

Summary
Agrobacterium tumefaciens transfers oncogenic DNA and effector proteins to plant cells during the course of infection. Substrate translocation across the bacterial cell envelope is mediated by a type IV secretion (TFS) system composed of the VirB proteins, as well as VirD4, a member of a large family of inner membrane proteins implicated in the coupling of DNA transfer intermediates to the secretion machine. In this study, we demonstrate with novel cytological screens – a two-hybrid (C2H) assay and bimolecular fluorescence complementation (BiFC) – and by immunoprecipitation of chemically cross-linked protein complexes that the VirE2 effector protein interacts directly with the VirD4 coupling protein at cell poles of A. tumefaciens. Analyses of truncation derivatives showed that VirE2 interacts via its C terminus with VirD4, and, further, an NH2-terminal membrane-spanning domain of VirD4 is dispensable for complex formation. VirE2 interacts with VirD4 independently of the virB-encoded transfer machine and T pilus, the putative periplasmic chaperones AcvB and VirJ, and the T-DNA transfer intermediate. Finally, VirE2 is recruited to polar-localized VirD4 as a complex with its stabilizing secretion chaperone VirE1, yet the effector–coupling protein interaction is not dependent on chaperone binding. Together, our findings establish for the first time that a protein substrate of a type IV secretion system is recruited to a member of the coupling protein superfamily.
Introduction
Type IV secretion (TFS) systems are broadly defined as the bacterial conjugation machines and evolutionarily related protein translocation systems (Salmond, 1994). These medically important secretion pathways mediate the widespread transfer of virulence genes among pathogenic bacteria, promote biofilm formation (Ghigo, 2001), and deliver DNA and protein effectors to the eukaryotic cytosol during infection (Fischer et al., 2002; Burns, 2003). Mechanistic studies are just emerging for the TFS systems dedicated to protein trafficking. However, the conjugation systems, e.g. the Escherichia coli F plasmid, the broad-host-range RP4 (IncP) and R388 (IncW) plasmids, and the Agrobacterium tumefaciens T-DNA transfer system, have long served as archetypes for unravelling the molecular details of TFS machine assembly and function (Baron et al., 2002).
Conjugation systems are composed of three functionally distinct protein subsets, as illustrated for the A. tumefaciens T-DNA transfer system. This system delivers oncogenic transfer-DNA (T-DNA) and proteins to plant cells during the course of infection (Zhu et al., 2000). One set of proteins, composed of the VirD2 relaxase, VirD1 and the VirC proteins, assemble as the relaxosome at origin-of-transfer (oriT)-like sequences flanking the T-DNA (Yanofsky et al., 1986). The VirD2 relaxase initiates conjugal DNA processing by generating single-stranded nicks at the T-DNA border sequences, and then remains covalently associated with the 5′ end of the DNA strand destined for transfer (T-strand). Conjugative plasmids generally are considered to co-transfer with the T-strand to recipient cells, though to date experimental evidence exists only for trafficking of the VirD2 relaxase to plant cells (Zhu et al., 2000; Llosa et al., 2002). The components of the relaxosome directing early conjugal processing reactions are termed the Dtr (DNA transfer and replication) proteins (Lessl and Lanka, 1994).
A second set of transfer (Tra) proteins assemble both as a transenvelope structure, the presumptive substrate transfer channel, and as an extracellular T pilus for establishment of donor-recipient cell contacts (Christie, 1997). The subunits of the T-DNA transfer channel/pilus include the inner membrane ATPases VirB4 and VirB11, a polytopic inner membrane protein VirB6, the bitopic membrane proteins VirB8 and VirB10, an outer membrane complex composed of VirB7 lipoprotein and VirB9, the outer membrane protein VirB3, and the VirB2 and VirB5 pilus subunits (Baron et al., 2002). Many conjugation systems encode homologues of these VirB proteins, whereas others most notably of the Gram-positive bacteria, encode structural subunits bearing little ancestral relatedness to the VirB proteins (Christie, 2001). The channel/pilus transfer proteins, e.g. the VirB proteins, direct productive mating pair formation and thus are termed the Mpf proteins (Grahn et al., 2000).
The third protein subset is actually a homomultimer of an integral inner membrane protein (Gomis-Ruth et al., 2001). Several compelling lines of evidence suggest that members of this large protein family, exemplified by VirD4 of the T-DNA transfer system, are DNA substrate specificity determinants for cognate conjugation machines (Llosa et al., 2002). In addition to genetic evidence for substrate specificity switching by chimeric conjugation machines (Cabezon et al., 1997; Hamilton et al., 2000), several VirD4 homologues, e.g. TraD of F plasmid, TraG of RP4, and TrwB of R388, bind ssDNA and ATP, and interact with relaxases and other Dtr proteins (Dash et al., 1992; Disque-Kochem and Dreiseikelmann, 1997; Moncalian et al., 1999; Szpirer et al., 2000; Hormaeche et al., 2002; Schroder et al., 2002). These proteins are designated as ‘coupling proteins’ because they are thought to link the relaxosome to the Mpf structure.
Recently, a crystal structure presented TrwB deleted of its NH2-terminal periplasmic domain as a spherical homohexameric ring of 110 Å in diameter and 90 Å in height with a central channel of 20 Å in diameter (Gomis-Ruth et al., 2001). Modelling of the NH2-terminal domain led to an overall structure strikingly similar to F1-ATPase (Gomis-Ruth et al., 2001). Moreover, purified forms of TrwB, TraG, and the Helicobacter pylori HP0524 coupling proteins also form homooligomers detectable by electron microscopy (Hormaeche et al., 2002; Schroder et al., 2002). This structural information suggests that the coupling proteins might recognize DNA substrates through interactions with relaxosome proteins and then translocate the conjugal DNA intermediate across the inner membrane, possibly by cycles of ATP binding and hydrolysis (Llosa et al., 2002). In support of this model, the coupling proteins bear structural and sequence similarities with the SpoIIIE and FtsK DNA translocases that move chromosomal DNA across membranes during sporulation and cell division respectively (Moncalian et al., 1999; Errington et al., 2001). Intriguingly, whereas these latter translocases localize at specific sites within bacterial cells in accordance with their translocase functions, the VirD4 coupling protein recently was shown to localize at the poles of A. tumefaciens (Kumar and Das, 2002).
Conjugation systems and related type IV systems also translocate protein substrates independently of DNA. The A. tumefaciens T-DNA transfer system delivers VirE2, VirE3, and VirF proteins to plant and yeast cells (Vergunst et al., 2000; Schrammeijer et al., 2003), the F and ColIb-P9 plasmid Tra systems export primase and the F system transfers TraC SSB to bacterial recipients (Rees and Wilkins, 1990; Wilkins and Thomas, 2000), the H. pylori Cag system transfers CagA protein to mammalian cells (Backert et al., 2000; Odenbreit et al., 2000; Stein et al., 2000), and the Legionella pneumophilus dot/icm system exports the DotA, RalF and LidA proteins (Nagai and Roy, 2001; Nagai et al., 2002; Conover et al., 2003). In the systems studied, the requirements for protein transfer include the Mpf proteins and the coupling protein, but not relaxosome subunits or an oriT sequence. Thus, a question of central importance for TFS-mediated protein trafficking is whether the coupling protein functions more broadly than previously envisaged by recruiting and, possibly, translocating protein substrates across the inner membrane. In the present study, we use a combination of novel cytological two-hybrid screens and biochemical approaches to demonstrate that the VirE2 effector protein interacts via its C terminus with the VirD4 coupling protein at the cell poles of A. tumefaciens. Our findings support a proposal that coupling proteins function minimally to recruit both DNA and protein substrates to the TFS apparatus.
Results
VirD4-dependent localization of VirE2 in vivo
Native VirD4 localizes at the cell poles of A. tumefaciens (Kumar and Das, 2002), and here we further show that VirD4 fused at its C terminus to GFP displays a polar localization. Both wild-type A348 and the virD4 null mutant Mx355, producing VirD4-GFP from the IncP replicon pKA62 (Table 1), exhibited strong fluorescent foci at the cell poles (Fig. 1A). By contrast, cells separately producing GFP from an IncP plasmid and VirD4 either from its native position on the pTi plasmid (A348(pZDB69); Fig. 1A) or from a PvirB promoter on an IncP plasmid (Mx355(pKA79); data not shown) were exclusively uniformly fluorescent, confirming that VirD4 must be fused to GFP for detection of fluorescent foci at the cell poles. Next, we asked whether a protein substrate is recruited in a VirD4-dependent manner to the cell poles. For this study, we fused GFP to the NH2 terminus of the VirE2 effector protein to monitor cellular localization. Of considerable interest, A348(pZDB73) cells producing GFP-VirE2 and native VirD4 displayed polar fluorescence, whereas Mx355(pZDB73) cells producing GFP-VirE2 in the absence of VirD4 were exclusively uniformly fluorescent (Fig. 1A).
Table 1.
