Domain of Tks5 with PI(3,4)P2 Stabilizes Invadopodium Precursors
Because the PX domain of Tks5 binds PI(3)P and PI(3,4)P2 [12] and PI(3,4)P2, but not PI(3)P, localizes at podosomes in Src-transformed NIH3T3 cells [14], we hypothesized that the inter-action of the PX domain of Tks5 with PI(3,4)P2 is required for invadopodium precursor stability and maturation. To test this, we generated a PI(3,4)P2 binding-deficient Tks5-R42A/R93A mutant (Figure S3A) [12, 14]. We performed KD-rescue experiments, in which we depleted endogenous Tks5 and rescued with either GFP or RNAi-resistant GFP-Tks5 or GFP-Tks5-R42A/R93A. Western blot analysis (Figure 3A) showed efficient endogenous Tks5 KD (~85%–90%) in all Tks5 siRNA samples. GFP-Tks5 was expressed at ~30%–40% of endogenous Tks5 and was found to be sufficient to rescue the invadopodia number and degradation area to levels seen with control siRNA (Figures S3B–S3G). The mutant was also expressed at similar levels (~30%–40% of endogenous Tks5). Consistent with observations made for invadopodia/podosomes in other cell types [13–15], we also found that Tks5 KD in MTLn3 cells leads to a dramatic decrease in the number of mature invadopodia (cortactin- and Tks5-positive puncta colocalizing with degradation hole) and degradation area. This decrease was rescued with wild-type Tks5 but not with the lipid binding-deficient Tks5-R42A/R93A mutant (Figures S3F and S3G). These results demonstrate that the interaction of Tks5 with PI(3,4)P2 through its PX domain is necessary for precursor maturation into functional invadopodia.
Figure 3. Binding of the PX Domain of Tks5 with PI(3,4)P2 Is Required for Invadopodium Precursor Stability and Maturation.

(A) Western blot showing Tks5 KD-rescue in all the four conditions.
(B–E) Stills from Movie S7 for the four Tks5 KD-rescue conditions, showing precursor stability. Note that EGF stimulates invadopodium precursor formation in all conditions (column “3 min after EGF”) but that precursors are stable only in conditions (B) and (D) and disappear in conditions (C) and (E) (column “30 min after EGF”). Scale bar represents 10 μm.
(F) Quantification of cells showing newly forming invadopodium precursors that were stable (i.e., remained until the end of the movie, 30 min). Pairwise statistical comparisons were made using Fisher’s exact test in GraphPad Prism. n = 48, 34, 37, and 59 cells analyzed for the four Tks5 KD-rescue conditions, respectively. The difference between Tks5 KD+GFP and Tks5KD+R42A/R93A was not significant (p = 0.154).
See also Figure S3.
To visualize invadopodium precursor stability directly, we performed high-resolution live-cell time-lapse imaging of cells under the four Tks5 KD-rescue conditions. Cells were stimulated with EGF to induce precursor initiation and followed for 30 min to assess their stability (Figures 3B–3E; Movie S7). Tks5 KD led to a decrease in the number of cells with new invadopodium precursors that were stable, which was rescued with the wild-type Tks5, but not with Tks5-R42A/R93A mutant (Figure 3F). These results are the first direct evidence that the interaction of the PX domain of Tks5 with PI(3,4)P2 is required for invadopodium precursor stability.
We observed that the Tks5-R42A/R93A mutant localizes at invadopodium precursors (Figure S3E). Quantification showed a 75% reduction in the mean fluorescence intensity of mutant compared to wild-type Tks5 (Figure S3H), indicating that although the mutant can get recruited to precursors, possibly via N-WASP binding [14], the precursor cannot grow further unless it is stabilized by Tks5-PI(3,4)P2 interaction.
PI(3,4)P2 Arrival Kinetics during Invadopodium Precursor Assembly
We next examined PI(3,4)P2 localization in MTLn3 cells with a PI(3,4)P2-specific probe, TAPP1-PH [28]. We found that PI(3,4)P2 localizes at the invadopodium core (Figure 4A), with 15% more enrichment at the core compared to the surrounding region (Figure 4E). Since Tks5 also binds PI(3)P [12], we checked PI(3)P localization using a PI(3)P-specific probe, 2xHrs-FYVE [29], and found that PI(3)P did not localize to the invadopodium core (Figures 4B and 4E). We also examined PI(4,5)P2 localization using a PI(4,5)P2-specific probe, YFP-PH-PLCδ1 [29]. PI(4,5)P2 localization was not observed at the invadopodium core, but the signal was enriched at the plasma membrane and sometimes adjacent to invadopodia (Figure 4C), although quantification showed no significant PI(4,5)P2 accumulation around the core (Figure 4E). We confirmed the endogenous PI(3,4)P2 and PI(4,5)P2 localizations with phospholipid-specific antibodies [30, 31] and found similar results (Figures S4A and S4B).
