

Abstract
Quantum chemical calculations are used to explore the origins of regioselectivity for proton-, Pt(II)- and Pd(II)-promoted cyclizations of 1,5-hexadienes, 5-aminoalkenes, and allylic acetimidates. The strain associated with achieving carbonium ion-like transition state geometries is shown to be a key factor in controlling 5-exo vs. 6-endo selectivity.
Introduction
The cyclization of carbocations derived from 1,5-hexadienes is a reaction that is broadly applied to the synthesis of complex carbocyclic structures. Transition metal promoted cyclization of 1,5-hexadienes generally leads to products containing or derived from 6-membered rings, i.e., 6-endo cyclization. For example, after activation of the less substituted alkene by the transition metal and generation of a carbocyclic intermediate, ring-opening to form Cope products (Fig. 1a),1,2 attack by appended nucleophiles (Fig. 1b)3 or H-abstraction (to form cyclic alkenes)3,4 can occur. Similar reactivity has been observed for related allylic trichloroacetimidates.1g However, for 5-ami-nopentenes, only 5-exo cyclization has been described (6-endo products are thought to arise from rearrangements of species formed by initial 5-exo cyclization).5 Although empirical guidelines exist, no mechanistic model rationalizes and/or predicts the regio-direction of such electrophilic cyclization reactions. A hypothesis invoking a stereocontrolling role for nonclassical (i.e., bridged/carbonium) ion intermediates stimulated the following computational investigation. As will be discussed, a predictive model has emerged.
Fig. 1.

Selected reactions involving cationic cyclizations of 1,5-hexadienes.
Nature initiates such cyclizations via alkene protonation (Fig. 1c) or alkylation (i.e., via a prior cyclization event; Fig. 1d), generally to form the most substituted carbocation, and then generates the product (5- or 6-membered ring) with the most substituted carbocation center.6 The issue of 5-exo vs. 6-endo selectivity was debated for many years in the context of sterol biosynthesis, but it now seems clear that apparent 6-endo cyclizations to form secondary carbocations are less favorable than 5-exo cyclizations to form tertiary carbocations followed by rearrangements that avoid secondary carbocations as minima (e.g., Fig. 1d).6 Biosynthetic cyclizations have, not surprisingly, been mimicked in the laboratory in attempts to synthesize complex hydrocarbons.7
Herein, we report the results of quantum chemical calculations8 that unveil the factors controlling 5-exo vs. 6-endo selectivity for Pt(II)- and Pd(II)-promoted cyclization of 1,5-hexadienes.9 This investigation focused on the viability of cyclic 3-center 2-electron bonding arrays (i.e., nonclassical or carbonium ions) as intermediates, transition state structures, or species found elsewhere along reaction coordinates (Scheme 1).10 The presence of nonclassical delocalization causes the strain associated with bicyclic structures to be expressed in a fashion that affects competing 5-exo and 6-endo reaction pathways. We contrast the role of this selectivity control element for cyclizations of hydrocarbon substrates (π-nucleophiles) and those of allylic acetimidates and aminoalkenes (lone pair nucleophiles).
Scheme 1.

Cyclization pathways. Classical (carbenium) structures in blue; nonclassical (carbonium) structures in red.
Results and discussion
First we focused on the structures and relative energies of the cationic intermediates produced via 6-endo and 5-exo cyclizations promoted by Pt(II) (modeled using [Pt(PH3)3]2+) and protonation (Scheme 1). No evidence of cyclic 3-center 2-electron bonding10 was found for any of the computed minima for Pt-containing systems (e.g., Fig. 2, top, resembling A and B), though nonclassical minima (C and D) were found for some protonated systems (R1 = H/R2 = CH3/R3 = H [Fig. 2, bottom], R1 = H/R2 = H/R3 = CH3, R1 = H/R2 = CH3/R3 = CH3).11 When R1 = CH3, A and B are tertiary carbocations (classical/carbenium ions), not in need of nonclassical delocalization, and when R1 = R2 = R3 = H, nonclassical delocalization leads to primary carbocation character at the R2/R3-bearing carbon (obviously, to be avoided).
Fig. 2.

Representative structures of computed minima and a transition state structure.