Plasmids constructed for these studies.a
| Plasmid | Relevant characteristics |
|---|---|
| virD4 and virE2 constructs | |
| pKA9 | Crbr, pBSIIKS+ with PvirB-virD4 |
| pKA21 | Kanr, pXZ153 (IncP) with PvirB-virD4 |
| pKA22 | Kanr, pAP1 with PT7-virD4ΔN87 |
| pKA38 | Crbr, pBSIIKS+ with PvirB-virD4ΔN87 |
| pKA43 | Kanr, pXZ153 (IncP) with PvirB-virD4ΔN87 |
| divIVA constructs | |
| pKA6 | Camr, pACYC184-based replicon with PBAD-divIVA-virD4 |
| pKA76 | Crbr, pMMB22 (IncQ) with Ptac-divIVA |
| pKV36 | Crbr, pMMB22 (IncQ) with Ptac-divIVA-virD4ΔN87 |
| pKV42 | Crbr, Kanr, co-integrate of pZD76 with Ptac-divIVA-virD4 and pZD73 with PvirB-GFP-virE2 |
| pKV43 | Crbr, Kanr, co-integrate of pKV36 with Ptac-divIVA-virD4ΔN87 and pZD73 with PvirB-GFP-virE2 |
| pZD6 | Crbr, pMMB22 (IncQ) with Ptac-divIVA-GFP |
| pZD76 | Crbr, pMMB22 (IncQ) with Ptac-divIVA-virD4 |
| pXZ66 | Kanr, pBKSK+ with Plac-virE2-GFP |
| GFP constructs | |
| pKA59 | Crbr, pBSIIKS+ with PvirB-virD4-GFP |
| pKA62 | Kanr, pXZ153 (IncP) with PvirB-virD4-GFP |
| pKA75 | Crbr, pBSIIKS+ with PvirB-virD4ΔN87-GFP |
| pKA77 | Crbr, Kanr, co-integrate of pKA21 with PvirB-virD4 and pZD72 with PvirB-GFP-virE2 |
| pKA78 | Crbr, Kanr, co-integrate of pKA43 with PvirB-virD4ΔN87 and pZD72 with PvirB-GFP-virE2 |
| pKA79 | Crbr, Kanr, co-integrate of pKA21 with PvirB-virD4 and pZD69 with PvirB-GFP |
| pZD12 | Kanr, pBSIIKS +NdeI with PvirB-virE1-GFP |
| pZD69 | Crbr, pBSIIKS+NdeI with PvirB-GFP |
| pZD72 | Crbr, pBSIIKS+NdeI with PvirB-GFP-virE2 |
| pZD73 | Kanr, pBSIIKS+NdeI with PvirB-GFP-virE2 |
| pZD120 | Kanr, pBSIIKS+NdeI with PvirB-GFP-virE2Δ524–533 |
| pXZ63 | Crbr, pBSIIKS+NdeI with Plac-GFP |
| pXZ65 | Crbr, pBSIIKS+NdeI with Plac-GFP-virE2 |
| BiFC constructs | |
| pKA64 | Crbr, pBSIIKS+ with PvirB-virD4-N’GFP |
| pKV35 | Crbr, pBSIIKS+ with PvirB-N’GFP |
| pKV38 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2 |
| pKV39 | Crbr, pBSIIKS+ with PvirB-GFP’C |
| pZD88 | Kanr, pBSIIKS+NdeI with PvirB-virE1-N’GFP |
| pZD89 | Crbr, pBSIIKS+NdeI with PvirB-GFP’C-virE2 |
| pZD109 | Crbr, pBSIIKS+NdeI with PvirB-GFP’C |
| pZD834 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2Δ1–331 |
| pZD836 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2Δ1–437 |
| pZD838 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2Δ1–84 |
| pZD846 | Crbr, pBSllKS+ NdeI with PvirB-GFP’C-virE2Δ437–533 |
| pZD122 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2Δ1–483 |
| pZD827 | Crbr, pBSIIKS+ with PvirB-GFP’C-virE2.i31–529 |
See Experimental procedures for details of plasmid constructions.
Fig. 1.

VirD4-dependent localization of GFP-VirE2 to A. tumefaciens cell poles.
A. A348 (WT) and Mx355 (virD4 null mutant) cells producing proteins indicated above each panel photographed 10 h after induction with 200 μM AS by fluorescence microscopy. The proteins indicated were synthesized from the following IncP plasmids: D4-GFP (pKA62); GFP (pZDB69); GFP-E2 (pZDB73) and D4 + GFP-E2 (pKA77). The number below each panel represents the percentage of cells with polar fluorescence out of a total of at least 1000 cells examined; the ‘−’ denotes no detectable polar fluorescence.
B. Immunodetection of fusion proteins produced in Mx355 derivatives at 10 h post induction. The proteins listed above each lane were synthesized from the IncP plasmids listed in (A); for D4 (pKA21). Blots were developed with the antisera listed at the right. The reactive species (~60-kDa) in all lanes detected by anti-VirE2 antisera is native VirE2 produced from pTi.
Agrobacterium tumefaciens begins transcribing its vir genes at detectable levels within 2 h following exposure to the phenolic inducer, acetosyringone (AS), and transcriptional activity increases exponentially for the next 8–10 h (Chen and Winans, 1991). Interestingly, within 4 h of vir gene induction (t = 4), nearly all A348(pKA62) cells producing VirD4-GFP (from the IncP plasmid) displayed polar foci. At this time, only ~10% of A348(pZDB73) cells producing GFP-VirE2 (from the IncP replicon) and VirD4 (from pTi) showed polar foci, whereas at t = 10 this value was estimated at ~25%. In A. tumefaciens, the copy number of this IncP replicon is approximately five times that of the pTi plasmid (Ward et al., 1991). We therefore hypothesized that the comparatively low frequency of GFP-VirE2-producing cells displaying polar fluorescence might be attributable to a limiting level of polar-localized VirD4 synthesized from the pTi plasmid. To test this hypothesis, we produced both GFP-VirE2 and VirD4 from a multicopy IncP replicon. Most of the corresponding A348(pKA77) cells displayed polar fluorescence, ~ 60% at t = 4 and ~ 80% at t = 10 (Fig. 1A). Immunoblot studies confirmed that the fusion proteins under study are stable, and, as expected, virD4 expression from the IncP plasmid yields higher steady-state levels of VirD4 than native gene expression from the pTi plasmid (Fig. 1B). Throughout these studies, we confirmed that cells displaying polar fluorescence were devoid of inclusion bodies by phase-contrast or Nomarski microscopy (Ding et al., 2002). We conclude that GFP-VirE2 targets to the cell poles in a VirD4-dosage-dependent manner.
Evidence for a direct interaction between VirD4 and VirE2
Next, we sought to determine whether VirE2 interacts directly with VirD4. First, we exploited a cytology-based two-hybrid (C2H) screen developed previously for visualization of protein–protein interactions in living bacteria (Ding et al., 2002). In this assay, a protein of interest fused to a cell division protein, e.g. B. subtilis DivIVA (Edwards et al., 2000), can target GFP to the midcell and cell poles if fused to an interacting partner protein. Accordingly, A348(pZD76, pZDB73) co-producing DivIVA-VirD4 and GFP-VirE2 showed the expected pattern of GFP fluorescence. Nearly all of these cells exhibited polar fluorescence and ~ 15% also showed midcell fluorescence. Similar results were obtained with Mx355(pKV42) producing both fusion proteins in the absence of pTi-encoded native VirD4 (Fig. 2A). Because VirD4 itself localizes to the cell poles, the significant result is that the synthesis of DivIVA-VirD4 correlates with a redistribution of GFP-VirE2 to the midcell. That only a fraction of cells showed detectable midcell fluorescence is consistent with our previous findings utilizing DivIVA as the localizing protein for the C2H screen (Ding et al., 2002). VirD4 also self-associates (Kumar and Das, 2002), and we correspondingly detected a similar ratio of polar to midcell fluorescence among A348(pZD76, pKA62) cells producing DivIVA-VirD4 and VirD4-GFP (Fig. 2A). As expected, A348(pKA76, pZDB73) cells co-producing DivIVA and GFP-VirE2 displayed fluorescence patterns similar to A348(pZDB73) cells producing only GFP-VirE2, namely predominantly homogeneous fluorescence and a low percentage of polar foci. A348(pKA76, pKA62) cells co-producing DivIVA and VirD4-GFP displayed patterns similar to A348(pKA62), namely polar foci without any detectable midcell fluorescence.
Fig. 2.

Cytology-based dihybrid screens for demonstrating VirE2–VirD4 complex formation.
A. C2H: A348 (WT) cells producing the proteins indicated above each panel were examined 4 h post induction by fluorescence microscopy. The proteins indicated were synthesized from the following plasmids: DivIVA-D4 + GFP-E2 (pZD76, pZDB73); DivIVA + GFP-E2 (pKA76, pZDB73); DivIVA-D4 + D4-GFP (pZD76, pKA62); DivIVA + D4-GFP (pKA76, pKA62). The number below each panel represents the percentage of cells with polar fluorescence. Typically, ~ 15% of cells exhibiting DivIVA-dependent targeting in the C2H screen show fluorescence at the midcell.
B. BiFC: KE1 (ΔvirE) and Mx355 cells producing the proteins indicated adjacent to each panel were examined 4 and 10 h postinduction, respectively, by fluorescence microscopy. The proteins indicated were synthesized from the following plasmids introduced into the strains singly or in combination: E1-N’GFP (pZDB88); GFP’C-E2 (pZDB89); D4-N’GFP (pKAB64); N’GFP + GFP’C (pKVB35, pKVB39); D4-N’GFP (pKAB64); D4-N’GFP + GFPC’ (pKAB64, pKVB39).
C. Immunodetection of fusion proteins produced in KE1 (top panel) and Mx355 (bottom panel) at 10 h post induction. The proteins listed above each lane were synthesized from the following plasmids introduced into the strains singly or in combination: E1-N’GFP (pZDB88); GFP’C-E2 (pZDB89); D4-N’GFP (pKAB64); D4-N’GFP + GFPC’-E2 (pKAB64, pZDB89); and DivIVA-D4 + GFP-E2 (pKV42). The dark arrowheads point to distinct proteins that exhibit similar mobilities, e.g., VirD4-N’GFP (~ 92-kDa) and GFP-VirE2 (~ 90-kDa) (immunoreactive with anti-GFP antiserum), and VirD4-N’GFP and DivIVA-VirD4 (~ 93 kDa) (immunoreactive with anti-VirD4 antiserum).