Figure 4. PI(3,4)P2 Accumulation at the Invadopodium Precursor Is a Late Event.

(A–D) Cells were transfected with GFP-TAPP1-PH (A), Venus-2xHrs-FYVE (B), GFP-PLCδ1-PH (C), or GFP-Akt-PH (D), plated on 405-gelatin matrix, and stained with cortactin and Tks5 antibodies to identify invadopodia. Insets show zoom images of invadopodia in each panel. Scale bars represent 10 μm.
(E) Quantification of phospholipid fluorescence intensities (means ± SEM) at the invadopodium core/surrounding region. n = 21 (TAPP1-PH), 24 (2xHrs-FYVE), 21 (PLCδ1-PH), and 19 (Akt-PH) invadopodia.
(F) Time-lapse montage of PI(3,4)P2 accumulation kinetics during invadopodium precursor assembly by TIRF microscopy. Red circles indicate the first frame of cortactin and PI(3,4)P2 appearance. Time 0 corresponds to the appearance of the cortactin punctum.
(G) Quantification of PI(3,4)P2 accumulation delay with respect to cortactin. Red lines indicate mean with 95% confidence interval. n = 44 precursors.
See also Figure S4.
Because the amount of PI(4,5)P2 (in the mM range) in cells is orders of magnitude higher than the amount of PI(3,4,5)P3 and PI(3,4)P2 (both in the μM range) [32], it is conceivable that any changes in PI(4,5)P2 levels, due to PI(3,4,5)P3 and subsequent PI(3,4)P2 production, would be negligible compared to changes observed in PI(3,4)P2 and PI(3,4,5)P3. To investigate this further, we looked at PI(3,4,5)P3 localization at the invadopodium using two PI(3,4,5)P3-specific markers, Akt-PH [33] and Grp1-PH [29]. Both markers showed a distinct PI(3,4,5)P3 ring around the core and a ratio of fluorescence intensities for the core/surrounding region less than 1 (Figures 4D, 4E, and S4C). Therefore, unlike PI(3,4)P2, which is enriched at the core, PI(3,4,5)P3 is found enriched in a ring around the core, and PI(4,5)P2 patches are seen in close proximity to invadopodia.
To investigate the timing of PI(3,4)P2 recruitment to the plasma membrane during precursor assembly, we used total internal reflection fluorescence (TIRF) microscopy to image TagRFP-cortactin- and GFP-TAPP1-PH-transfected cells after EGF stimulation. We found no enrichment of PI(3,4)P2 at the invadopodium precursor; rather, it was homogeneously distributed throughout the first 3 min of precursor assembly (Figure S4D). To investigate the possibility that PI(3,4)P2 accumulation at the precursor is a late event, we extended our TIRF imaging rate to frames every 1 min and found that PI(3,4)P2 started accumulating at the precursors approximately 4 min after cortactin arrival (Figures 4F and 4G). These results indicate that low basal levels of PI(3,4)P2 support initial precursor assembly, followed by an enrichment of PI(3,4)P2 at the invadopodium as it matures.
PI(3,4)P2 Sequestration Leads to a Dose-Dependent Decrease in the Number of Invadopodia and Matrix Degradation
To investigate whether PI(3,4)P2 is required for invadopodium formation, we used the overexpression of TAPP1-PH as a strategy for PI(3,4)P2 sequestration and thereby depletion of PI(3,4)P2. Similar strategies have previously been used for the sequestration of PI(4,5)P2 and PI(3,4,5)P3 [33–35]. We found that cells expressing high levels of TAPP1-PH and thereby sequestering most of the free PI(3,4)P2 did not form any invadopodia and failed to degrade matrix (a phenotype similar to Tks5 KD; Figures S3F and S3G), whereas cells expressing low to moderate levels of TAPP1-PH formed normal invadopodia and degraded matrix (Figures 5A–5C). TAPP1-PH overexpression was also shown to inhibit podosomes in Src-transformed cells [14]. These results indicate that, like Tks5, free PI(3,4)P2 levels are important for invadopodium formation and maturation. To specifically check the role of PI(3,4) P2 in precursor formation, we measured invadopodium precursors in low- and high-TAPP1-PH-expressing cells and found no difference (Figure 5D), suggesting that PI(3,4)P2 primarily plays a role in stabilizing the precursor, not in its initiation.