As shown in Table 1, for all protonated systems except that with a tetrasubstituted C=C double bond (entry 8), the intermediate derived from 6-endo cyclization is lower in energy than that from 5-exo cyclization. This preference is largest for systems where nonclassical intermediates are found (entries 3, 4 and 7). Considering these intermediates as distorted bicyclo-[3.1.0]- and bicyclo[2.1.0]-alkanes suggests that the large energy difference between the 5-exo and 6-endo intermediates is related to ring strain. The computed ring strain of bicyclo[2.1.0]pentane is ~24 kcal mol−1 higher than bicyclo[3.1.0]hexane, some residue of which must be present in the delocalized intermediate.12,13 Relative stabilities of nonclassical 6-endo and 5-exo carbonium geometries can therefore be rationalized by inherent bicyclo-strain energies. Differences in hyperconjugation, steric repulsion between the two exocyclic methyl groups (see Fig. 2, ) and torsional strain (apparent from R1–C5–C6–R2 dihedral angles) also play a role, but the nature of the bicyclic architecture is most useful in predicting reaction outcome.
Table 1.
Free energies (kcal mol−1; gas phase) of intermediates formed via activation with [Pt(PH3)3]2+ or H+ relative to lowest energy intermediate for each system. R1, R2 and R3 correspond to labels in Scheme 1
| 6-endo
|
5-exo
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Entry | X | R1 | R2 | R3 | A | C | D | B |
| 1 | H | H | H | H | 0.0 | 3.8 | ||
| Pt | 0.0 | 5.3 | ||||||
| 2 | H | Me | H | H | 0.0 | 2.0 | ||
| Pt | 1.3 | 0.0 | ||||||
| 3 | H | H | Me | H | 0.0 | 12.6 | ||
| Pt | 0.0 | 5.5 | ||||||
| 4 | H | H | H | Me | 0.0 | 14.0 | ||
| Pt | 0.4 | 0.0 | ||||||
| 5 | H | Me | Me | H | 0.0 | 1.4 | ||
| Pt | 0.0 | 2.4 | ||||||
| 6 | H | Me | H | Me | 0.0 | 1.6 | ||
| Pt | 2.0 | 0.0 | ||||||
| 7 | H | H | Me | Me | 0.0 | 14.4 | ||
| Pt | 0.0a | 12.2a | ||||||
| 8 | H | Me | Me | Me | 0.8 | 0.0 | ||
| Pt | 5.5 | 0.0 | ||||||
These structures resemble 4- and 5-membered carbocycles with exocyclic 3º carbocation groups; structural data is available in the ESI.
For the Pt-containing systems, each minimum is classical (i.e., is a carbenium ion) and the 5-exo cyclization isomers are preferred (entries 2, 4, 6 and 8), likely a result of steric inter-actions between the hydrocarbon backbone and the [Pt(PH3)3]+ group (this group is directly attached to the ring in A, but separated from it by a methylene in B).14
Transition state structures were also calculated for systems with tri- and tetrasubstituted C=C double bonds (Fig. 2 and 3), models of 1,5-hexadienes commonly used in Pt(II)- and Pd(II)-promoted cyclizations (e.g., Fig. 1a/b).1,3,4 As shown in Fig. 3, all predicted barriers for Pt(II)-promoted cyclization are small and all reactions are predicted to be downhill, while Pd(II)-promoted cyclizations are predicted to have higher barriers and to be uphill. Nonetheless, in all cases shown, there is a predicted kinetic preference for 6-endo cyclization, even when there is a thermodynamic preference for the 5-exo product (i.e., Pt2+ with R1 = R2 = R3 = CH3). A rationale for this observation is outlined below.
Fig. 3.
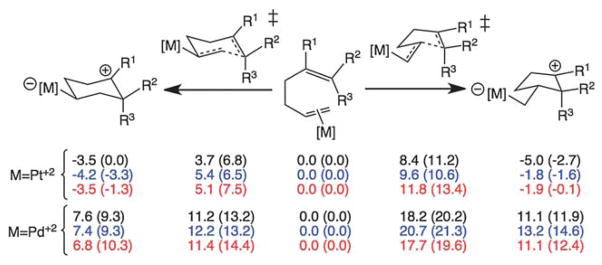
Computed reaction enthalpies (free energies in parentheses; kcal mol−1; in DCE) and barriers for [Pt(PH3)3]2+ and [PdCl2(NCMe)] promoted 6-endo and 5- exo cyclizations. Black are for R1 = R2 = R3 = CH3; blue are for R1 = R3 = CH3/R2 = H; red are for R1 = R2 = CH3/R3 = H. Preferences for 6-endo cyclization with [Pt(PH3)3]2+ are also predicted for systems with a methyl group added to the terminal carbon of the Pt-complexed C=C π-bond (and R1 = R2 = R3 = H or R1 = R2 = R3 = CH3); see ESI† for details.