Second, we assayed for VirE2–VirD4 complex formation with another novel cytology-based screen recently applied to the study of protein–protein interactions in mammalian cells. Termed biomolecular fluorescence complementation (BiFC), this assay is based on reconstitution of intact, fluorescent YFP protein by fusion of its halves to interacting partner proteins (Hu et al., 2002). We adapted this assay for GFP by fusion of its halves, designated N’GFP and GFP’C, to Vir proteins (see Experimental procedures). Initially, we screened for an interaction between VirE2 and its secretion chaperone VirE1 (Fig. 2B). The ΔvirE mutant strain KE1 bearing either pZDB89 (GFP’C-VirE2) or pZDB88 (VirE1-N’GFP) lacked detectable fluorescence. In striking contrast, KE1(pZDB88, pZDB89) co-producing both fusion proteins exhibited strong fluorescence at t = 4, and fluorescence intensity continued to increase through t = 10. All cells were uniformly fluorescent throughout this time period, consistent with the predominantly cytoplasmic location of this chaperone–effector protein complex (Christie et al., 1988; Zhao et al., 2001). KE1 also produces VirD4 from the Ti plasmid, and we detected polar foci in a fraction (~ 5%) of cells suggestive of recruitment of the VirE1-VirE2 complex to VirD4. However, the high background of uniform fluorescence interfered with detection of these foci, and stronger evidence for VirD4 recruitment of a chaperone-effector complex to the coupling protein is presented below. As additional controls for these experiments, we assayed for BiFC of strains KE1(pKVB35, pKVB39) (N’GFP; GFP’C) and KE1(pZDB88, pKVB39) (VirE1-N’GFP; GFP’C). These strains exhibited only a faint level of fluorescence upon prolonged vir gene induction of >12h (data not shown).
Next, we screened for an interaction between VirE2 and VirD4 by BiFC. Mx355(pKAB64, pZDB89) co-producing VirD4-N’GFP and GFP’C-VirE2 displayed strong fluorescent foci at the cell poles. These foci were detectable in ~ 50% of cells at t = 4 and nearly all cells at t = 10 (Fig. 2B). As above, Mx355 strains producing VirD4-N’GFP or GFP’C-VirE2 separately or with the complementing half-GFP fusion proteins were non- or weakly fluorescent. (Fig. 2B, data not shown). Finally, although we have found that the two halves of GFP produced individually are intrinsically unstable, all of the GFP-half fusion proteins examined in these studies were stably produced, insofar as immunoreactive species of the expected sizes were readily detected in cell extracts (Fig. 2C and data not shown). The two halves of the fluorescent protein must be in close apposition to yield BiFC (Hu et al., 2002), thus our findings strongly support a proposal that the coupling protein interacts directly with this secretion substrate. Furthermore, the results show for the first time that BiFC based on reconstitution of fluorescent GFP from its halves can be used to study protein–protein interactions in bacteria.
Finally, we assayed for complex formation between native forms of VirD4 and VirE2 by a coupled DSP-cross-linking and immunoprecipitation assay (see Experimental procedures). As shown in Fig. 3A, the anti-VirE2 antiserum, but not preimmune serum, co-precipitated a presumptive complex of VirE2 and VirD4 from extracts of wild-type A348 and of Mx355(pKA21) producing VirD4 from an IncP replicon. The anti-VirE2 antiserum failed to precipitate VirD4 from extracts of At12516 (virE2−). Reciprocally, the anti-VirD4 antiserum, but not preimmune serum, co-precipitated VirD4 and VirE2 from A348 and Mx355(pKA21) extracts. The anti-VirD4 antiserum failed to precipitate VirE2 from extracts of Mx355 (virD4−). We recovered the VirE2–VirD4 complex at a detectable level only if membranes were first treated with DSP. These biochemical findings are compatible with a prediction that the VirE2 secretion substrate interacts weakly or transiently with the VirD4 coupling protein. Additionally, we note that at least in laboratory growth conditions, wild-type cells accumulate a predominantly cytosolic VirE1-VirE2 complex that is removed during harvesting of membrane fractions used for the cross-linking/immunoprecipitation studies. Only a very small amount, estimated at ≤5%, of the total cellular VirE2 is membrane-associated (Christie et al., 1988), and of this only a small fraction was found to cross-link with VirD4 (Fig. 3A). The possible physiological importance of these different cellular pools of VirE2 is discussed below (see Discussion).
Fig. 3.
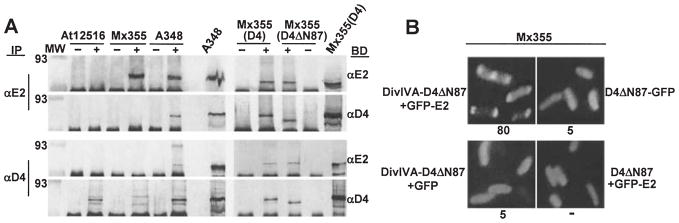
A. Co-immunoprecipitation of VirD4 and VirD4ΔN87 with VirE2. Isolated membranes were cross-linked with DSP and detergent-solubilized complexes were immunoprecipitated (IP) with preimmune (−) or immune (+) antisera listed at the left of the immunoblots. Blots were developed (BD) with antisera listed at the right. Strains: A348 (WT), At12516 (virE2−), Mx355 (virD4−); Mx355(D4) and Mx355(D4ΔN87) are the virD4 null mutant producing VirD4 and VirD4ΔN87 from plasmids pKA21 and pKA43, respectively. Lanes headed with slanted strain names, A348 and Mx355(D4), were loaded with total solubilized membrane protein extracts to show presence of VirE2 (~ 60-kDa), VirD4 (~ 76-kDa), and VirD4ΔN87 (~ 65-kDa). Immunoreactive bands at the bottoms of all blots correspond to the IgG heavy chain.
B. C2H screen for a VirD4ΔN87 interaction with GFP-VirE2. Strain Mx355 producing proteins indicated adjacent to each panel were examined 4 h post induction by fluorescence microscopy. Proteins were produced from the following plasmids: DivIVA-D4ΔN87 + GFP-E2 (pKV43); D4ΔN87-GFP (pKAB75); DivIVA-D4ΔN87 + GFP (pKA43, pZDB69); D4ΔN87 + GFP-E2 (pKA43, pZDB72).
Interaction of VirD4ΔN87 with VirE2
Previous studies have supplied some evidence that coupling proteins can still interact with components of the relaxosome even when deleted of their NH2-terminal membrane-spanning domains (Sastre et al., 1998; Llosa et al., 2002; Schroder et al., 2002). To test whether the cytoplasmic domain of VirD4 interacts with VirE2, we fused VirD4ΔN87 at it C terminus to GFP. A VirD4 derivative lacking its periplasmic loop does not target to A. tumefaciens poles (Kumar and Das, 2002). Thus, as expected, Mx355(pKAB75) cells producing VirD4ΔN87-GFP were uniformly fluorescent, as were Mx355(pKA78) cells co-producing VirD4ΔN87 and GFP-VirE2 (Fig. 3B). To assay for complex formation, we therefore localized VirD4ΔN87 to cell poles and the midcell by fusion to DivIVA. As shown in Fig. 3B, most (~ 80%) Mx355(pKV43) cells producing DivIVA-VirD4ΔN87 and GFP-VirE2 displayed fluorescent foci at the cell poles and a fraction (~ 10%) also showed midcell fluorescence. Both the percentages of cells with GFP localization and the ratio of cells with polar versus polar and midcell localization closely resembled values obtained with targeting mediated by full-length VirD4 (Fig. 3B).
Furthermore, in the complementary biochemical assay, the anti-VirD4 antiserum, but not preimmune serum, co-precipitated a presumptive complex of VirD4ΔN87 and VirE2 from DSP-treated membranes of strain Mx355(pKA43) (Fig. 3A). Conversely, the anti-VirE2 antiserum, but not preimmune serum, co-precipitated VirE2 and VirD4ΔN87 (Fig. 3A). Together, these findings indicate that VirE2 interacts with a domain of VirD4 exposed to the cytoplasm.
Localization of a VirD4 interaction domain to the C terminus of VirE2
A recent study suggested that the C terminus of VirE2 carries a signal required for export to plant cells (Simone et al., 2001). To test whether the C terminus mediates the VirE2–coupling protein interaction, we introduced VirE2′ truncation derivatives fused at their NH2 termini to GFP’C into a strain producing VirD4-N’GFP. As shown in Fig. 4A, cells producing VirD4-N’GFP and GFP’C fused to VirE2 derivatives carrying the last 100 residues fluoresced brightly at the cell poles. Cells producing VirD4-N’GFP and GFP’C fused to the last 50 residues of VirE2 lacked BiFC; however, in contrast to the other fusion proteins characterized, GFP’C-VirE2Δ1–483 was unstable as determined by immunoblot analysis (Fig. 4B). Cells producing GFP’C fused to VirE2′ fragments deleted of the C-terminal 100 or 9 residues lacked detectable fluorescence, as did cells producing GFP’C fused to full-length VirE2 bearing a 31-residue peptide insertion four residues from the C terminus (Fig. 4A). These findings indicate that the C-terminal 100 residues of VirE2 are both necessary and sufficient for targeting to VirD4 in vivo, and they further implicate the extreme C terminus in this targeting activity. Consistent with this proposal, we found that cells producing VirE2 fused at its C terminus to GFP were exclusively uniformly fluorescent even when VirD4 was produced at an elevated level from an IncP replicon (data not shown). We suspect that in this configuration GFP sterically blocks the C terminus of VirE2 from interacting with the coupling protein (see Discussion). The interaction studies described in Fig. 4 were carried out with wild-type A348, and similar results were obtained with other host strains, including Mx355 (virD4−) and KE1 (virE1−,virE2−) respectively (data not shown).
Fig. 4.

Localization of a VirD4 interaction domain to the C terminus of VirE2.
A. A348 producing VirD4-N’GFP and GFP’C fused to the VirE2 derivatives listed at the left and schematically represented were examined at 10 h (top two panels) or 18 h (bottom six panels) post induction by fluorescence microscopy. The proteins indicated were produced from the following plasmids: D4-N’GFP (pKAB64); GFPC’-E2 (pZDB89); GFPC’-E2Δ1–84 (pZDB838); GFPC’-E2Δ1–331 (pZDB834); GFPC’-E2Δ1–437 (pZDB836); GFPC’-E2Δ1–483 (pZDB122); GFPC’-E2Δ437–533 (pZDB846); GFPC’-E2i31–529 (pZDB827); GFPC’ (pKVB39); GFP-VirE2Δ524–533 (pZDB120). ‘ +’, fluorescent foci. ‘−’, no detectable foci, although these cells exhibited weak uniform fluorescence at t = 18.
B. Immunodetection of fusion proteins produced in A348 derivatives at 10 h postinduction. The proteins listed above each lane were synthesized from the IncP plasmids listed in (A). Blots were developed with the antisera listed at the right. The reactive species (~60-kDa) in all lanes, except At12516 (virE2−), reactive with the anti-VirE2 antisera is native VirE2 produced from pTi.