Figure 5. PI(3,4)P2 Sequestration Causes Significant Reductions in Invadopodium Formation and Matrix Degradation.

(A) Cells were transiently transfected with GFP-TAPP1-PH, plated on 405-gelatin, and stained with cortactin and Tks5 antibodies. An area with cells having varying expression levels of TAPP1-PH is shown using rainbow lookup table (LUT). Cell outlines with no (black), low (purple), moderate (cyan), and high (red) TAPP1-PH levels are shown. Scale bar represents 10 μm.
(B and C) Scatterplots showing the effect of TAPP1-PH overexpression on cells’ ability to form invadopodia (B) and degrade matrix (C). Red lines are linear regression fit. n = 73 cells.
(D) Quantification of invadopodium precursors per cell (means ± SEM) in low (lower 25th percentile) and high (upper 25th percentile) TAPP1-PH-expressing cells, indicating that PI(3,4)P2 sequestration does not affect precursor formation. n = 20 (low) and 20 (high) cells.
SHIP2 Localizes at the Invadopodium Core, and Its Arrival Coincides with PI(3,4)P2 Accumulation
PI(3,4)P2 is primarily made in cells via dephosphorylation of PI(3,4,5)P3 by a 5′-inositol phosphatase [21] (Figure S5A). Recently, it was reported that SHIP2 is highly expressed in breast cancer cells, enhances metastasis [36], and leads to an increase in invadopodia and degradation area [23]; however, the localization of endogenous SHIP2 at invadopodia and whether SHIP2-mediated PI(3,4)P2 regulation is localor global have not been studied. Therefore, we checked whether MTLn3 cells express SHIP2. Western blots showed that these cells do indeed express SHIP2, but not SHIP1 (Figure 6A), a closely related isoform. They also do not express synaptojanin-2 (Figure S5B), another 5′-phosphatase, implicated previously in the regulation of invadopodia and podosomes [14, 22]. Next, we examined the localization of endogenous SHIP2 using a SHIP2 antibody and found that SHIP2 localizes at the invadopodium core (Figure 6B). This localization was further confirmed by localization of a GFP-SHIP2 construct (Figure 6C).
Figure 6. SHIP2 Arrival Coincides with the Beginning of PI(3,4)P2 Accumulation at the Invadopodium Precursor.
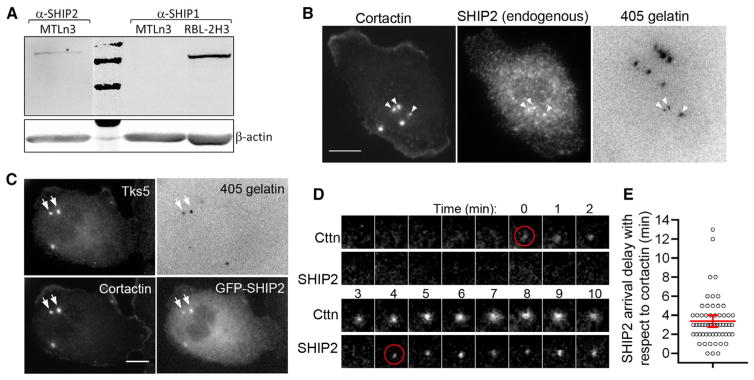
(A) MTLn3 whole-cell lysate was analyzed for the SHIP2 and SHIP1 expression levels by western blotting. Rat hematopoietic RBL-2H3 whole-cell lysate was used as a positive control for SHIP1. Second lane in the top membrane shows 250, 150, and 100 kDa marker bands.
(B) Endogenous SHIP2 distribution checked by staining cells with cortactin and SHIP2 antibodies. White arrowheads show the colocalization of SHIP2, cortactin, and degradation holes. Scale bar represents 10 μm.
(C) Cells were transfected with GFP-SHIP2 and stained with cortactin and Tks5 antibodies. White arrows show colocalization of SHIP2, cortactin, Tks5, and degradation holes. Scale bar represents 10 μm.
