

Abstract
Copy number variations (CNVs) are one of the main sources of variability in the human genome. Many CNVs are associated with various diseases including cardiovascular disease. In addition to hybridization-based methods, next-generation sequencing (NGS) technologies are increasingly used for CNV discovery. However, respective computational methods applicable to NGS data are still limited. We developed a novel CNV calling method based on outlier detection applicable to small cohorts, which is of particular interest for the discovery of individual CNVs within families, de novo CNVs in trios and/or small cohorts of specific phenotypes like rare diseases. Approximately 7,000 rare diseases are currently known, which collectively affect ∼6% of the population. For our method, we applied the Dixon’s Q test to detect outliers and used a Hidden Markov Model for their assessment. The method can be used for data obtained by exome and targeted resequencing. We evaluated our outlier- based method in comparison to the CNV calling tool CoNIFER using eight HapMap exome samples and subsequently applied both methods to targeted resequencing data of patients with Tetralogy of Fallot (TOF), the most common cyanotic congenital heart disease. In both the HapMap samples and the TOF cases, our method is superior to CoNIFER, such that it identifies more true positive CNVs. Called CNVs in TOF cases were validated by qPCR and HapMap CNVs were confirmed with available array-CGH data. In the TOF patients, we found four copy number gains affecting three genes, of which two are important regulators of heart development (NOTCH1, ISL1) and one is located in a region associated with cardiac malformations (PRODH at 22q11). In summary, we present a novel CNV calling method based on outlier detection, which will be of particular interest for the analysis of de novo or individual CNVs in trios or cohorts up to 30 individuals, respectively.
Introduction
Many genomic studies have revealed a high variability of the human genome, ranging from single nucleotide variations and short insertions or deletions to larger structural variations and aneuploidies. Structural variations include copy number variations (CNVs), which cause gains (duplications) or losses (deletions) of genomic sequence. These copy number changes are usually defined to be longer than ∼500 bases, including large variations with more than 50 kilobases [1], [2]. Recent studies have identified CNVs associated with a number of complex diseases such as Crohn’s disease, intellectual disability and congenital heart disease [3]–[6].
Congenital heart disease (CHD) are the most common birth defect in human with an incidence of around 1% in all live births [7], [8]. They comprise a heterogeneous group of cardiac malformations that arise during heart development. The most common cyanotic form of CHD is Tetralogy of Fallot (TOF), which accounts for up to 10% of all heart malformations [9]. TOF is characterized by a ventricular septal defect with an overriding aorta, a right ventricular outflow tract obstruction and a right ventricular hypertrophy [10]. It is a well-recognized subfeature of syndromic disorders such as DiGeorge syndrome (22q11 deletion), Down syndrome, Holt-Oram syndrome and Williams-Beuren syndrome [11]. Deletions at the 22q11 locus account for up to 16% of TOF cases [12] and copy number changes at other loci were identified in several syndromic TOF patients [13]–[15]. However, the majority of TOFs are isolated, non-syndromic cases caused by a multifactorial inheritance with genetic-environmental interactions, which is also the situation for the majority of CHDs [16]. Using SNP arrays, three recent studies also identified CNVs in large cohorts of non-syndromic TOF patients [17]–[19]. Observing the overlap between these studies with hundreds of cases revealed only one locus (1q21.1) affected in 11 patients (Figure 1), which underlines the heterogeneous genetic background of non-syndromic TOF.
Figure 1. Overlap of three recent CNV studies in TOF patients.

All three studies are based on SNP arrays. Loci with detected CNVs are depicted according to their respective cytoband. For 1q21.1, which was identified in all three studies, the RefSeq genes that are affected in at least one patient in each of the publications are listed in the order of their genomic position. Genes that are expressed in mouse heart development (E8.5– E12.0, Mouse Atlas of Gene Expression at http://www.mouseatlas.org/mouseatlas_index_html) are marked in bold. # denotes the number of individuals.
