

Abstract
The tubulin cytoskeleton plays a key role in maintaining the characteristic quiescent discoid shape of resting platelets. Upon activation, platelets undergo a dramatic change in shape; however, little is known of how the microtubule system contributes to regulating platelet shape and function. Here we investigated the role of the covalent modification of α-tubulin by acetylation in the regulation of platelet physiology during activation. Superresolution microscopy analysis of the platelet tubulin cytoskeleton showed that the marginal band together with an interconnected web of finer tubulin structures collapsed upon platelet activation with the glycoprotein VI (GPVI)-agonist collagen-related peptide (CRP). Western blot analysis revealed that α-tubulin was acetylated in resting platelets and deacetylated during platelet activation. Tubacin, a specific inhibitor of the tubulin deacetylase HDAC6, prevented tubulin deacetylation upon platelet activation with CRP. Inhibition of HDAC6 upregulated tubulin acetylation and disrupted the organization of the platelet microtubule marginal band without significantly affecting platelet volume changes in response to CRP stimulation. HDAC6 inhibitors also inhibited platelet aggregation in response to CRP and blocked platelet signaling events upstream of platelet Rho GTPase activation. Together, these findings support a role for acetylation signaling in controlling the resting structure of the platelet tubulin marginal band as well as in the coordination of signaling systems that drive platelet cytoskeletal changes and aggregation.
Keywords: acetylation, HDAC6, platelets, tubulin
platelets are the primary cellular mediators of hemostasis (13, 18, 32). As discoid cellular fragments formed from the appendages of megakaryocytes, platelets patrol the circulation as guardians of vascular integrity. When platelets detect signals of vessel damage, they adhere to subendothelial matrix proteins, activate and aggregate with other platelets to potentiate thrombus formation to stem the leakage of blood. During this process, platelets change shape from discs to irregularly shaped spheres with finger-like pseudopods and form lamellipodial sheets to facilitate spreading. The characterization of the molecular and cellular regulators of platelet cytoskeletal organization will provide a better understanding of the role that changes in platelet morphology upon activation play in platelet physiology and function.
The discoid shape of quiescent platelets is maintained by a circumferential marginal band of microtubules that scaffolds the platelet inner periphery (44). This marginal band is made up of multiple dynamic, continually polymerizing tubulin coils of mixed polarity (31). During platelet activation, the marginal band disappears, collapsing towards the platelet center, in a process thought to promote platelet shape change; however, the exact manner in which marginal band dynamics regulate platelet structure and function is not well described. Moreover, the molecular signaling processes that regulate marginal band structure and platelet morphology are largely unexplored.
Despite the roles of microtubules in platelet generation (30, 34), platelet aggregation (26, 36, 37), thrombus formation (27), and clot retraction (35), little is known about how tubulin dynamics are regulated in platelets. In nucleated cells, a number of covalent modifications are hypothesized to regulate tubulin function; notably the acetylation of α-tubulin at Lys 40 has been suggested to have roles in maintaining microtubule stability (19). Like phosphorylation, the reversible nature of protein acetylation catalyzed by acetyltransferases and deacetylases serves as a signaling switch in diverse cellular processes (21, 45). Previous studies of the platelet marginal band have reported that platelet α-tubulin is heavily acetylated (31). More recently, platelets have been shown to express the tubulin deacetylase, HDAC6, and tubulin deacetylation has been demonstrated to regulate the kinetics of platelet spreading (33). However, the molecular mechanisms by which acetylation and HDAC6-mediated deacetylation of tubulin regulates platelet physiology remain unspecified.
Here, we demonstrate that upon activation, the platelet microtubule marginal band collapses towards the platelet center as platelets condense in volume. This centripetal redistribution of the marginal band is associated with the deacetylation of tubulin, which occurs downstream of Src tyrosine kinase activation but upstream of Rho GTPase signaling processes. Tubulin acetylation may play roles in marginal band organization and dynamics, as inhibitors of HDAC6 changed resting marginal band structure, altered marginal band collapse, and inhibited the efficiency of platelet aggregation. Together, our results provide evidence for a role of tubulin acetylation in the regulation of platelet marginal band structure and platelet function and suggest that acetylation may have roles in linking tubulin cytoskeletal processes to signaling events that underlie platelet activation.1
MATERIALS AND METHODS
Reagents.