(D) Time-lapse montage of SHIP2 arrival kinetics during invadopodium precursor assembly by TIRF microscopy. Red circles indicate the first frame of cortactin and SHIP2 appearance. Time 0 corresponds to the appearance of the cortactin punctum.
(E) Quantification of SHIP2 arrival delay with respect to cortactin. Red lines indicate mean with 95% confidence interval. n = 60 precursors.
See also Figure S5.
To investigate the SHIP2 arrival kinetics during invadopodium precursor formation, we imaged TagRFP-cortactin- and GFP-SHIP2-transfected cells stimulated with EGF by TIRF microscopy. We did not observe SHIP2 arrival during the first 3 min of precursor assembly (Figure S5C). Therefore, we extended our TIRF imaging rate to frames every 1 min and found that SHIP2 arrived at the invadopodium precursor approximately 3–4 min after cortactin arrival (Figures 6D and 6E), the time window coinciding with the start of PI(3,4)P2 accumulation. Simultaneous arrival of SHIP2 and PI(3,4)P2 was further confirmed by imaging GFP-SHIP2 and RFP-TAPP1-PH in the same cell (Figures S5D and S5E). Furthermore, we observed a 10% decrease in PI(3,4,5)P3 levels at the core/surrounding region after SHIP2 arrival (Figures S5F and S5G). Taken together, these results strongly suggest that newly forming PI(3,4)P2 at the invadopodium precursor is generated from PI(3,4,5)P3 by local SHIP2 activity arriving after the initial core protein assembly at the precursor.
SHIP2 Regulates Invadopodium Precursor Maturation by Modulating PI(3,4)P2 Levels Locally at the Invadopodium
To investigate whether SHIP2 phosphatase activity regulates invadopodium precursor formation and maturation, we treated cells with a specific SHIP2 inhibitor, AS1949490 [37]. We found that PI(3,4)P2 levels in the whole cell and at the invadopodia decreased by 50% (Figures S5H–S5J). Furthermore, cells treated with SHIP2 inhibitor showed no change in invadopodium precursors, but mature invadopodia and degradation area were significantly decreased (Figures 7A–7D). Similar results were obtained when SHIP2 was depleted with shRNA (Figures S6A–S6E). SHIP2 inhibition also showed a 33% decrease in average invadopodium lifetime (Figure 7E), by increasing the number of short-lived (1–10 min) invadopodia and decreasing the number of long-lived (>1 hr) invadopodia (Figure 7F). Moreover, SHIP2 inhibition led to a 50% decrease in Matrigel invasion of cells (Figure S6F), an effect that can be attributed to similar reduction observed in matrix degradation ability of these cells under SHIP2 inhibition (Figures 7D and S6E). As an alternative, to check the role of SHIP2 phosphatase activity in PI(3,4)P2 generation and matrix degradation, we overexpressed either wild-type SHIP2 or phosphatase-inactive SHIP2-D608A mutant [38] in MTLn3 cells. We found that both PI(3,4)P2 levels and matrix degradation were increased approximately 2.5-fold with wild-type SHIP2, but not with SHIP2-D608A mutant (Figures S6G and S6H).
Figure 7. SHIP2 Regulates Invadopodia Lifetime and Maturation.

(A) DMSO-treated or 10 μM SHIP2 inhibitor AS1949490-treated cells were plated on 405-gelatin and stained with cortactin and Tks5 antibodies to identify invadopodia. Scale bar represents 20 μm.
(B–D) Quantification of invadopodium precursors per cell; n = 91 (DMSO) and 67 (SHIP2 inhibitor) cells (B); mature invadopodia per cell, n = 91 (DMSO) and 67 (SHIP2 inhibitor) cells (C); and degradation area per field of view, n = 25 (DMSO) and 26 (SHIP2 inhibitor) fields of view (D). Values shown are means ± SEM.
(E) Average invadopodia lifetime (means ± SEM) in DMSO-treated and 10 μM SHIP2 inhibitor-treated cells. n = 225 (DMSO) and 208 (SHIP2 inhibitor) invadopodia in (E) and (F).
(F) Invadopodia lifetime distributions in cells treated with DMSO or 10 μM SHIP2 inhibitor.