VirE2 targeting to VirD4 independently of the T-strand-VirD2, virB-encoded Mpf and T-pilus, and VirE1 secretion chaperone
VirE2 translocation to plant cells requires in addition to VirD4 the VirB proteins (Stahl et al., 1998; Vergunst et al., 2000) and the VirJ/AcvB periplasmic proteins whose functions are related to biogenesis or function of this type IV secretion system (Kalogeraki and Winans, 1995; Pantoja et al., 2002). To define the contributions of Mpf-associated proteins to the VirE2–VirD4 interaction, we introduced the relevant expression constructs for C2H and BiFC screens into various A. tumefaciens mutants. As shown in Fig. 5A, first, we observed that the virD2 mutant Mx311, defective for processing of the T-strand, displayed VirD4-dependent targeting of VirE2 to the cell poles. This finding is consistent with previous work showing that VirE2 is exported independently of the T-DNA to plant cells (Citovsky et al., 1992; Vergunst et al., 2000). Second, the virB operon deletion mutant PC1000 and the acvBΔvirJ double mutant, both defective for Mpf-dependent substrate transfer, also displayed the VirE2–VirD4 interaction. VirD4 therefore retains the capacity to interact with VirE2 in the cytoplasm independently of any interactions with Mpf subunits required for substrate transfer.
Fig. 5.
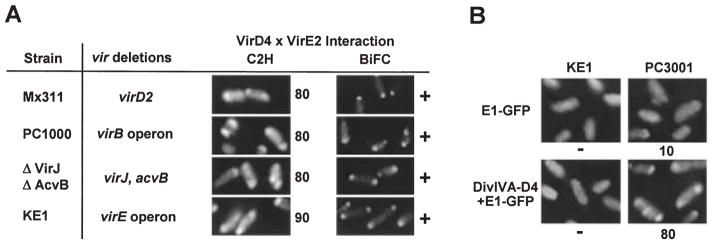
VirE2 targeting to VirD4 independently of T-strand-VirD2, Mpf proteins and VirE1 secretion chaperone.
A. Complex formation assessed with the C2H and BiFC screens. The A348 mutants listed at the left co-produced VirD4 and GFP-E2 from pKA77 [C2H], or D4-N’GFP and GFPC’-E2 from pKAB64 and pZDB89). Strains were examined 10 h post induction by fluorescence microscopy. The numbers to the right of panels depicting the C2H results correspond to the percentage of cells with polar fluorescence. For BiFC, ‘+’, fluorescent foci; ‘−’, no detectable foci.
B. VirD4 recruitment of VirE2 as a complex with VirE1. KE1 and PC3001 (ΔvirE1) producing E1-GFP from pZDB12, or DivIVA-D4 and E1-GFP from pZD76 + pZDB12 were examined 4 h after induction with 200 μM AS by fluorescence microscopy.
Translocation of VirE2 to plant cells also depends on complex formation with the VirE1 secretion chaperone (Sundberg et al., 1996). Interestingly, cells co-producing the tagged VirE2 and VirD4 proteins in the absence of VirE1 synthesis displayed polar fluorescence by C2H and BiFC (Fig. 5A). Furthermore, with the C2H screen, the percentage of polar foci correlated with the level of VirD4 production, e.g. ~25% for KE1(pZDB73) producing VirD4 from pTi and ~80–90% for KE1(pKA77) overproducing VirD4 from an IncP plasmid; these values are similar to those described above for cells co-producing these same proteins together with VirE1 (Fig. 1A). KE1(pKV42) cells co-producing DivIVA-VirD4 and GFP-VirE2 in the absence of VirE1 also displayed both polar and midcell fluorescence (data not shown). The VirE1 chaperone therefore is not required for recruitment of VirE2 to the coupling protein.
Finally, we previously reported that a VirE1-GFP fusion protein interacts with and directs the export of VirE2 (Zhou and Christie, 1999). Of considerable interest, on cytological examination we found that VirE1-GFP-producing cells display polar fluorescence by a VirD4-and VirE2-dependent mechanism. KE1(pZDB12) cells producing VirE1-GFP and VirD4 but no VirE2 were exclusively uniformly fluorescent even with prolonged incubation (>24 h) (Fig. 5B). Similarly, Mx355(pZDB12) cells producing VirE1-GFP and VirE2 but no VirD4 were uniformly fluorescent (data not shown). In striking contrast, an appreciable fraction (~10% at t = 4; ~30% at t = 10) of PC3001(pZDB12) cells producing VirE1-GFP, VirE2 and VirD4 (the latter two proteins from pTi) displayed polar fluorescence (Fig. 5B). An estimated 80% of PC3001(pZDB12, pZD76) cells producing VirE1-GFP, VirE2, and DivIVA-VirD4 (all from IncP) displayed polar fluorescence at t = 4. Approximately 15% of these cells also displayed midcell fluorescence. These findings support a proposal that VirE1 binding to VirE2 temporally precedes the delivery of VirE2 to the VirD4 coupling protein.
Discussion
The VirD4-like proteins are ubiquitous components of bacterial conjugation machines. There is strong genetic and biochemical evidence that these coupling proteins function as specificity determinants for DNA substrates by virtue of their capacity to recognize and bind protein components of the relaxosome (Cabezon et al., 1997; Hamilton et al., 2000; Hormaeche et al., 2002; Schroder et al., 2002). In addition, recent structural findings suggest the coupling proteins also function as inner membrane DNA translocases to direct the movement of DNA substrates across the inner membrane by an ATP-dependent mechanism (Errington et al., 2001; Gomis-Ruth et al., 2001; Llosa et al., 2002; Schroder et al., 2002). Coupling proteins also are common components of TFS systems shown to translocate protein substrates, but until now their role in this process was unspecified. Here, we show for the first time that a protein substrate of a TFS system interacts with a coupling protein. Our findings add to a general understanding of TFS-dependent protein trafficking pathways, as summarized below.
VirE2 processing and recruitment to the TFS
For VirE2 translocation, the effector must be first synthesized in a translocation-competent form and then delivered to the TFS machine. The virE1 chaperone gene plays an early regulatory role, contributing both translationally and post-translationally to synthesis of a stable form of the effector protein (Zhao et al., 2001). The VirE1 chaperone stabilizes VirE2 by binding to the NH2 terminus and also to a central region of VirE2 located between residues 320 and 390 (Deng et al., 1999; Sundberg and Ream, 1999; Zhou and Christie, 1999; Zhao et al., 2001). VirE2 forms homomultimers (Deng et al., 1999; Sundberg and Ream, 1999; Zhou and Christie, 1999) and binds ssDNA (see Ward and Zambryski, 2001), but these properties appear to be important for effector functions following delivery of VirE2 to the plant cell, because chaperone binding clearly prevents formation of higher-order VirE2 multimers or aggregates in vitro (Deng et al., 1999) and in the bacterium (Zhao et al., 2001). Additionally, contrary to an early prediction (Christie et al., 1988), there is now strong evidence that VirE2 is exported independently of any association with T-DNA in A. tumefaciens (Binns et al., 1995; Vergunst et al., 2000). Thus, reminiscent of several type III secretion (TTS) chaperones (Feldman and Cornelis, 2003), VirE1 functions early in the VirE2 translocation pathway by (i) stabilizing a presumably translocation-competent form of VirE2 and (ii) preventing the effector from prematurely interacting with T-DNA or protein transfer intermediates (Deng et al., 1999; Sundberg and Ream, 1999).
Our present studies defined requirements for presentation of VirE2 to the secretion machine. Most importantly, we showed that VirE2 interacts with the VirD4 coupling protein. Our cytological evidence for this interaction is that GFP-VirE2 localizes to A. tumefaciens cell poles in a VirD4-dosage-dependent manner, and, furthermore, GFP-VirE2 is relocalized to the midcell upon placement of VirD4 at this site by fusion to DivIVA. The fluorescence patterns observed with the presumptive GFP-VirE2–DivIVA–VirD4 C2H interaction closely resemble patterns detected with DivIVA-GFP (Edwards et al., 2000; Ding et al., 2002), and with other C2H interactions we have tested, e.g. VirE1–VirE2, VirB8–VirB10, VirB10–VirB10 (Ding et al., 2002), and VirD4–VirD4 (Fig. 2A). Additionally, our adaptation of the BiFC screen (Hu et al., 2002) for GFP as the protein–protein interaction sensor molecule also yielded the predicted results, namely, homogeneous fluorescence expected of soluble VirE1-VirE2 complex formation (Zhao et al., 2001), and bright fluorescent foci expected of the VirE2–VirD4 interaction at the cell poles. Finally, we confirmed that the native forms of VirE2 and VirD4 interact by a coupled cross-linking-immunoprecipitation assay. We have been unable to obtain evidence for interactions between full-length VirD4 or its cytoplasmic domain (VirD4ΔN87) and VirE2 by use of a dihybrid screen in the heterologous yeast host (data not shown). Perhaps this is not surprising, though, given the predicted biochemical properties of this coupling protein, e.g. integral inner membrane protein, homohexameric structure, ATP-binding/hydrolysis activity and results of our immunoprecipitation studies suggesting this is probably a weak-affinity or transient interaction.
It is also important to note that, in fact, only a small fraction of total cellular VirE2 is recovered as a complex with VirD4. Thus, at least for cells induced with AS in laboratory media, VirE2 accumulates as distinct pools, a cytoplasmic VirE1–VirE2 complex constituting the major fraction, a membrane-associated form at low levels (~ 5%) (Christie et al., 1988), and the VirE2–VirD4 complex at even lower levels (<1%). Although this is problematic for detecting the effector–coupling protein interaction, the accumulation VirE2 in different cellular compartments is probably an important feature of this translocation pathway. Conceivably, the cytosolic VirE1–VirE2 complex contributes in an undefined way to the assembly or function of this translocation system. Alternatively, it might simply serve as a reservoir of translocation-competent substrate for recruitment on demand to the TFS machine, e.g. upon establishment of productive contact between the bacterium and plant target cells.