All reactions are predicted to be reversible, but experimentally the intermediates are, in general, rapidly trapped through subsequent downhill reactions. If this trapping occurs faster than equilibration of the intermediates and is essentially irreversible, the kinetic preference should be manifested. For example, systems with appended OH nucleophiles (related to those in Fig. 1b) were examined computationally (see ESI†). For these systems, trapping of the cation formed in the initial cyclization event by C–O bond formation is predicted to be very exergonic and barrierless if the OH group is in the vicinity of the carbocation center. Thus, although thermodynamic preferences for ether products arising from initial 5-exo cyclization were predicted for some systems, the product distributions for these reactions are expected to reflect the kinetic preference for initial 6-endo cyclization, consistent with the fact that only products of 6-endo cyclization have been observed for Pt-promoted cyclizations of this type.3,9,15
The predicted 6-endo preference may result from the fact that the transition state structures for Pt(II)-promoted 1,5-diene cyclization (e.g., Fig. 2, right) resemble nonclassical cations with bridging alkyl groups (although the partial C–C bonds in these structures are longer than expected for nonclassical minima, e.g., Fig. 2, bottom). Thus, although bridged carbocations are not intermediates in Pt(II)- and Pd(II)-promoted cyclizations, they do appear along reaction coordinates for cyclization.16
Pd(II) catalysts also have been shown to promote exclusive 6-endo cyclizations en route to Cope products (Fig. 1a).1,3,4 Previous computational studies showed the 6-endo cyclization to be feasible,1f,g but barriers were not compared with those for competing 5-exo pathways. As shown in Fig. 3, the 6-endo cyclization is kinetically and thermodynamically favored over the 5-exo, consistent with experimental observations, and again nonclassical transition state structures were generally observed (see ESI†).1,3,4
The possibility of a correlation between nonclassical transition state geometries and high 6-endo selectivity for 1,5-diene cyclization prompted the examination of two alternative types of systems (Fig. 4, top). Enamine-containing systems were examined to mitigate the issues of strain associated with nonclassical transition structure geometries (vide supra) for 5-exo cyclization, while maintaining sp2 hybridization for the carbon next to the attacking atom. In contrast to the consistent 6-endo preferences shown in Fig. 3, kinetic preferences for both 5-exo and 6-endo cyclization are predicted for enamine nucleophiles, depending on the substitution of the metal-complexed π-bond. When the sp2 carbon is replaced by an sp3 carbon (e.g., aminoalkenes 2a and 2b), kinetic preferences for both 5-exo and 6-endo cyclization are again predicted, and are again associated with the substitution of the metal-complexed π-bond. The use of Pd(II) instead of Pt(II) in these systems had a small effect on the energetics and did not change the predicted selectivities. To our knowledge, Pd(II) and Pt(II) promoted cyclizations of amines onto connected internal alkenes have not yet been reported in the literature.5
Fig. 4.

Free energy barriers and ender/exergonicities (kcal mol−1; in DCE; exergonicities in parentheses) for systems with nitrogen nucleophiles. For 1a/b and 2a/b, bold values are for [M] = [Pt(PH3)3]2+ and plain text values are for [M] = [Pd(PH3)3]2+. For 3a–d, [M] = [PdCl2(NCMe)]. Barriers for 2b are negative because they are based on an extended rather than productive conformation of the reactant.
Allyl acetimidates were also examined (Fig. 4, bottom), since these have been utilized extensively in synthetic contexts.1g The key difference between these systems and those with C–C π-bonds is that the acetimidate reacts via its nitrogen lone pair rather than its π-bond. Similar barriers and exergonicities to those reported previously for 6-endo cyclizations in a study of Pd-promoted Cope-like rearrangements1g were found for our model systems. Again, kinetic preferences are predicted to depend on the presence or absence of an alkyl substituent on the metal-complexed π-bond, i.e., a 5-exo preference is possible.17
Conclusions
These data together indicate that π and lone pair nucleophiles react differently! Since π-nucleophiles transition through bicyclo-structures, the ring strains of bicyclo[3.1.0] versus bicyclo [2.1.0] carbonium structures lead to a 6-endo preference for cyclizations onto metal-complexed π-bonds. In contrast, the less delocalized nature of transition state structures for attack by nucleophilic lone pairs releases the constraints of a bicyclic transition structure and enables 5-exo/6-endo selectivities to be controlled by substitution patterns on the metal-bound alkene.18 Thus, we arrive at the following guidelines for predicting the kinetic selectivity of cyclization onto C=C π-bonds:
If the electrophilic π-bond is strongly activated such that a discrete carbocation is formed (e.g., by protonation), then the most stable carbocation formed through cyclization onto the carbocation center is expected to predominate.