(G) A sequential model of invadopodium precursor assembly. Cofilin, cortactin, actin, and N-WASP arrive together to form the invadopodium precursor, followed by Tks5 arrival, which stabilizes the precursor by binding to basal PI(3,4)P2 at the plasma membrane. This is followed by a burst in actin barbed-end activity [13] and recruitment of SHIP2 at the precursor, which results in local PI(3,4)P2 production at the precursor. Increased PI(3,4)P2 production supports a growing invadopodium precursor, leading to its maturation and matrix degradation.
See also Figures S5 and S6.
Taken together, these results, consistent with the late arrival (3–4 min after precursor initiation) of SHIP2, suggest that SHIP2 phosphatase activity plays a critical role in modulating PI(3,4)P2 levels, invadopodium stability, and maturation, but not in precursor initiation.
The Invadopodium Is a Highly Dynamic Structure
Because some invadopodium precursors were found to initiate at the proximal tip of focal adhesions (Figures S1C–S1D; Movie S5), we further investigated the role of focal adhesions in invadopodium stability by performing high-resolution time-lapse analysis. We found that stable invadopodia were associated with a ring of focal adhesion proteins (vinculin and talin) around the invadopodium core (Figures S6I and S6J; Movie S8); therefore, we think that focal adhesion rings are important for invadopodium anchoring and stability, similar to observations made for invadopodia in head and neck cancer cells [11]. Interestingly, we saw oscillations in focal adhesion ring proteins occurring in synchrony with cortactin oscillations (time period, 10 min) (Figure S6K), and similar to cortactin-cofilin oscillations observed in invadopodia in MDA-MB-231 cells during the dynamic extension/retraction cycle of invasive protrusions [3]. Therefore, not only are the recruitment order and timing of invadopodium core proteins and PI(3,4)P2, observed here in invadopodia, different from those in podosomes [14, 39], there are key differences in the dynamics of these two structures: podosomes do not move laterally in the cell membrane and have not been shown to display oscillations in assembly and protrusion [14], whereas invadopodia in carcinoma cells do both (Figures S1F and S6K; Movie S8; [3, 9]).
Discussion
A Model for Carcinoma Cell-Specific Invadopodium Assembly and Regulation
Based on the results reported here for carcinoma cells, we propose a novel sequential model of invadopodium assembly (Figure 7G). During invadopodium precursor initiation, cortactin acts as a scaffold to bring cofilin and N-WASP together to form the initial invadopodium precursor core. Since actin was found to arrive together with cortactin, the cortactin-actin complex may also act as a scaffold to bring in cofilin and N-WASP. This initial assembly step is followed by the arrival of Tks5, possibly via binding to N-WASP [14]. PI(3,4)P2 does not participate during this initial Tks5 recruitment to precursor complex, as the PI(3,4)P2 binding-deficient Tks5 mutant retains the ability to localize to precursors and PI(3,4)P2 sequestration does not affect precursor formation. Because PI(3,4)P2 is distributed homogeneously at the plasma membrane and no PI(3,4)P2 enrichment is seen at the site of precursor assembly until 3–4 min after the start of precursor formation, we propose that initially precursor binds weakly to the plasma membrane via the interaction of the PX domain of Tks5 with basal PI(3,4)P2 and is possibly aided by the association of N-WASP with PI(4,5)P2 [40]. This is followed by an increase in actin barbed-end activity and polymerization [8] and recruitment of SHIP2 to the invadopodium precursor. SHIP2 recruitment leads to PI(3,4)P2 enrichment at the invadopodium precursor, which stabilizes the precursor core by association with PI(3,4)P2 via Tks5 binding. This last step has the effect of priming the precursor for maturation and matrix degradation as described in previous studies (Figure 7G; [2, 3, 8, 13]).
This new invadopodium-specific model is attractive because it helps explain the more dynamic behavior of invadopodia in carcinoma cells as compared to podosomes in other cell types. For example, since the binding interaction between Tks5 and PI(3,4)P2 is reversible and this destabilizes the precursor, anchorage of the precursor to the membrane as well as core protein composition can oscillate under the influence of changes in EGFR and NHE1 signaling, as shown previously [3]. In addition, the proposed model is consistent with the recently reported role of PI3K signaling in invadopodium formation [23, 33], another rapidly reversible signaling pathway.