As an alternative to the conventional SNP arrays, next-generation sequencing (NGS) technologies have been widely used to detect single or short sequence variations. The obtained sequence data can also be used to find larger CNVs. Depending on the sequencing technologies, there are different computational approaches for detecting copy numbers from NGS data. For exome sequencing or targeted resequencing, the read-depth or depth of coverage approach is widely used. It assumes that the mapped reads are randomly distributed across the reference genome or targeted regions. Based on this assumption, the read-depth approach analyses differences from the expected read distribution to detect duplications (higher read depth) and deletions (lower read depth) [20]. Applying this approach, several tools have been developed to identify CNVs from exome sequencing data, such as FishingCNV, CONTRA, ExomeCNV, ExomeDepth, XHMM, CoNVEX and CoNIFER [21]–[27].
Here, we aimed to identify copy number alterations in a small cohort of non-syndromic TOF patients based on targeted resequencing data. Assuming a heterogeneous genetic background with individual disease-relevant CNVs, we developed a novel CNV calling method based on outlier detection using Dixon’s Q test and assessment of outliers using a Hidden Markov Model (HMM). For evaluation, we applied our method to a small cohort of HapMap samples and compared it to results obtained with ExomeDepth and CoNIFER. Subsequently, our method and CoNIFER were used to detect CNVs in the TOF patients. Two copy number gains were identified by both methods and are duplications in the PRODH gene located at the 22q11 locus. In addition, our outlier-based method found a gain in NOTCH1 as well as in ISL1. All four CNVs could be validated by quantitative real-time PCR.
Materials and Methods
Ethics Statement
Studies on TOF patients were performed according to institutional guidelines of the German Heart Institute in Berlin, with approval of the ethics committee of the Charité Medical Faculty and informed written consent of patients and/or parents, kin, caretakers, or guardians on the behalf of the minors/children participants involved in our study.
TOF Samples and DNA Targeted Resequencing
Targeted resequencing was performed for eight TOF patients, which are unrelated sporadic cases with a well-defined coherent phenotype and no further anomalies. Blood samples (TOF-23, TOF-24, TOF-25, TOF-26, TOF-27) and cardiac tissue from the right ventricle (TOF-01, TOF-02, TOF-18) were collected in collaboration with the German Heart Institute in Berlin and the National Registry of Congenital Heart Disease in Berlin and used for the extraction of genomic DNA. 3–5 µg of genomic DNA were used for Roche NimbleGen sequence capturing using 365 K arrays. For array design, 867 genes and 167 microRNAs (12,910 exonic targets representing 4,616,651 target bases) were selected based on knowledge gained in various projects [28]–[30]. DNA enriched after NimbleGen sequence capturing was sequenced using the Illumina Genome Analyzer (GA) IIx (36 bp paired-end reads). Sequencing was performed by Atlas Biolabs (Berlin) according to manufacturers’ protocols.
On average, sequencing resulted in 13,331,661 read pairs per sample (Table 1). Average read depths of 75× and base quality scores of 34 (Phred scores) were reached in the captured regions over all samples (Table 1 and Figure 2).
Table 1. Number and quality of 36-end reads obtained from targeted resequencing in TOF patients using Illumina’s Genome Analyzer IIx platform.
| Captured regions | ||||||
| Sample | Number ofreads | Number of readpairs | Phred qualityscore | Mediancoverage | Meancoverage | Target bases with ≥10xcoverage |
| TOF-01 | 31,942,782 | 15,971,391 | 33.3 | 40 | 47 | 93.85% |
| TOF-02 | 26,970,680 | 13,485,340 | 32.7 | 66 | 76 | 97.70% |
| TOF-18 | 25,476,308 | 12,738,154 | 35.4 | 71 | 80 | 98.35% |
| TOF-23 | 20,885,192 | 10,442,596 | 35.0 | 60 | 69 | 97.41% |
| TOF-24 | 25,483,166 | 12,741,583 | 34.7 | 51 | 58 | 96.72% |
| TOF-25 | 30,551,674 | 15,275,837 | 34.6 | 84 | 92 | 98.91% |
| TOF-26 | 27,878,750 | 13,939,375 | 34.7 | 75 | 84 | 98.34% |
| TOF-27 | 24,118,022 | 12,059,011 | 34.6 | 78 | 90 | 98.00% |
Figure 2. Base qualities versus coverage values.