Cytological dihybrid screens are ideal for characterizing the dynamics of protein–protein interactions in vivo, especially complexes that assemble at a specific site in a cell (Ding et al., 2002; Hu et al., 2002). For example, in this study, the assignment of a TFS coupling protein to the cell poles (Kumar and Das, 2002) permitted an examination of a question of long-standing interest in the field of protein secretion – does the secretion chaperone contribute a piloting or targeting function necessary for substrate delivery to the translocon? By monitoring polar targeting of GFP-VirE2 in a virE1 mutant strain, we showed that VirE2 retains its capacity to interact with VirD4 even in the absence of its chaperone, thus arguing against a role for the chaperone in presentation of this substrate to the translocase. However, we further discovered that VirE1-GFP is recruited to the cell poles in a VirD4- and VirE2-dependent manner. This finding is completely consistent with the genetic and biochemical data described above indicating that the chaperone acts very early in this translocation pathway through stabilization and ‘bodyguard’ functions. VirE1 is the first identified chaperone of any TFS substrate, but as noted above, it shares several features with the TTS chaperones. Our findings thus extend our knowledge of how secretion chaperones function in diverse macromolecular secretion pathways.
A C-terminal secretion signal for TFS substrates
Our cytological studies of GFP fused to VirE2 truncation derivatives supplied evidence for a C-terminal secretion signal. All GFP fusions to VirE2 truncation derivatives bearing an intact C terminus, including a fusion to the C-terminal 100 residues of VirE2, displayed a VirD4 interaction. Conversely, GFP fusions to VirE2 derivatives lacking these C-terminal residues failed to interact with VirD4 (Fig. 4A). The C terminus of VirE2 therefore carries information necessary and sufficient for complex formation with VirD4. In view of these findings, it is of interest that a Chou-Fasmann secondary structure analysis suggests that the C terminus of VirE2 is unstructured and thus might dangle from the rest of the protein (Sen et al., 1989). Accessibility of an intact C terminus might be an important feature for substrate docking, as suggested by studies showing that small C-terminal deletions (Fig. 4A), or additions of GFP (VirE2-GFP) (Zhou and Christie, 1999) a FLAG epitope (Simone et al., 2001), or an i31 peptide (VirE2.i31.529) (Zhou and Christie, 1999) at or near the C terminus block VirE2 delivery to plant cells. Conversely, modifications of the NH2 terminus of VirE2, e.g. addition of a FLAG epitope (Simone et al., 2001), an i31 peptide (Zhou and Christie, 1999), or Cre recombinase (Vergunst et al., 2000), are completely tolerated. The GFP-VirE2 fusion protein characterized in this study does not complement a virE2− mutation with respect to restoration of virulence, raising the possibility that GFP blocks translocation at a step following recruitment to VirD4. However, this question needs further study because Cre-VirE2, a protein that also does not substitute for wild-type VirE2 function in plants, is nevertheless translocated to plant and yeast cells as shown by Cre-mediated recombination at lox sites (Vergunst et al., 2000; Schrammeijer et al., 2003).
Two independent lines of study support the notion that VirE2 and other protein substrates of this TFS system carry C-terminal secretion signals. First, early evidence that VirE2 functions as an effector upon delivery to the plant cells was derived from the finding that an avirulent virE2 mutant incites wild-type tumours on VirE2-producing transgenic plants (Citovsky et al., 1992). By use of transgenic plants in infection assays, the Binns laboratory recently showed that the extreme C-terminal 18 residues of VirE2 is necessary for TFS-dependent translocation across the A. tumefaciens envelope, but this region is completely dispensible for VirE2 effector function when produced in transgenic plants (Simone et al., 2001). Second, the Hooykaas laboratory has reported that a C-terminal fragment of VirE2 (A. Vergunst, 23rd Crown Gall Conference, St Paul, MN) and also of VirF (Vergunst et al., 2000) can mediate the transfer of Cre recombinase to plant cells by a virB/virD4-dependent mechanism. Very interestingly, the Hooykaas (Vergunst et al., 2000; Schrammeijer et al., 2003) and Binns (Simone et al., 2001) laboratories also have reported that VirE2 and other protein substrates, e.g. VirF and VirE3, of the virB/virD4-encoded TFS pathway carry a conserved Arg-Pro-Arg motif at their C termini. This motif begins 10 residues from the end of VirE2 and thus its removal might explain our finding that GFP-VirE2Δ524–533 fails to target to VirD4 (Fig. 4A). It is intriguing to propose that this motif mediates targeting to the coupling protein for all substrates of this TFS system. Consistent with such a model, VirD2 relaxase, a protein that is co-translocated with the T-strand to plant cells, carries an Arg-Pro-Arg sequence near its C terminus, and we recently determined that GFP-VirD2 targets to VirD4 in a low percentage (~ 5%) of cells (K. Atmakuri, unpubl. obs). Further studies more precisely defining the nature of the presumptive C-terminal secretion signal of these and other TFS substrates are underway in several laboratories.
VirE2 translocation across the cell envelope
Upon recruitment to the coupling protein, in the next step of this translocation pathway, how are DNA or protein substrates of TFS systems delivered across the inner membrane? Several lines of evidence strongly favour the notion that VirD4 and the VirB proteins co-ordinate their activities by assembling as a supramolecular structure that spans the cell envelope for substrate transfer. First, early genetic studies established that all of these proteins are required for substrate transfer (Berger and Christie, 1994), and, more recently, genetic evidence was presented for functional interactions between VirD4, the VirB11 ATPase (Sagulenko et al., 2001) and the polytopic membrane protein VirB6 (Jakubowski et al., 2003). Second, VirD4 and several of the VirB proteins considered to be structural components of the transfer channel, e.g. VirB7 through VirB10 also were recently shown to co-fractionate on passage of detergent-solubilized cell extracts through gel filtration columns (Krall et al., 2002). Finally, cell biology studies have identified the cell pole as a likely site for assembly and action of the TFS machine. In addition to polar VirD4 (Kumar and Das, 2002), and substrate recruitment to this site (this study), the virB-encoded T-pilus assembles at the cell poles (Lai et al., 2000), and A. tumefaciens cells are often seen attached in a polar manner to plant cells during infection (Matthysse, 1987).
At this juncture, we favour one of two models describing this stage of translocation. According to the first, VirD4 physically interacts with inner membrane VirB proteins to form a transfer channel for both DNA and protein substrates. Intriguingly, homologues of VirD4 and VirB11 present as homohexameric rings with channel diameters large enough to accommodate DNA and unfolded or partially folded protein substrates (Yeo et al., 2000; Gomis-Ruth et al., 2001); these proteins thus might form a stacked ring channel structure and utilize the energy of ATP hydrolysis to drive substrate transfer. The second model is a slightly modified version of the ‘shoot and pump’ model of F. de la Cruz and co-workers (Llosa et al., 2002). On the assumption that the T-DNA is co-transferred to recipient cells with a relaxase protein covalently associated at its 5′ end, this model proposes that the TFS machine is a dedicated protein transporter that recognizes and ‘shoots’ the relaxase to the recipient cell, whereas the coupling protein co-ordinately acts as a molecular motor to ‘pump’ the T-DNA across the cell envelope (Llosa et al., 2002). To accommodate our present findings, we suggest the coupling protein could function as a general recruitment factor for both DNA (via recognition of relaxosome subunits) and protein substrates of TFS systems. For translocation of a bona fide protein substrate, the first step is substrate recruitment to the coupling protein via an interaction with a C-terminal secretion signal, and the second is substrate delivery to the Mpf proteins for protein trafficking. In future studies, it should be feasible to discriminate between these two models by defining a temporal order of substrate contacts with TFS machine components.
Experimental procedures
Bacterial strains, plasmids and growth conditions
Escherichia coli strain DH5α served as the host for general plasmid construction and maintenance, and strain BL21(DE3) was used for protein overexpression for antibody production (see below). Agrobacterium tumefaciens A348 is strain C58 bearing the octopine-type pTiA6NC plasmid (Zhu et al., 2000). A348 mutants used in this study are: Mx355 (virD4−) and Mx311 (virD2−) (Stachel and Nester, 1986); KE1 (ΔvirE operon) (McBride and Knauf, 1988); PC3001 (virE1−) (Zhao et al., 2001); At12516 (virE2−) (Fullner and Nester, 1996); PC1000 (ΔvirB operon) (Fernandez et al., 1996); A348ΔvirJΔacvB, a gift from S. C. Winans and V. S. Kalogeraki. Escherichia coli was maintained on Luria–Bertani (LB) medium at 37°C and A. tumefaciens strains were grown in LB supplemented with mannitol and glutamate at 28°C (Zhou and Christie, 1999). Conditions for induction of the A. tumefaciens vir genes in ABIM medium [glucose-containing minimal medium (pH 5.5), 1 mM phosphate, 200 μM acetosyringone (AS)] have been described previously (Zhou and Christie, 1999). Medium was supplemented with antibiotics (in μg μl−1) as follows: for E. coli, kanamycin (50), carbenicillin (100), and gentamicin (20); and for A. tumefaciens, kanamycin (100), carbenicillin (100), gentamicin (100), tetracycline (5), spectinomycin (300) and streptomycin (300).
Table 1 lists relevant properties of plasmids constructed for the present study. ColE1 plasmids were introduced into A. tumefaciens by ligation to the IncP plasmids pSW213 (Chen and Winans, 1991) or pXZ153 (Zhou and Christie, 1999), or pBBR1MCS2 which is compatible with IncP, IncQ and IncW plasmids (Kovach et al., 1994). Such co-integrate plasmids were given the ColE1 plasmid name plus a B to indicate ligation to a broad-host-range (BHR) replicon. For introduction of two ColE1 plasmids into the same A. tumefaciens strain, plasmids were first ligated to compatible BHR replicons, then the resulting cointegrate plasmids were introduced into A. tumefaciens with appropriate antibiotic selection.