If the electrophilic π-bond is activated by complexation to an electron deficient metal, then…
6-endo products are expected if the nucleophile is a π-bond.
Both 5-exo and 6-endo products are possible if the nucleophile is a lone pair, but the major product usually will be that from attack on the least electron-rich end of the electrophilic π-bond.
We look forward to the application of the concepts described herein to other systems and are eager to see if these simple selectivity guidelines prove to be truly general.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the NSF (D.J.T.); the UC MEXUS program, the UCD & Humanities Graduate Research Award in Chemistry, UCD’s R. B. Miller and David and Ruth Volman Graduate Fellowships (O.G.); a Graduate Assistance in Areas of National Need (GAANN) Fellowship (J.G.H.); DGTIC-UNAM for computing time (F.C.G.); and the National Institute of General Medicine (GM 60578) (M.R.G.).
Footnotes
Electronic supplementary information (ESI) available: Computational details and additional references.
Contributor Information
Michel R. Gagné, Email: mgagne@unc.edu.
Dean J. Tantillo, Email: djtantillo@ucdavis.edu.
Notes and references
- 1.Pd(II): Overman LE. Angew Chem, Int Ed Engl. 1984;23:579–586.Overman LE, Jacobsen EJ. J Am Chem Soc. 1982;104:7225–7231.Overman LE, Renaldo AF. Tetrahedron Lett. 1983;24:3757–3760.Bluthe N, Malacria M, Gore J. Tetrahedron Lett. 1983;24:1157–1160.Overman LE, Renaldo AF. J Am Chem Soc. 1990;112:3945–3949.Siebert MR, Tantillo DJ. J Am Chem Soc. 2007;129:8686–8687. doi: 10.1021/ja072159i.Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031–5044. doi: 10.1021/ja0676962.
- 2.Au(I): Felix RJ, Dieter W, Gutierrez O, Tantillo DJ, Gagné MR. Nat Chem. 2012;4:405–409. doi: 10.1038/nchem.1327.
- 3.Pt(II): Mullen CA, Gagné MR. J Am Chem Soc. 2007;129:11880–11881. doi: 10.1021/ja073573l.Koh JH, Gagné MR. Angew Chem, Int Ed. 2004;43:3459–3461. doi: 10.1002/anie.200453913.Pd(II): Overman LE, Renaldo AF. Tetrahedron Lett. 1983;24:2235–2238.
- 4.Rare cases of Pd(II)-promoted 5-exo cyclizations (generally involving oxygen-substituted reactants): Bray KL, Lloyd-Jones GC, Munoz MP, Slatford PA, Tan EHP, Tyler-Mahon AR, Worthington PA. Chem–Eur J. 2006;12:8650–8663. doi: 10.1002/chem.200600924.Nelson B, Hiller W, Pollex A, Hiersemann N. Org Lett. 2011;13:4438–4441. doi: 10.1021/ol201795w.InBr3 promoted cyclizations of hexa-1-yn-5-ene-derived systems favor 6-endo cyclization: Qiu WW, Surendra K, Yin L, Corey EJ. Org Lett. 2011;13:5893–5895. doi: 10.1021/ol202621g.
- 5.(a) Bender CF, Widenhoefer RA. J Am Chem Soc. 2005;127:1070–1071. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]; (b) Michael FE, Cochran BM. J Am Chem Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]; (c) Cochran BM, Michael FE. J Am Chem Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997. [DOI] [PubMed] [Google Scholar]; (d) Liskin DV, Sibbald PA, Rosewall CF, Michael FE. J Org Chem. 2010;75:6294–6296. doi: 10.1021/jo101171g. [DOI] [PubMed] [Google Scholar]
- 6.For leading references, see: Tantillo DJ. Nat Prod Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c.A seminal paper on 5-exo vs. 6-endo cyclizations in steroid biosynthesis: Hess BA., Jr J Am Chem Soc. 2002;124:10286–10287. doi: 10.1021/ja026850r.A seminal paper invoking bridged nonclassical structures during cyclizations leading to steroids: Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv Chim Acta. 1955;38:1890–1904.