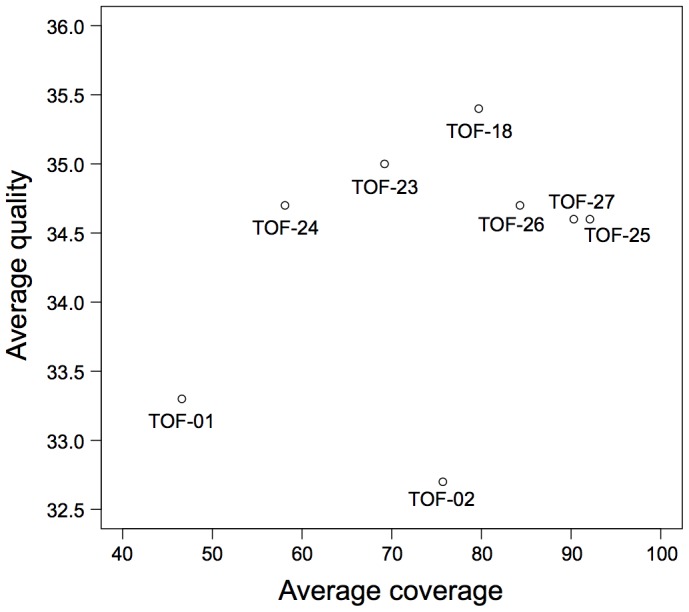
Scatterplot indicates the average base qualities (Phred scores) and depths of coverage for samples targeted resequenced by Illumina’s Genome Analyzer IIx platform (36 bp paired-end reads).
HapMap Samples
We used exome sequencing data from eight HapMap individuals (NA18507, NA18555, NA18956, NA19240, NA12878, NA15510, NA18517, NA19129). The exomes were captured using Roche NimbleGen EZ Exome SeqCap Version 1 and sequencing was performed using an Illumina HiSeq 2000 platform with 50 bp paired-end reads. The exome sequence data are available from the Short Read Archive at the NCBI (SRA039053). The reads were further trimmed to 36 bp.
Outlier-based CNV Calling Method
Our CNV calling method was developed for exome or targeted resequencing data of small sets of samples (at least 3 and at most 30) assuming that the bias in the captured regions is similar in all samples enriched and sequenced with the same technology. Based on a heterogeneous genetic background in the cohort, it was further assumed that a unique disease-related copy number change is only present in very few samples.
First, read mapping and calculation of copy number values were performed for each sample separately. The sequenced reads were mapped to the targeted regions of the reference genome using BWA v.0.5.9 in paired-end mode (‘sampe’) with default parameters [31]. Up- and downstream, the targeted regions (usually exons) were extended by 35 bp (read length minus one base pair) to correctly capture the coverage at the start and end of a region. After mapping, the extended regions with their mapped reads were joined chromosome-wise and the tool mRCaNaVaR v0.34 [32] was used to split the joined regions into non-overlapping windows of 100 bp in length. The copy number value C for each window W∈{1,…,n} of a sample S∈{1,…,n} was then calculated by mRCaNaVaR using the following formula:
 |
with additional GC correction [32] (Figure 3A). Reads spanning the border of two windows were assigned to the left window. In general, our method calculates a copy number value using mrCaNaVaR, which can accurately predict CNVs with at least 4x coverage [32].
Figure 3. Outlier-based CNV calling method.

(A) Read mapping and calculation of copy number value per window. Reads are mapped to extended targeted regions, which are then joined chromosome-wise. mrCaNaVaR is used to split the joined regions into windows. For each window, its copy number value is calculated by mrCaNaVaR, where 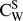 represents the value for window W in sample S. (B) Dixon’s Q test is applied for each window over all samples to identify outliers. Here, sample 1 represents an outlier (loss, L) for the first, second, third and fifth window, while sample 2 represents an outlier (gain, G) for the fourth window. (C) Assessment of outliers using a Hidden Markov Model (HMM). In the given example, the fourth window of sample 1 is considered as normal (N). After applying the HMM, it will also be considered as a loss. Similarly, the fourth window of sample 2 is considered as normal after applying the HMM. A region is called as a copy number alteration, if at least five continuous windows show the same kind of change, i.e. either gain or loss.