Construction of plasmids with virD4 and virD4ΔN87
virD4 was PCR-amplified as a 2.17 kb fragment using the oligonucleotide primers 5′-CGGTGAACATATGAATTCCAGCAA-3′ and 5′-TCACTCGAGGCATCAGCCTG-3′ with underlined bases indicating NdeI and XhoI restriction sites respectively. Plasmid pMY1153 carrying the complete virD operon of pTiA6NC (Yanofsky et al., 1986) served as template. PCR-amplified virD4 was confirmed by sequencing, and the corresponding NdeI–XhoI fragment carrying virD4 was substituted for virB1 in pPC914KS+ (Berger and Christie, 1994) to obtain pKA9 expressing PvirB-virD4. A ~2.75-kb XbaI–KpnI fragment carrying PvirB-virD4 from pKA9 was introduced into similarly digested pXZ153 to obtain pKA21. Strain Mx355 (virD4−) transformed with pKA21 exhibited wild-type virulence, establishing that PvirB-virD4 expression yields a fully functional VirD4 protein (data not shown).
VirD4ΔN87, a derivative lacking the NH2-terminal 87 residues comprising a periplasmic loop and flanking trans-membrane α-helices, was PCR-amplified with the primer 5′-CTCATCATACATATGCGCAATC-3′ (NdeI underlined) and the XhoI-containing primer mentioned above. Amplified virD4ΔN87 (1.9-kb) was substituted as an NdeI–XhoI fragment for wild-type virD4 in pKA9 to obtain pKA38 expressing PvirB-virD4ΔN87. A 2.5-kb XbaI–KpnI fragment carrying PvirB-virD4ΔN87 from pKA38 was introduced into pXZ153 to obtain pKA43. As expected, PvirB-virD4ΔN87 expression failed to complement the virD4 null mutation of strain Mx355 (data not shown).
Construction of fusion proteins for the C2H assay
DivIVA fusions. Plasmid pZD6 expressing Ptac-divIVA-GFP is a derivative of the IncQ plasmid pMMB22 (Ding et al., 2002). Plasmid pKA6 expressing PBAD-divIVA-virD4 was obtained by substituting virE1 of pZD1398 (Ding et al., 2002) with a 2.1 kb NdeI-XhoI fragment carrying virD4 from pKA9. For pZD76 expressing Ptac-divIVA-virD4, a 2.1 kb XbaI-XhoI fragment carrying virD4 from pKA6 was substituted for GFP of pZD6. For pKV36 expressing Ptac-divIVA-virD4ΔN87, a ~ 1.9 kb NdeI-XhoI fragment from pKA38 carrying virD4ΔN87 was substituted for virD4 in pZD76. For pKA76 expressing Ptac-divIVA, pZD76 was digested with XbaI and XhoI to excise virD4, the restriction sites were made blunt-ended, and the plasmid was religated. A translational stop codon resides immediately downstream of the divIVA coding sequence.
GFP fusions. Plasmid pZD69 expressing PvirB-GFP was constructed by introducing a 0.8 kb NdeI-KpnI fragment from pXZ63 (Rashkova et al., 2000) released by partial digestion into similarly digested pPC914KS+ (Berger and Christie, 1994). For pZD72 expressing PvirB-GFP-virE2, a 2.5 kb NcoI-KpnI fragment containing a fragment of GFP through the end of virE2 from pXZ65 was introduced into similarly digested pZD69. pXZ65, expressing Plac-GFP-virE2, was constructed by introducing a 1.6 kb NdeI/KpnI fragment containing virE2 from pPC725 (Zhou and Christie, 1999) into SmaI/KpnI-digested pXZ63. pZD73 is pZD72 with a Kanr cassette from pUC4K (Pharmacia) inserted into a unique ScaI site located in the Crbr gene. To construct pKA59 expressing PvirB-virD4-GFP, we first removed a BamHI site from pKA9 by digestion, blunt-ending, and religation. We then introduced a BamHI site at the 3′ end of virD4, immediately upstream of its stop codon, by PCR amplification using the NdeI-containing primer mentioned above for pKA9 and the primer 5′-ACGAATTGCACTCGAGTTAAATTGGATCCGGTGCGGCCGCATTTCGCAGGC-3′ (BamHI underlined). Finally, GFP from pXZ63 was introduced as a 0.8 kb BamHI-KpnI fragment to yield pKA59. A similar strategy was used to construct pKA75 expressing PvirB-virD4ΔN87-GFP, first removing a BamHI site from pKA38, then introducing a BamHI site immediately upstream of the virD4ΔN87 stop codon by PCR amplification with primers 5′-CTCATCATACATATGCGCAATC-3′ (NdeI site underlined) and the BamHI-containing primer mentioned above for pKA59, and, finally, introducing GFP as a 0.8 kb BamHI-KpnI fragment from pXZ63.
Construction of fusion proteins for the BiFC assay
We PCR-amplified the halves of GFP such that the 5′ half (termed N’GFP) terminates at codon 154, and the 3′ half (GFP’C) begins at the ATG codon at position 153. Codon 154 served as the junction for the YFP halves in the original BiFC assay (Hu et al., 2002). We first constructed pZD88 expressing PvirB-virE1-N’GFP. This was achieved by PCR-amplification of a fragment composed of virE1 from a unique SphI site near its 5′ end to codon 154 of GFP (GFP’N) by use of oligonucleotide primers 5′-GCCATCATCAAGCCGCATGCGAAC-3′ (SphI underlined) and 5′-CCGGTACCTCATGCCATGATGTATACATTGTGTG-3′ (KpnI underlined; codon 154 in bold) and template plasmid pZD12 (expresses PvirB-virE1-GFP) (Ding et al., 2002). Next, the SphI-KpnI fragment from pZD86 containing virE1′-N’GFP was substituted for a corresponding fragment of pZD12 bearing virE1’ fused to full-length GFP to yield pZD88 (PvirB-virE1-N’GFP). A similar strategy was used to construct pZD89 expressing PvirB-GFP’C-virE2. First, primers 5′-TTCATATGGCAGACAAACAAAAGAATGGAATC-3′ (NdeI underlined; Met codon 153 in bold) and 5′-TAAAAGATCTCTGTGCCAGTGATCCC–3′ (BglII underlined) were used for amplification of GFP’C fused to a fragment of virE2 from its start codon to an internal BglII site from template plasmid pZD72 (expresses PvirB-GFP-virE2). Next, the NdeI-BglII fragment containing GFP’C-virE2′ was substituted for a corresponding fragment of pZD72 to yield pZD89 (PvirB-GFP’C-virE2). To construct a versatile vector for expressing GFP’C gene fusions, we amplified the GFP’C fragment with flanking NcoI and NdeI sites with the oligonucleotide primers 5′-TTCCATGGCAGACAAACAAAAGAATGGAATC-3′ (NcoI underlined, Met codon 153 in bold) and 5′-TTCATATGGGGATCCTTGTATAGTTCATCCATG-3′ (NdeI underlined) with plasmid pZD89 as a template. We then introduced the NcoI–NdeI fragment carrying GFP’C into a derivative of pPC914KS+ to yield plasmid pZD109 expressing PvirB-GFP’C and downstream restriction sites for in-frame insertions of vir genes.
Plasmids pXZ728, pXZ724, and pXZ726 are pBSIISK+ plasmids carrying virE2 alleles with 31-residue insertions and a novel BamHI site (introduced by Tn3lacZ/In mutagenesis) immediately after codons 84, 331, and 437 respectively (Zhou and Christie, 1999). To produce GFP’C fused to VirE2 C-terminal fragments, virE2 gene fragments corresponding to codons 85–533, 332–533, and 438–533 were isolated as BamHI-XhoI fragments from the above pXZ plasmids and introduced into plasmid pZD109 to yield pZD838, pZD834 and pZD836 respectively. For pZD122 expressing PvirB-GFP’C-virE2Δ1–483, first, a virE2′ fragment corresponding to codons 484 through 533 (the end of the gene) was PCR-amplified with primers 5′-TGGATCCACATATGAGGCTGCCAGCCGATGCAGCGGG-3′ (NdeI underlined) and 5′-TTCTCGAGTCAAAAGCTGTTGACGCTTTGGCTACG-3′ (XhoI underlined) using template plasmid pZD72. Next, the NdeI-XhoI fragment bearing the C-terminal 50 residues of virE2 was introduced downstream of GFP in pZD109. For pZD846 expressing PvirB-GFP’C-virE2Δ437–533, the 1.4-kb NdeI-BamHI containing the relevant virE2′ fragment from pXZ726 was substituted for full-length virE2 in pZD109. Similarly, for pZD827 expressing PvirB-GFP’C-virE2.i31–529, the NdeI/XhoI fragment from pXZ727 containing virE2.i31–529 was substituted for full-length virE2 in pZD109.
For plasmid pKA64 expressing PvirB-virD4-N’GFP, the ~ 2.2-kb NdeI fragment extending from the start-site of virD4 through an internal NdeI site in GFP of pKA59 was substituted for the corresponding fragment of pZD88 (PvirB-virE1-N’GFP; an NdeI site is at the beginning of virE1). Similarly, for pKV38 expressing PvirB-virD4ΔN87-N’GFP, the 1.9-kb NdeI fragment from pKA75 was substituted for the corresponding fragment of pZD88. For pKV35 expressing PvirB-N’GFP, the ~ 0.4-kb NcoI-KpnI fragment from pZD88 was substituted for the 0.6-kb NcoI-KpnI fragment of pZD71 (a Kanr-derivative of pZD69 expressing PvirB-GFP). For pKV39 expressing PvirB-GFP’C, initially a ~ 1.9-kb NcoI-KpnI fragment harbouring GFP’C-E2 from pZD109 was introduced into the corresponding sites of a derivative of pPC914KS+ (Berger and Christie, 1994) to obtain pKV38. This plasmid was then digested with NdeI-EcoRV to remove virE2, the NdeI site was made blunt-ended and the plasmid religated.
Microscopy and image analysis
Freshly transformed cells were used for the cytology studies. A. tumefaciens strains were incubated in the presence of AS (200 μM) for expression from the PvirB promoter and IPTG (25 μM) for expression from the Ptac promoter. Cells were examined for GFP localization by fluorescence microscopy. Each experiment was replicated at least three times. Results of the C2H assay are presented as a percentage of cells showing a discernible fluorescence pattern. Images of cells were acquired with an Olympus BX60 microscope equipped with a 100× oil immersion phase-contrast objective as described previously (Ding et al., 2002). Throughout these studies, cells with GFP foci were routinely examined for the possible production of inclusion bodies by phase contrast or Nomarski microscopy.