Differences between the Carcinoma Cell-Specific Invadopodium Model and Previous Work
In podosomes, the assembly order of the core relative to the arrival of Tks5 is different from that observed here for invadopodia. In fibroblast podosomes, Tks5 forms a complex with Grb2 and PI(3,4)P2 in advance of the recruitment and assembly of the core proteins [14]. In the smooth muscle cell podosomes, Tks5 is proposed to be a key scaffold for the recruitment of the core proteins [39]. However, our results in carcinoma cell invadopodia indicate that the cortactin-actin complex acts as the initial scaffold that brings together N-WASP and cofilin to form the invadopodium precursor core complex. Numerous studies have demonstrated that cortactin can directly bind to cofilin [8] and N-WASP [41]. Only after the precursor assembles does Tks5 get recruited to it, possibly via N-WASP interaction [14].
Another difference with previous work is the absence of PI(4,5)P2 localization at the invadopodium core. The absence of PI(4,5)P2 at the invadopodium core is consistent with the observation made for the podosomes of Src-transformed NIH 3T3 cells [14]. However, PI(4,5)P2 localization was reported at invadopodia in human MDA-MB-231 cells [35]. On closer examination of Figure 1 in [35], we found that our data are consistent with that previous study in the sense that PI(4,5)P2 does not localize at the invadopodium core but PI(4,5)P2 patches are seen in close proximity around the invadopodium core, as seen in our study. This suggests that PI(4,5) P2 may play a role in recruitment of PI(4,5)P2 binding proteins, such as N-WASP, to the vicinity of the assembling precursor core but that it does not directly function in the final anchoring of the core complex to the membrane.
SHIP2 Recruitment and the Role of Other 5′-Phosphatases in PI(3,4)P2 Generation at Invadopodia
SHIP2 has been shown to interact with a number of molecules—p130Cas [42], RhoA [43], filamin [44], intersectin [38], and vinexin [45]. Any one of these molecules is a potential candidate for SHIP2 recruitment to invadopodium precursors. Among these molecules, RhoA is not active at the invadopodium core in MTLn3 cells [2], and knockdown of p130Cas using siRNA showed no difference in invadopodia formation and cells’ ability to degrade matrix (data not shown). Therefore, we propose that filamin, intersectin, vinexin, or yet another unidentified SHIP2 binding partner might be recruiting SHIP2 to the invadopodium precursor; this will need to be further explored in future studies.
Here we report for the first time that SHIP2 localizes at the invadopodium core and regulates PI(3,4)P2 levels locally at the invadopodium. Our results indicate that SHIP2 arrival at the invadopodium precursor coincides with the beginning of PI(3,4)P2 enrichment at the precursor. We also found that the carcinoma cells studied here do not express either SHIP1, a closely related SHIP2 isoform, or synaptojanin-2, another 5′-phosphatase. Based on these results in carcinoma cells, we propose that SHIP2 5′-phosphatase activity is a key regulator of PI(3,4)P2 levels locally at invadopodia. However, we did find synaptojanin-2 expression in metastatic human MDA-MB-231 cells (Figure S5B), another triple-negative breast carcinoma cell. Therefore, it is possible that different molecular subtypes of carcinoma cells may employ additional 5′-phos-phatases or a combination of 5′-phosphatases to regulate PI(3,4)P2 levels at invadopodia. Future studies are necessary to evaluate the role of other 5′-phosphatases [21], their coordination in PI(3,4)P2 generation at invadopodia, and their individual contributions to the invasive and metastatic phenotype.
Supplementary Material
Acknowledgments
We thank the investigators listed in the Supplemental Experimental Procedures for generous sharing of cells and reagents; Xiaoming Chen for the Tks5-R42A/R93A mutant; the Analytical Imaging Facility and Gruss Lipper Biophotonics Center at Albert Einstein College of Medicine for microscopy help; and members of the Condeelis, Segall, Cox, and Hodgson laboratories for guidance. This work was funded by NIH grants CA150344 and U54-CA112967 (F.B.G.) and a postdoctoral fellowship from Susan G. Komen for the Cure KG111405 to V.P.S.
Footnotes
Supplemental Information includes six figures, Supplemental Experimental Procedures, and eight movies and can be found with this article online at http://dx.doi.org/10.1016/j.cub.2013.08.044.