represents the value for window W in sample S. (B) Dixon’s Q test is applied for each window over all samples to identify outliers. Here, sample 1 represents an outlier (loss, L) for the first, second, third and fifth window, while sample 2 represents an outlier (gain, G) for the fourth window. (C) Assessment of outliers using a Hidden Markov Model (HMM). In the given example, the fourth window of sample 1 is considered as normal (N). After applying the HMM, it will also be considered as a loss. Similarly, the fourth window of sample 2 is considered as normal after applying the HMM. A region is called as a copy number alteration, if at least five continuous windows show the same kind of change, i.e. either gain or loss.
Second, Dixon’s Q test was applied for each window at the same position over all samples to identify gains or losses considered as outliers (Figure 3B). This test was introduced in 1950 for the analysis of extreme values and for the rejection of outlying values [33]. We used the formulas for r10 and r20 [34], also known as type10 and type20 in the R package ‘outliers’ v0.14 (http://www.R-project.org). Type10 (recommended for 3–7 samples) can only detect a single outlying window at the same genomic position over all samples, while type20 (recommended for 8–30 samples) can identify exactly two outlying windows, meaning the Q test will not detect outliers if more than 2 outliers are present. For each window, we first applied type20, however, if no two significant outliers (samples) were found, type10 was used to detect at most one outlier. Note that our method can also be applied using type10 and type20 independently. Outliers were regarded as significant with a p-value of less than or equal to 0.01. In general, the higher the p-value cutoff, the higher the number of detected outliers but also the number of false positives, i.e. the p-value is a tuning parameter for sensitivity of our method.
In the third and final step, the samples were again considered separately. For each sample, a Hidden Markov Model [35] was applied to get the most likely state of each window (i.e. gain, loss or normal). The initial transition and emission probabilities of the HMM are given in Table S1 and the values were recomputed using the Baum-Welch algorithm [36] implemented in the R package ‘HMM’ v1.0. The most likely sequence of the hidden states was then found by the Viterbi algorithm [37] also implemented in the R package ‘HMM’. Finally, a region was called as copy number gain or loss if at least five continuous windows were considered as a gain or loss, respectively (Figure 3C). This results in a minimum size of 500 bp for detectable CNVs.
We have included a script, written in R 2.15.1 (http://www.R-project.org), for our CNV calling method based on outlier detection in exome and/or targeted resequencing data (Script S1).
CNV Validation
Genomic DNA was extracted from whole blood or cardiac biopsies using standard procedures. Quantitative real-time PCR was carried out using GoTag qPCR Master Mix (Promega) on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) according to the manufacturer’s instructions and with normalization to the RPPH1 gene. Primer sequences are available on request. As a reference, genomic DNA from the HapMap individual NA10851 was obtained from the Coriell Cell Repositories (New Jersey, USA).
Results and Discussion
We applied our outlier-based CNV calling method to eight HapMap control samples and intersected our exome-based calls from five of the samples with previously generated calls from high-resolution microarray-based comparative genomic hybridization (array-CGH) [2]. In addition to our method, we used the two publicly available tools ExomeDepth and CoNIFER [23], [27]. Other tools such as CONTRA, FishingCNV, CoNVEX and ExomeCNV could not be applied to this dataset since they need either matched or non-matched controls.
CoNIFER (copy number inference from exome reads) is a method that combines the read-depth approach with singular value decomposition (SVD) normalization to identify rare and common copy number alterations from exome sequencing data [27]. Applying our method with type10 Dixon’s Q test (assuming at most one outlier), we found 40 CNVs over the five HapMap controls (Table S2), out of which 37 regions were also identified in the array-CGH data, showing a high positive predictive value of 93%. With type20 (assuming at most two outliers), we found 65 copy number changes (Table S3), out of which 55 regions are present in the array-CGH data, resulting in a positive predictive value of 85%. Using CoNIFER, 32 CNVs were identified in the five HapMap exome controls and only 26 of these regions are also present in the array-CGH data [27], which corresponds to a positive predictive value of 81% (Table 2). Comparing our results to those obtained from CoNIFER, we found that with type10 16 out of 40 regions (40%) are overlapping with regions called by CoNIFER by at least one base pair. Vice versa, 11 out of 32 regions (34%) overlap with our calls. With type20, 24 out of our 65 called regions (37%) overlap with those from CoNIFER and oppositely, 47% of the regions (15 out of 32) overlap with our calls. In general, CNV regions identified by CoNIFER are longer than those found by our method, meaning that regions called by CoNIFER can correspond to more than one of our CNVs, which explains the different overlap proportions.