Protein cross-linking, membrane solubilization and immunoprecipitation
Agrobacterium tumefaciens cultures (500 ml) were induced for vir gene expression in ABIM for 18 h at 22°C. Cells were harvested, lysed by French-Press treatment, and total membranes were recovered as previously described (Rashkova et al., 2000). Membranes were resuspended in 50 mM sodium phosphate buffer pH 7.4 containing 20% sucrose, 0.1 mg ml−1 DNase I, 0.1 mg ml−1 RNase A and EDTA-free protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN). Approximately 2 mg of total membrane in 200 μl of 10 mM sodium phosphate buffer (pH 7) was cross-linked with dithiobis (succinimidyl propionate) (DSP) (20 mg ml−1 stock, final concentration, 0.5 μg μl−1) for 30 min on ice, and the reaction was quenched with 0.5 M L-lysine pH 8.5 (final concentration, 62.5 mM) for 15 min on ice. The cross-linked sample was washed once with 50 mM Tris-HCl, pH 7.4, and the total membranes were pelleted at 4°C by centrifugation for 1 h at 125 000 g in a Beckman table-top ultracentrifuge. The pellet was solubilized with 1.5% N-Lauoryl sarcosine in 50 mM Tris-HCl, pH 7.4 and 1 mM EDTA, at 4°C for 4 h with gentle rocking. The solubilized material was centrifuged at 15 000 g for 15 min and used for immunoprecipitation.
The solubilized material was rocked gently at 4°C for 12 h with 10 mg Protein A-Sepharose CL-4B beads (Amersham Biosciences; Piscataway, NJ) to remove non-specific binding proteins. The samples were centrifuged for 5 min at 2000 g at 4°C, and the supernatant was divided in two for incubation with preimmune or immune sera. Samples were incubated with 10 mg of Protein A-Sepharose CL-4B beads for 12 h at 4°C with slow rocking. The samples were centrifuged at 2000 g, washed three times with 50 mM Tris-HCl, pH 7.4, 0.15% N-Lauroyl sarcosine, 1 mM EDTA, and resuspended in 5× Laemmli’s buffer. The samples were boiled for 5 min at 100°C and analysed by SDS-PAGE and immunostaining.
Proteins were resolved by sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE) or with a Tricine-SDS-PAGE system as previously described (Fernandez et al., 1996). Vir proteins were visualized by SDS-PAGE, transfer of the proteins to nitrocellulose membranes, and development of immunoblots with goat anti-rabbit antibodies conjugated to alkaline phosphatase.
Generation of anti-VirD4 antiserum. BL21(DE3,pKA22) was induced overnight with IPTG (0.1 mM) for PT7-his-virD4ΔN87 expression using the pAP1 expression vector (Burgess et al., 2000). Cells were lysed by French-Press treatment and total cellular proteins were subjected to SDS-PAGE. Overproduced His-VirD4ΔN87 protein was excised from the gels and sent to Cocalico Biologicals. (Reamstown, PA) for antibody production in New Zealand White rabbits. Anti-VirE2 and anti-GFP antibodies were generated previously (Zhou and Christie, 1999; Rashkova et al., 2000).
Acknowledgments
We thank T. Kerppola for sending strains and information about the BiFC assay, S. C. Winans and V. S. Kalogeraki for the ΔacvB, ΔvirJ strain and W. Margolin for the anti-GFP antiserum. We thank V. Krishnamoorthy for excellent technical assistance, and the members of the laboratory for helpful discussions and manuscript critiques. We gratefully acknowledge the financial support of the NIH, GM48746, for studies in this laboratory.
References
- Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- Baron C, O’Callaghan D, Lanka E. Bacterial secrets of secretion: Euro Conference on the biology of type IV secretion processes. Mol Microbiol. 2002;43:1359–1365. doi: 10.1046/j.1365-2958.2002.02816.x. [DOI] [PubMed] [Google Scholar]
- Berger BR, Christie PJ. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol. 1994;176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns A, Beaupre C, Dale E. Inhibition of VirB-mediated transfer of diverse substrates from Agrobacterium tumefaciens by the IncQ plasmid RSF1010. J Bacteriol. 1995;177:4890–4899. doi: 10.1128/jb.177.17.4890-4899.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RR, Arthur TM, Pietz BC. Mapping protein–protein interaction domains using ordered fragment ladder far-western analysis of hexahistidine-tagged fusion proteins. Methods Enzymol. 2000;328:141–157. doi: 10.1016/s0076-6879(00)28396-1. [DOI] [PubMed] [Google Scholar]
- Burns DL. Type IV transporters of pathogenic bacteria. Curr Opin Microbiol. 2003;6:1–6. doi: 10.1016/s1369-5274(02)00006-1. [DOI] [PubMed] [Google Scholar]
- Cabezon E, Sastre JI, de la Cruz F. Genetic evidence of a coupling role for the TraG protein family in bacterial conjugation. Mol Gen Genet. 1997;254:400–406. doi: 10.1007/s004380050432. [DOI] [PubMed] [Google Scholar]
- Chen CY, Winans SC. Controlled expression of the transcriptional activator gene virG in Agrobacterium tumefaciens by using the Escherichia coli lac promoter. J Bacteriol. 1991;173:1139–1144. doi: 10.1128/jb.173.3.1139-1144.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ. The Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. J Bacteriol. 1997;179:3085–3094. doi: 10.1128/jb.179.10.3085-3094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ. Type IV secretion: Intercellular transfer of macromolecules by systems ancestrally-related to conjugation machines. Mol Microbiol. 2001;40:294–305. doi: 10.1046/j.1365-2958.2001.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Ward JE, Winans SC, Nester EW. The Agrobacterium tumefaciens virE2 gene product is a single-stranded-DNA-binding protein that associates with T-DNA. J Bacteriol. 1988;170:2659–2667. doi: 10.1128/jb.170.6.2659-2667.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citovsky V, Zupan J, Warnick D, Zambryski P. Nuclear localization of Agrobacterium VirE2 protein in plant cells. Science. 1992;256:1802–1805. doi: 10.1126/science.1615325. [DOI] [PubMed] [Google Scholar]
- Conover GM, Derre I, Vogel JP, Isberg RR. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol. 2003;48:305–321. doi: 10.1046/j.1365-2958.2003.03400.x. [DOI] [PubMed] [Google Scholar]
- Dash PK, Traxler BA, Panicker MM, Hackney DD, Minkley EG., Jr Biochemical characterization of Escherichia coli DNA helicase I. Mol Microbiol. 1992;6:1163–1172. doi: 10.1111/j.1365-2958.1992.tb01555.x. [DOI] [PubMed] [Google Scholar]
- Deng W, Chen L, Peng WT, Liang X, Sekiguchi S, Gordon MP, Nester EW. VirE1 is a specific molecular chaperone for the exported single-stranded-DNA-binding protein VirE2 in Agrobacterium. Mol Microbiol. 1999;31:1795–1807. doi: 10.1046/j.1365-2958.1999.01316.x. [DOI] [PubMed] [Google Scholar]
- Ding Z, Zhao Z, Jakubowski S, Krishnamohan A, Margolin W, Christie PJ. A novel cytology-based, two-hybrid screen for bacteria applied to protein–protein interaction studies of a type IV secretion system. J Bacteriol. 2002;184:5572–5582. doi: 10.1128/JB.184.20.5572-5582.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disque-Kochem C, Dreiseikelmann B. The cytoplasmic DNA-binding protein TraM binds to the inner membrane protein TraD in vitro. J Bacteriol. 1997;179:6133–6137. doi: 10.1128/jb.179.19.6133-6137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DH, Thomaides HB, Errington J. Promiscuous targeting of Bacillus subtilis cell division protein DivIVA to division sites in Escherichia coli and fission yeast. EMBO J. 2000;19:2719–2727. doi: 10.1093/emboj/19.11.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J, Bath J, Wu LJ. DNA transport in bacteria. Nat Rev Mol Cell Biol. 2001;2:538–545. doi: 10.1038/35080005. [DOI] [PubMed] [Google Scholar]
- Feldman MF, Cornelis GR. The multitalented type III chaperones: all you can do with 15 kDa. FEMS Microbiol Lett. 2003;219:151–158. doi: 10.1016/S0378-1097(03)00042-9. [DOI] [PubMed] [Google Scholar]
- Fernandez D, Dang TAT, Spudich GM, Zhou XR, Berger BR, Christie PJ. The Agrobacterium tumefaciens virB7 gene product, a proposed component of the T-complex transport apparatus, is a membrane-associated lipoprotein exposed at the periplasmic surface. J Bacteriol. 1996;178:3156–3167. doi: 10.1128/jb.178.11.3156-3167.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Haas R, Odenbreit S. Type IV secretion systems in pathogenic bacteria. Int J Med Microbiol. 2002;292:159–168. doi: 10.1078/1438-4221-00199. [DOI] [PubMed] [Google Scholar]
- Fullner KJ, Nester EW. Temperature affects the T-DNA transfer machinery of Agrobacterium tumefaciens. J Bacteriol. 1996;178:1498–1504. doi: 10.1128/jb.178.6.1498-1504.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigo JM. Natural conjugative plasmids induce bacterial biofilm development. Nature. 2001;412:442–445. doi: 10.1038/35086581. [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth FX, Moncalian G, Perez-Luque R, Gonzalez A, Cabezon E, de la Cruz F, Coll M. The bacterial conjugation protein TrwB resembles ring helicases and F1- ATPase. Nature. 2001;409:637–641. doi: 10.1038/35054586. [DOI] [PubMed] [Google Scholar]