References
- 1.Beaty BT, Sharma VP, Bravo-Cordero JJ, Simpson MA, Eddy RJ, Koleske AJ, Condeelis J. β1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol Biol Cell. 2013;24:1661–1675. S1–S11. doi: 10.1091/mbc.E12-12-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L, Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011;21:635–644. doi: 10.1016/j.cub.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magalhaes MA, Larson DR, Mader CC, Bravo-Cordero JJ, Gil-Henn H, Oser M, Chen X, Koleske AJ, Condeelis J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J Cell Biol. 2011;195:903–920. doi: 10.1083/jcb.201103045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–737. doi: 10.1016/j.jocn.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE. Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J Cell Biol. 2012;197:421–437. doi: 10.1083/jcb.201108143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 8.Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, Desmarais V, van Rheenen J, Koleske AJ, Condeelis J. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol. 2009;186:571–587. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller SC, Ghersi G, Akiyama SK, Sang QX, Howard L, Pineiro-Sanchez M, Nakahara H, Yeh Y, Chen WT. A novel protease-docking function of integrin at invadopodia. J Biol Chem. 1999;274:24947–24952. doi: 10.1074/jbc.274.35.24947. [DOI] [PubMed] [Google Scholar]
- 11.Branch KM, Hoshino D, Weaver AM. Adhesion rings surround invadopodia and promote maturation. Biol Open. 2012;1:711–722. doi: 10.1242/bio.20121867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–16851. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- 13.Mader CC, Oser M, Magalhaes MA, Bravo-Cordero JJ, Condeelis J, Koleske AJ, Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH-src cells. J Cell Biol. 2008;182:157–169. doi: 10.1083/jcb.200801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seals DF, Azucena EF, Jr, Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, Courtneidge SA. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–165. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P. Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci. 2009;122:2727–2740. doi: 10.1242/jcs.046680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blouw B, Seals DF, Pass I, Diaz B, Courtneidge SA. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur J Cell Biol. 2008;87:555–567. doi: 10.1016/j.ejcb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artym VV, Matsumoto K, Mueller SC, Yamada KM. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur J Cell Biol. 2011;90:172–180. doi: 10.1016/j.ejcb.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linder S, Wiesner C, Himmel M. Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol. 2011;27:185–211. doi: 10.1146/annurev-cellbio-092910-154216. [DOI] [PubMed] [Google Scholar]
- 20.Oser M, Dovas A, Cox D, Condeelis J. Nck1 and Grb2 localization patterns can distinguish invadopodia from podosomes. Eur J Cell Biol. 2011;90:181–188. doi: 10.1016/j.ejcb.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, Mitchell CA. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419:29–49. doi: 10.1042/BJ20081673. [DOI] [PubMed] [Google Scholar]
- 22.Chuang YY, Tran NL, Rusk N, Nakada M, Berens ME, Symons M. Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res. 2004;64:8271–8275. doi: 10.1158/0008-5472.CAN-04-2097. [DOI] [PubMed] [Google Scholar]
- 23.Hoshino D, Jourquin J, Emmons SW, Miller T, Goldgof M, Costello K, Tyson DR, Brown B, Lu Y, Prasad NK, et al. Network analysis of the focal adhesion to invadopodia transition identifies a PI3K-PKCα invasive signaling axis. Sci Signal. 2012;5:ra66. doi: 10.1126/scisignal.2002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowden ET, Coopman PJ, Mueller SC. Invadopodia: unique methods for measurement of extracellular matrix degradation in vitro. Methods Cell Biol. 2001;63:613–627. doi: 10.1016/s0091-679x(01)63033-4. [DOI] [PubMed] [Google Scholar]
- 25.Sharma VP, Entenberg D, Condeelis J. High-resolution live-cell imaging and time-lapse microscopy of invadopodium dynamics and tracking analysis. Methods Mol Biol. 2013;1046:343–357. doi: 10.1007/978-1-62703-538-5_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaverina I, Stradal TE, Gimona M. Podosome formation in cultured A7r5 vascular smooth muscle cells requires Arp2/3-dependent de-novo actin polymerization at discrete microdomains. J Cell Sci. 2003;116:4915–4924. doi: 10.1242/jcs.00818. [DOI] [PubMed] [Google Scholar]