Table 2. Exome sequencing-based CNV calls in HapMap samples.
| Method | Numberof CNVs | Validation dataset | Number ofoverlapping CNVs | Positive predictivevalue | Sensitivity |
| Outlier-based calling method with type10 | 40 | 3,330 arrayCGH calls | 37 | 93% | 1.1% |
| Outlier-based calling method withtype20 including type10 | 65 | 55 | 85% | 1.7% | |
| CoNIFER | 32 | 26 | 81% | 0.8% | |
| ExomeDepth | 1,555 | 253 | 16% | 7.6% |
Overall, our method was able to detect more copy number changes and has a higher proportion of true positives compared to CoNIFER. However, there is still a large number of CNVs observed in the array-CGH data, which were identified by neither of the two exome-based methods (Table 2). This can for example be explained by their location in segmental duplications and polymorphic but not duplicated regions [27].
ExomeDepth uses a beta-binomial model for the read count data to identify CNVs from exome sequencing data [24]. We applied ExomeDepth with default parameters to the eight HapMap samples and intersected the found CNVs from five of the samples with previously generated calls from array-CGH. In summary, ExomeDepth found 1,555 CNVs in the five samples (median number of 286 CNVs per sample). Out of these, only 253 CNVs overlapped with 3,330 array-CGH calls, which suggest a positive predictive value of 16% and sensitivity of 7.6% (Table 2).
Interestingly, all the five rare CNVs in the five HapMap samples (see Krumm et al. 2012, Table S2 [27]) were found by our method, CoNIFER and ExomeDepth. Moreover, ExomeDepth identified more CNVs as compared to CoNIFER and to our method (Table 2), however; the positive predictive value is very low. Therefore, we decided not to use ExomeDepth for detecting CNVs in the TOF patients.
To identify copy number alterations in TOF patients, we applied our outlier-based method as well as CoNIFER to targeted resequencing data of our eight cases. Using our method, we found four copy number gains in three genes, namely ISL1, NOTCH1 and PRODH. CoNIFER only identified two gains in PRODH, which overlap with the two regions found by our method (Table 3 and Figure 4A). We further validated all four regions identified by our method using quantitative real-time PCR (Figure 4B–D). ISL1 is a homeobox transcription factor that marks cardiovascular progenitors [38] and is known to be associated with human congenital heart disease [39]. NOTCH1 is a transmembrane receptor involved in the NOTCH signaling pathway, which plays a crucial role in heart development [40]. Mutations in NOTCH1 are associated with a spectrum of congenital aortic valve anomalies [41], [42] and a copy number loss was identified in a patient with TOF [17] (locus 9q34.3, Figure 1). The mitochondrial protein PRODH catalyzes the first step in proline degradation and is located in the 22q11.2 locus. Deletions in this region are associated with the DiGeorge syndrome and 80% of cases harbor cardiovascular anomalies [43]. A copy number gain and two losses in the 22q11.2 locus overlapping PRODH were also identified in sporadic TOF patients [17], [18] (Figure 1).
Table 3. Targeted resequencing-based CNV calls in TOF patients.
| Method | Type of variation | Position (hg19) | Length in bp | Gene | Sample |
| Outlier-based calling method withtype20 including type10 | Gain | chr5∶50,689,340–50,689,940 | 601 | ISL1 | TOF-23 |
| Gain | chr9∶139,402,477–139,404,228 | 1,752 | NOTCH1 | TOF-01 | |
| Gain | chr22∶18,900,412–18,901,127 | 716 | PRODH | TOF-02 | |
| Gain | chr22∶18,910,691–18,918,575 | 7,885 | PRODH | TOF-02 | |
| CoNIFER | Gain | chr22∶18,900,414–18,905,939 | 5,526 | PRODH | TOF-02 |
| Gain | chr22∶18,910,575–18,923,866 | 13,292 | PRODH | TOF-02 |
Figure 4. CNVs in TOF patients.