- Grahn AM, Haase J, Bamford DH, Lanka E. Components of the RP4 conjugative transfer apparatus form an envelope structure bridging inner and outer membranes of donor cells: Implications for related macromolecule transport systems. J Bacteriol. 2000;182:1564–1574. doi: 10.1128/jb.182.6.1564-1574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CM, Lee H, Li PL, Cook DM, Piper KR, von Bodman SB, et al. TraG from RP4 and TraG and VirD4 from Ti plasmids confer relaxosome specificity to the conjugal transfer system of pTiC58. J Bacteriol. 2000;182:1541–1548. doi: 10.1128/jb.182.6.1541-1548.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormaeche I, Alkorta I, Moro F, Valpuesta JM, Goni FM, De La Cruz F. Purification and properties of TrwB, a hexameric, ATP-binding integral membrane protein essential for R388 plasmid conjugation. J Biol Chem. 2002;277:46456–46462. doi: 10.1074/jbc.M207250200. [DOI] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- Jakubowski S, Krishnamoorthy V, Christie PJ. Agrobacterium tumefaciens VirB6 proteins participates in assembly of VirB7 and VirB9 complexes required for type IV secretion. J Bacteriol. 2003;185:2867–2878. doi: 10.1128/JB.185.9.2867-2878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeraki VS, Winans SC. The octopine-type Ti plasmid pTiA6 of Agrobacterium tumefaciens contains a gene homologous to the chromosomal virulence geneAcvb. J Bacteriol. 1995;177:892–897. doi: 10.1128/jb.177.4.892-897.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach ME, Phillips RW, Elzer PH, Roop RM, II, Peterson KM. pBBR1MCS: a broad-host-range cloning vector. Biotechniques. 1994;16:800–802. [PubMed] [Google Scholar]
- Krall L, Wiedemann U, Unsin G, Weiss S, Domke N, Baron C. Detergent extraction identifies different VirB protein subassemblies of the type IV secretion machinery in the membranes of Agrobacterium tumefaciens. Proc Natl Acad Sci USA. 2002;99:11405–11410. doi: 10.1073/pnas.172390699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RB, Das A. Polar location and functional domains of the Agrobacterium tumefaciens DNA transfer protein VirD4. Mol Microbiol. 2002;43:1523–1532. doi: 10.1046/j.1365-2958.2002.02829.x. [DOI] [PubMed] [Google Scholar]
- Lai EM, Chesnokova O, Banta LM, Kado CI. Genetic and environmental factors affecting T-pilin export and T-pilus biogenesis in relation to flagellation of Agrobacterium tumefaciens. J Bacteriol. 2000;182:3705–3516. doi: 10.1128/jb.182.13.3705-3716.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessl M, Lanka E. Common mechanisms in bacterial conjugation and Ti-mediated T-DNA transfer to plant cells. Cell. 1994;77:321–324. doi: 10.1016/0092-8674(94)90146-5. [DOI] [PubMed] [Google Scholar]
- Llosa M, Gomis-Ruth FX, Coll M, de la Cruz F. Bacterial conjugation: a two-step mechanism for DNA transport. Mol Microbiol. 2002;45:1–8. doi: 10.1046/j.1365-2958.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- Matthysse AG. Characterization of nonattaching mutants of Agrobacterium. J Bacteriol. 1987;169:313–323. doi: 10.1128/jb.169.1.313-323.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KE, Knauf VC. Genetic analysis of the virE operon of the Agrobacterium Ti plasmid pTiA6. J Bacteriol. 1988;170:1430–1437. doi: 10.1128/jb.170.4.1430-1437.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncalian G, Cabezon E, Alkorta I, Valle M, Moro F, Valpuesta JM, et al. Characterization of ATP and DNA binding activities of TrwB, the coupling protein essential in plasmid R388 conjugation. J Biol Chem. 1999;274:36117–36124. doi: 10.1074/jbc.274.51.36117. [DOI] [PubMed] [Google Scholar]
- Nagai H, Roy CR. The DotA protein from Legionella pneumophila is secreted by a novel process that requires the Dot/Icm transporter. EMBO J. 2001;20:5962–5970. doi: 10.1093/emboj/20.21.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Pantoja M, Chen L, Chen Y, Nester EW. Agrobacterium type IV secretion is a two-step process in which export substrates associate with the virulence protein VirJ in the periplasm. Mol Microbiol. 2002;45:1325–1335. doi: 10.1046/j.1365-2958.2002.03098.x. [DOI] [PubMed] [Google Scholar]
- Rashkova S, Zhou XR, Christie PJ. Self-assembly of the Agrobacterium tumefaciens VirB11 traffic ATPase. J Bacteriol. 2000;182:4137–4145. doi: 10.1128/jb.182.15.4137-4145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees CED, Wilkins BM. Protein transfer into the recipient cell during bacterial conjugation: studies with F and RP4. Mol Microbiol. 1990;4:1199–1205. doi: 10.1111/j.1365-2958.1990.tb00695.x. [DOI] [PubMed] [Google Scholar]
- Sagulenko Y, Sagulenko V, Chen J, Christie PJ. Role of Agrobacterium VirB11 ATPase in T-pilus assembly and substrate selection. J Bacteriol. 2001;183:5813–5825. doi: 10.1128/JB.183.20.5813-5825.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond GPC. Secretion of extracellular virulence factors by plant pathogenic bacteria. Ann Rev Phytopath. 1994;32:181–200. [Google Scholar]
- Sastre JI, Cabezon E, de la Cruz F. The carboxyl terminus of protein TraD adds specificity and efficiency to F-plasmid conjugative transfer. J Bacteriol. 1998;180:6039–6042. doi: 10.1128/jb.180.22.6039-6042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrammeijer B, Dulk-Ras Ad A, Vergunst AC, Jurado Jacome E, Hooykaas PJ. Analysis of Vir protein translocation from Agrobacterium tumefaciens using Saccharomyces cerevisiae as a model: evidence for transport of a novel effector protein VirE3. Nucleic Acids Res. 2003;31:860–868. doi: 10.1093/nar/gkg179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder G, Krause S, Zechner EL, Traxler B, Yeo HJ, Lurz R, et al. TraG-like proteins of DNA transfer systems and of the Helicobacter pylori type IV secretion system: inner membrane gate for exported substrates? J Bacteriol. 2002;184:2767–2779. doi: 10.1128/JB.184.10.2767-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Pazour GJ, Anderson D, Das A. Cooperative binding of Agrobacterium tumefaciens VirE2 protein to single-stranded DNA. J Bacteriol. 1989;171:2573–2580. doi: 10.1128/jb.171.5.2573-2580.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone M, McCullen CA, Stahl LE, Binns AN. The carboxy-terminus of VirE2 from Agrobacterium tumefaciens is required for its transport to host cells by the virB-encoded type IV transport system. Mol Microbiol. 2001;41:1283–1293. doi: 10.1046/j.1365-2958.2001.02582.x. [DOI] [PubMed] [Google Scholar]
- Stachel SE, Nester EW. The genetic and transcriptional organization of the vir region of the A6 Ti. EMBO J. 1986;5:1445–1454. doi: 10.1002/j.1460-2075.1986.tb04381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl LE, Jacobs A, Binns AN. The conjugal intermediate of plasmid RSF1010 inhibits Agrobacterium tumefaciens virulence and VirB-dependent export of VirE2. J Bacteriol. 1998;180:3933–3939. doi: 10.1128/jb.180.15.3933-3939.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg CD, Ream W. The Agrobacterium tumefaciens chaperone-like protein, VirE1, interacts with VirE2 at domains required for single-stranded DNA binding and cooperative interaction. J Bacteriol. 1999;181:6850–6855. doi: 10.1128/jb.181.21.6850-6855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C, Meek L, Carroll K, Das A, Ream W. VirE1 protein mediates export of the single-stranded DNA-binding protein VirE2 from Agrobacterium tumefaciens into plant cells. J Bacteriol. 1996;178:1207–1212. doi: 10.1128/jb.178.4.1207-1212.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpirer CY, Faelen M, Couturier M. Interaction between the RP4 coupling protein TraG and the pBHR1 mobilization protein Mob. Mol Microbiol. 2000;37:1283–1292. doi: 10.1046/j.1365-2958.2000.02077.x. [DOI] [PubMed] [Google Scholar]
- Vergunst AC, Schrammeijer B, den Dulk-Ras A, de Vlaam CM, Regensburg-Tuink TJ, Hooykaas PJ. VirB/D4-dependent protein translocation from Agro-bacterium into plant cells. Science. 2000;290:979–982. doi: 10.1126/science.290.5493.979. [DOI] [PubMed] [Google Scholar]
- Ward DV, Zambryski PC. The six functions of Agrobacterium VirE2. Proc Natl Acad Sci USA. 2001;98:385–386. doi: 10.1073/pnas.98.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J, Dale EEM, Binns AN. Activity of the Agrobacterium T-DNA transfer machinery is affected by virB genes products. Proc Natl Acad Sci USA. 1991;88:9350–9354. doi: 10.1073/pnas.88.20.9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BM, Thomas AT. DNA-independent transport of plasmid primase protein between bacteria by the I1 conjugation system. Mol Microbiol. 2000;38:650–657. doi: 10.1046/j.1365-2958.2000.02164.x. [DOI] [PubMed] [Google Scholar]
- Yanofsky MF, Porter SG, Young C, Albright LM, Gordon MP, Nester EW. The virD operon of Agrobacterium tumefaciens encodes a site-specific endonuclease. Cell. 1986;47:471–477. doi: 10.1016/0092-8674(86)90604-5. [DOI] [PubMed] [Google Scholar]
- Yeo HJ, Savvides SN, Herr AB, Lanka E, Waksman G. Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV system. Mol Cell. 2000;6:1461–1472. doi: 10.1016/s1097-2765(00)00142-8. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Sagulenko E, Ding Z, Christie PJ. Activities of virE1 and the VirE1 secretion chaperone in export of the multifunctional VirE2 effector via an Agrobacterium type IV secretion pathway. J Bacteriol. 2001;183:3855–3865. doi: 10.1128/JB.183.13.3855-3865.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XR, Christie PJ. Mutagenesis of Agrobacterium VirE2 single-stranded DNA-binding protein identifies regions required for self association and interaction with VirE1 and a permissive site for hybrid protein construction. J Bacteriol. 1999;181:4342–4352. doi: 10.1128/jb.181.14.4342-4352.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Oger PM, Schrammeijer B, Hooykaas PJ, Farrand SK, Winans SC. The bases of crown gall tumorigenesis. J Bacteriol. 2000;182:3885–3895. doi: 10.1128/jb.182.14.3885-3895.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]