- 27.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, Whisstock JC, Rowe T, Majerus PW, Mitchell CA. The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol Biol Cell. 2005;16:2218–2233. doi: 10.1091/mbc.E04-09-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furutani M, Tsujita K, Itoh T, Ijuin T, Takenawa T. Application of phosphoinositide-binding domains for the detection and quantification of specific phosphoinositides. Anal Biochem. 2006;355:8–18. doi: 10.1016/j.ab.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Sharma VP, DesMarais V, Sumners C, Shaw G, Narang A. Immunostaining evidence for PI(4,5)P2 localization at the leading edge of chemoattractant-stimulated HL-60 cells. J Leukoc Biol. 2008;84:440–447. doi: 10.1189/jlb.0907636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokogawa T, Nagata S, Nishio Y, Tsutsumi T, Ihara S, Shirai R, Morita K, Umeda M, Shirai Y, Saitoh N, Fukui Y. Evidence that 3′-phosphorylated polyphosphoinositides are generated at the nuclear surface: use of immunostaining technique with monoclonal antibodies specific for PI 3,4-P(2) FEBS Lett. 2000;473:222–226. doi: 10.1016/s0014-5793(00)01535-0. [DOI] [PubMed] [Google Scholar]
- 32.Stephens LR, Jackson TR, Hawkins PT. Agonist-stimulated synthesis of phosphatidylinositol(3,4,5)-trisphosphate: a new intracellular signalling system? Biochim Biophys Acta. 1993;1179:27–75. doi: 10.1016/0167-4889(93)90072-w. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi H, Yoshida S, Muroi E, Yoshida N, Kawamura M, Kouchi Z, Nakamura Y, Sakai R, Fukami K. Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J Cell Biol. 2011;193:1275–1288. doi: 10.1083/jcb.201009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Várnai P, Bondeva T, Tamás P, Tóth B, Buday L, Hunyady L, Balla T. Selective cellular effects of overexpressed pleckstrin-homology domains that recognize PtdIns(3,4,5)P3 suggest their interaction with protein binding partners. J Cell Sci. 2005;118:4879–4888. doi: 10.1242/jcs.02606. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Yoshida S, Muroi E, Kawamura M, Kouchi Z, Nakamura Y, Sakai R, Fukami K. Phosphatidylinositol 4,5-bisphosphate and PIP5-kinase Ialpha are required for invadopodia formation in human breast cancer cells. Cancer Sci. 2010;101:1632–1638. doi: 10.1111/j.1349-7006.2010.01574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad NK, Tandon M, Badve S, Snyder PW, Nakshatri H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis. 2008;29:25–34. doi: 10.1093/carcin/bgm213. [DOI] [PubMed] [Google Scholar]
- 37.Suwa A, Yamamoto T, Sawada A, Minoura K, Hosogai N, Tahara A, Kurama T, Shimokawa T, Aramori I. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br J Pharmacol. 2009;158:879–887. doi: 10.1111/j.1476-5381.2009.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakatsu F, Perera RM, Lucast L, Zoncu R, Domin J, Gertler FB, Toomre D, De Camilli P. The inositol 5-phosphatase SHIP2 regulates endocytic clathrin-coated pit dynamics. J Cell Biol. 2010;190:307–315. doi: 10.1083/jcb.201005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crimaldi L, Courtneidge SA, Gimona M. Tks5 recruits AFAP-110, p190RhoGAP, and cortactin for podosome formation. Exp Cell Res. 2009;315:2581–2592. doi: 10.1016/j.yexcr.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Prehoda KE, Scott JA, Mullins RD, Lim WA. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol. 2004;24:5269–5280. doi: 10.1128/MCB.24.12.5269-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prasad N, Topping RS, Decker SJ. SH2-containing inositol 5′-phosphatase SHIP2 associates with the p130(Cas) adapter protein and regulates cellular adhesion and spreading. Mol Cell Biol. 2001;21:1416–1428. doi: 10.1128/MCB.21.4.1416-1428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato K, Yazawa T, Taki K, Mori K, Wang S, Nishioka T, Hamaguchi T, Itoh T, Takenawa T, Kataoka C, et al. The inositol 5-phosphatase SHIP2 is an effector of RhoA and is involved in cell polarity and migration. Mol Biol Cell. 2012;23:2593–2604. doi: 10.1091/mbc.E11-11-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dyson JM, O’Malley CJ, Becanovic J, Munday AD, Berndt MC, Coghill ID, Nandurkar HH, Ooms LM, Mitchell CA. The SH2-containing inositol polyphosphate 5-phosphatase, SHIP-2, binds filamin and regulates submembraneous actin. J Cell Biol. 2001;155:1065–1079. doi: 10.1083/jcb.200104005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paternotte N, Zhang J, Vandenbroere I, Backers K, Blero D, Kioka N, Vanderwinden JM, Pirson I, Erneux C. SHIP2 interaction with the cytoskeletal protein Vinexin. FEBS J. 2005;272:6052–6066. doi: 10.1111/j.1742-4658.2005.04996.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.