(A) CNVs detected in PRODH by CoNIFER and our outlier-based CNV calling method. The duplications are depicted in the UCSC Genome Browser as blue bars. The positions of the two quantitative real-time PCR products selected for validation are shown as light and dark grey bars, respectively. (B) Quantitative real-time PCR validation of PRODH copy number gains. Measurement was performed at two different positions (light and dark grey bars, respectively) and normalized to the RPPH1 gene. The HapMap individual NA10851 was used as a reference. The plot shows a representative of two independent measurements, which were each performed in triplicates. (C–D) Validation of copy number gains in ISL1 and NOTCH1, respectively, that were only identified by our outlier-based CNV calling method.
In summary, we developed an outlier-based CNV calling method for a small cohort size of up to 30 individuals. The exploration of the human phenotype and its genetic and molecular background is the challenge of the next century and it is already clear that more precise phenotyping will lead to smaller cohort sizes. Here, novel approaches will be of exceptional relevance. Moreover, analyzing small patient cohorts is of special interest for rare diseases with only few available patient samples. Approximately 7,000 rare diseases are currently known and together affect about 6% of the population [44]. Our method is based on the assumption that individual CNVs (outliers) are disease-relevant and can be applied to exome as well as targeted resequencing data. Both sequencing techniques achieve a high read coverage over the targeted regions. Nevertheless, there are non-uniform patterns in the read depth resulting mainly from repetitive regions. Thus, the detection of copy number alterations is limited in these genomic regions, which is shown by the high number of false negatives compared to array-CGH [27].
We evaluated our method using publicly available data of eight HapMap samples and subsequently applied it to a small number of TOF patients. Compared to CoNIFER we identified more CNVs in both the HapMap samples as well as in our TOF cohort. In general, our method assumes a uniform read distribution over all exons of all individuals enriched and sequenced with the same technology to compare read counts between all samples to detect outliers. In contrast, CoNIFER considers the read depth across all individuals after SVD normalization. This difference is also reflected by the overlap of their calls in the eight HapMap samples. Although the general overlap is relatively low, we were able to identify all rare CNVs detected by CoNIFER. In addition to searching for rare CNVs, we also found a subset of common CNVs called by CoNIFER. This might be explained by variations present in only one or two of the eight individuals, but defined as common based on their frequency in a larger population.
In our TOF cohort comprising eight cases, we found four copy number gains in three patients, while CoNIFER only detected two of the gains in one patient. All four gains could be validated and in addition, the three genes affected by the CNVs are important regulators of heart development (NOTCH1, ISL1) or are located in a region associated with cardiac malformations (PRODH). Two of the variations also overlap with copy number alterations in TOF patients previously identified by array-CGH [17], [18]. Taken together, this illustrates the advantage of using an outlier-based detecting method in a small cohort with a heterogeneous genetic background. Thus, our method is of special interest for small cohorts of specific phenotypes like rare diseases. Moreover, it can be used for the discovery of individual CNVs within families and de novo CNVs in trios.
Supporting Information
Initial transition and emission probabilities of the HMM.
(PDF)
CNVs found in the five HapMap samples using type10 Dixon’s Q test in the outlier-based CNV calling method.
(PDF)
CNVs found in the five HapMap samples using type20 Dixon’s Q test in the outlier-based CNV calling method.
(PDF)
R script for CNV calling.
(TXT)
Acknowledgments
We are deeply grateful to the TOF patients and families for their cooperation. We thank the German Heart Institute Berlin (Berlin, Germany) and the National Registry of Congenital Heart Disease (Berlin, Germany) for sample contribution. We further thank Ilona Dunkel for sample preparation. We also thank Biostar (www.biostars.org) and Cross Validated Stack Exchange (www.stats.stackexchange.com) for providing supporting discussion platforms.