- 7.e.g., Johnson WS. Acc Chem Res. 1968;1:1–8.van Tamelen EE. Acc Chem Res. 1975;8:152–158.Wendt KU, Schultz GE, Corey EJ, Liu DR. Angew Chem, Int Ed. 2000;39:2812.Fraga MB. Nat Prod Rep. 2003;20:392–413. doi: 10.1039/b208084m.Ishibashi H, Ishihara K, Yamamoto H. J Am Chem Soc. 2004;126:11122–11123. doi: 10.1021/ja0472026.Ishirara K, Ishibashi H, Yamamoto H. J Am Chem Soc. 2002;124:3647–3655. doi: 10.1021/ja0124865.Nakamura S, Ishibara K, Yamamoto H. J Am Chem Soc. 2002;122:8131–8140.Skakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553.Surendra K, Corey EJ. J Am Chem Soc. 2012;134:11992–11994. doi: 10.1021/ja305851h.Examples where product cations are stabilized by nearby OR and SiR3 groups: Shenvi RA, Corey EJ. Org Lett. 2010;12:3548–3551. doi: 10.1021/ol101410g.
- 8.All structures were computed using M06-2X/6-31G(d) for organic structures and M06 for organometallic systems with the 6-31G(d) basis for main group atoms and the SDD basis set for Pt and Pd using GAUSSIAN09. Full references and additional details are provided in the ESI.†
- 9.For a computational study of Pt(II)-promoted cyclizations of 1,6-dienylphenols, see: Nowroozi-Isfahani T, Musaev DG, Morokuma K, Gagné MR. Organometallics. 2007;26:2540–2549.For related computational work, see ref. 1f, g and 2.
- 10.Leading references on nonclassical ions: Grob CA. Acc Chem Res. 1983;16:426–431.Brown HC. Acc Chem Res. 1983;16:432–440.Olah GA, Prakash GKS, Saunders M. Acc Chem Res. 1983;16:440–448.Walling C. Acc Chem Res. 1983;16:448–454.Brown HC. The Nonclassical Ion Problem. Plenum; New York: 1977. (with comments by P. v. R. Schleyer)
- 11.A computational study on related carbocations: Lee JK, Houk KN. Angew Chem, Int Ed Engl. 1997;36:103–105.
- 12.The 5-exo intermediate from entry 4 has significant delocalization between the C5–C6 bond and the R3 substituent (see ESI† for details).
- 13.Borst MLG, Ehlers AW, Lammertsma K. J Org Chem. 2005;70:8110–8116. doi: 10.1021/jo0513010. [DOI] [PubMed] [Google Scholar]
- 14.See ESI† for additional discussion on electron delocalization in 5-exo and 6-endo reactants.
- 15.For some systems with strongly Lewis basic tridentate phosphine ligands, an equilibrium could be established between the alkene complex and cyclized Pt-alkyl. See: Feducia JA, Gagné MR. J Am Chem Soc. 2008;130:592–599. doi: 10.1021/ja075518i.
- 16.Strain associated with bicyclic transition structures also appears to affect the Pt(II)-catalyzed cycloisomerization of 1,6-dienes, for which both 5-exo and 6-endo products are observed (Kerber WD, Gagné MR. Org Lett. 2005;7:3379–3381. doi: 10.1021/ol051277c.but the competing transition state structures adopt bicyclo[3.1.0]- and bicyclo[4.1.0]-like structures, the parent hydrocarbons of which have roughly equal strain energies.13
- 17.Syn-carbometallative pathways generally impose a 5-exo regio-preference, since chelation involving both the π-bond and nucleophilic lone pair (not possible with the ligand systems examined herein) is involved. See: Ye X, White PB, Stahl SS. J Org Chem. 2013;78:2083–2090. doi: 10.1021/jo302266t.and references therein.
- 18.For examples of cyclization selectivity based on substrate geometry, compare the linear substrates in Qian H, Widenhoefer RA. J Am Chem Soc. 2003;125:2056–2057. doi: 10.1021/ja0293002.and Wang X, Pei T, Han X, Widenhoefer RA. Org Lett. 2003;5:2699–2701. doi: 10.1021/ol034879+.which can cyclize via a bicyclo[3.1.0]-like transition state, with the branched substrates in Han X, Widenhoefer RA. J Org Chem. 2004;69:1738–1740. doi: 10.1021/jo035576w.for which the relevant carbonium ion-like transition states would be strained bicyclo[2.1.0]- and bicyclo[1.1.0]-like species. The latter substrates instead react via an oxygen lone pair.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.