Funding Statement
This work was supported by the European Community’s Seventh Framework Programme contracts (“CardioGeNet”) 2009-223463 and (“CardioNet”) People-2011-ITN-289600 (all to SRS), a Marie Curie PhD fellowship to VB, a PhD scholarship to CD by the Studienstiftung des Deutschen Volkes, and the German Research Foundation (Heisenberg professorship and grant 574157 to SRS). This work was also supported by the Competence Network for Congenital Heart Defects funded by the Federal Ministry of Education and Research (BMBF), support code FKZ 01GI0601. The funders had no role in study design, data collection and analysis.
References
- 1. Feuk L, Carson AR, Scherer SW (2006) Structural variation in the human genome. Nat Rev Genet 7: 85–97 10.1038/nrg1767 [DOI] [PubMed] [Google Scholar]
- 2. Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, et al. (2010) Origins and functional impact of copy number variation in the human genome. 464: 704–712 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19812545&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, et al. (2006) A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. 79: 439–448 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16909382&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Vries BBA, Pfundt R, Leisink M, Koolen DA, Vissers LELM, et al. (2005) Diagnostic genome profiling in mental retardation. 77: 606–616 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16175506&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, et al. (2007) Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 28: 2778–2784 10.1093/eurheartj/ehl560 [DOI] [PubMed] [Google Scholar]
- 6. Erdogan F, Larsen LA, Zhang L, Tümer Z, Tommerup N, et al. (2008) High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet 45: 704–709 10.1136/jmg.2008.058776 [DOI] [PubMed] [Google Scholar]
- 7. Hoffman JIE, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol 39: 1890–1900. [DOI] [PubMed] [Google Scholar]
- 8. Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A (2008) Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr 153: 807–813 10.1016/j.jpeds.2008.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferencz C, Rubin JD, McCarter RJ, Brenner JI, Neill CA, et al. (1985) Congenital heart disease: prevalence at livebirth. The Baltimore-Washington Infant Study. American journal of epidemiology 121: 31–36 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=3964990&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 10.Apitz C, Webb G (2009) ScienceDirect.com - The Lancet - Tetralogy of Fallot. Available: http://www.sciencedirect.com/science/article/pii/s0140-6736(09)60657-7.
- 11. Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112: 707–720 10.1161/CIRCRESAHA.112.300853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, et al. (1998) Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol 32: 492–498. [DOI] [PubMed] [Google Scholar]
- 13. Cuturilo G, Menten B, Krstic A, Drakulic D, Jovanovic I, et al. (2011) 4q34.1-q35.2 deletion in a boy with phenotype resembling 22q11.2 deletion syndrome. 170: 1465–1470 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21833498&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 14. Luo H, Xie L, Wang S-Z, Chen J-L, Huang C, et al. (2012) Duplication of 8q12 encompassing CHD7 is associated with a distinct phenotype but without duane anomaly. 55: 646–649 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=22902603&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 15. Luo C, Yang Y-F, Yin B-L, Chen J-L, Huang C, et al. (2012) Microduplication of 3p25.2 encompassing RAF1 associated with congenital heart disease suggestive of Noonan syndrome. 158A: 1918–1923 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=22786616&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 16. Nora JJ (1968) Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation 38: 604–617. [DOI] [PubMed] [Google Scholar]
- 17. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, et al. (2009) De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41: 931–935 10.1038/ng.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Silversides CK, Lionel AC, Costain G, Merico D, Migita O, et al. (2012) Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet 8: e1002843 10.1371/journal.pgen.1002843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, et al. (2012) Contribution of Global Rare Copy-Number Variants to the Risk of Sporadic Congenital Heart Disease. The American Journal of Human Genetics 91: 489–501 10.1016/j.ajhg.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alkan C, Coe BP, Eichler EE (2011) Genome structural variation discovery and genotyping. Nat Rev Genet 12: 363–376 10.1038/nrg2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi Y, Majewski J (2013) FishingCNV: a graphical software package for detecting rare copy number variations in exome-sequencing data. Bioinformatics 29: 1461–1462 10.1093/bioinformatics/btt151 [DOI] [PubMed] [Google Scholar]
- 22. Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, et al. (2012) CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28: 1307–1313 10.1093/bioinformatics/bts146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sathirapongsasuti JF, Lee H, Horst BAJ, Brunner G, Cochran AJ, et al. (2011) Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. 27: 2648–2654 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=21828086&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, et al. (2012) A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28: 2747–2754 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, et al. (2012) Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91: 597–607 10.1016/j.ajhg.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amarasinghe KC, Li J, Halgamuge SK (2013) CoNVEX: copy number variation estimation in exome sequencing data using HMM. BMC Bioinformatics 14 Suppl 2S2 10.1186/1471-2105-14-S2-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, et al. (2012) Copy number variation detection and genotyping from exome sequence data. 22: 1525–1532 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=22585873&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaynak B, Heydebreck von A, Mebus S, Seelow D, Hennig S, et al. (2003) Genome-wide array analysis of normal and malformed human hearts. Circulation 107: 2467–2474 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=12742993&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 29. Toenjes M, Schueler M, Hammer S, Pape UJ, Fischer JJ, et al. (2008) Prediction of cardiac transcription networks based on molecular data and complex clinical phenotypes. Mol Biosyst 4: 589–598 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=18493657&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 30. Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, et al. (2011) The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet 7: e1001313 10.1371/journal.pgen.1001313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. 25: 1754–1760 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19451168&retmode=ref&cmd=prlinks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, et al. (2009) Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067 10.1038/ng.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon WJ (1950) Analysis of extreme values. Available: http://www.jstor.org/stable/10.2307/2236602.
- 34.Rorabacher DB (1991) Statistical treatment for rejection of deviant values: critical values of Dixon’s ‘Q’ parameter and related subrange ratios at the 95% confidence level - Analytical Chemistry (ACS Publications). Available: http://pubs.acs.org/doi/abs/10.1021/ac00002a010.
- 35. Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. 77: 257–286 Available: http://ieeexplore.ieee.org/lpdocs/epic03/wrapper.htm?arnumber=18626. [Google Scholar]
- 36.Baum LE, Petrie T, Soules G, Weiss N (1970) A maximization technique occurring in the statistical analysis of probabilistic functions of Markov chains. Available: http://www.jstor.org/stable/10.2307/2239727.
- 37.Viterbi A (1967) Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. Available: http://ieeexplore.ieee.org/xpls/abs_all.jsp?arnumber=1054010.
- 38. Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, et al. (2009) Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. 460: 113–117 Available: http://pubget.com/site/paper/19571884?institution=. [DOI] [PubMed] [Google Scholar]
- 39. Stevens KN, Hakonarson H, Kim CE, Doevendans PA, Koeleman BPC, et al. (2009) Common Variation in ISL1 Confers Genetic Susceptibility for Human Congenital Heart Disease. 5: e10855–e10855 Available: http://pubget.com/site/paper/20520780?institution=. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nemir M, Pedrazzini T (2008) Functional role of Notch signaling in the developing and postnatal heart. 45: 10–10 Available: http://pubget.com/site/paper/18410944?institution=. [DOI] [PubMed] [Google Scholar]
- 41. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, et al. (2005) Mutations in NOTCH1 cause aortic valve disease. Nature 437: 270–274 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- 42. Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, et al. (2006) Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. 345: 1460–1465 Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=16729972&retmode=ref&cmd=prlinks. [DOI] [PubMed] [Google Scholar]
- 43. Momma K (2010) Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 105: 1617–1624 10.1016/j.amjcard.2010.01.333 [DOI] [PubMed] [Google Scholar]
- 44. Humphreys G (2012) Coming together to combat rare diseases. Bull World Health Organ 90: 406–407 10.2471/BLT.12.020612 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Initial transition and emission probabilities of the HMM.
(PDF)
CNVs found in the five HapMap samples using type10 Dixon’s Q test in the outlier-based CNV calling method.
(PDF)
CNVs found in the five HapMap samples using type20 Dixon’s Q test in the outlier-based CNV calling method.
(PDF)
R script for CNV calling.
(TXT)