Tubacin was from Enzo Life Sciences (Farmingdale, NY). Collagen-related peptide (CRP) was from Dr. Richard Farndale (Cambridge, UK). Y-27632 and TC-H 106 were from Tocris Bioscience (Bristol, UK). EHT 1864, PP2, BAY 61-3606, and trichostatin A (TSA) and anti-α-tubulin (T6199) and anti-acetylated tubulin (T7451) antisera were from Sigma-Aldrich (St. Louis, MO). Alexa Fluor secondary antibodies were from Invitrogen. HDAC1 (5356), HDAC2 (5113), HDAC4 (7628), HDAC7, pPAK (2606), PAK (2615), LIMK1 pThr508 (3841), LIMK1 (3842), phospho-MEK1/2 (9154), MEK1/2 (9126), pERK, tERK, and phospho-Akt (4056 and 4058) antibodies were from Cell Signaling Technologies. 4G10 monoclonal antibody was from Millipore. Anti-HDAC6, Akt and secondary goat anti-rabbit and anti-mouse horseradish peroxidase (HRP) secondary antibodies were from Santa Cruz Biotechnology.
Preparation of human washed platelets.
Human venous blood was drawn from healthy donors into sodium citrate and acid/citrate/dextrose as previously described (6, 25). Written informed consent was obtained from all study participants and the protocol was approved by the Oregon Health & Science University Institutional Review Board. Platelet-rich plasma (PRP) was prepared by centrifugation of anticoagulated blood at 200 g for 10 min. Platelets were further purified from PRP by centrifugation at 1,000 g in the presence of prostacyclin (0.1 μg/ml). Purified platelets were resuspended in modified HEPES/Tyrode buffer (in mM: 129 NaCl, 0.34 Na2HPO4, 2.9 KCl, 12 NaHCO3, 20 HEPES, 5 glucose, 1 MgCl2; pH 7.3) containing 0.1 μg/ml prostacyclin. Platelets were washed once by centrifugation and resuspended in modified HEPES/Tyrode buffer at indicated concentrations.
Western blotting.
Western blot experiments were carried out as previously described (19). Briefly, platelet solutions were denatured in an equal volume of Laemmli sample buffer (Bio-Rad, Hercules, CA) with 0.5 M dithiothreitol (100°C, 5 min), separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and blotted with indicated antibodies and HRP-conjugated secondary antibodies. Protein was detected using ECL (Thermo Scientific).
Platelet aggregation.
For platelet aggregation studies, 300 μl of purified human platelets (2 × 108/ml) were pretreated with inhibitors for 10 min as indicated. Platelet aggregation was triggered by CRP at indicated concentrations and monitored under continuous stirring at 1,200 rpm at 37°C by measuring changes in light transmission using a PAP-4 aggregometer as previously described (6, 25).
HDAC activity assay.
Purified human platelets were treated with inhibitors as described before lysis into assay buffer and colorimetric analysis of total HDAC activity with a BioVision HDAC Colorimetric Activity Assay Kit according to the manufacturer's instructions using a Molecular Devices microplate reader system.
Static platelet assays.
To examine platelet morphology in solution, platelets (4 × 107/ml) were fixed in 4% paraformaldehyde before attachment to poly-l-lysine-coated coverglass. Inhibitors or vehicle was added to platelets in solution at the indicated concentrations for 10 min prior to stimulation at 37°C and fixation. Platelets were imaged using Köhler illuminated Nomarski differential interference contrast (DIC) optics with a Zeiss ×63 oil immersion 1.40 numerical aperture (NA) plan-apochromat lens on a Zeiss Axiovert 200M microscope as previously described (3, 6).
Optical quantification of platelet volume.
For volume quantification studies, purified human platelets (5 × 106/ml) were treated with inhibitors prior to fixation in 4% paraformaldehyde. Platelets were immobilized on poly-l-lysine-coated coverglass and mounted on microscope slides using Fluoromount G (Southern Biotech). Through-focus DIC imagery at ×63 with an objective lens NA of 1.4 and an illumination condenser lens NA of 0.9 in a Köhler configuration was carried out on a Zeiss Axio Imager 2 microscope. This system possesses an axial resolution of 0.41 μm at the central green wavelengths of the tungsten lamp. Optical sectioning of the sample was achieved by acquiring images in 0.1-μm axial increments with a total axial extent of 10 μm in the three-dimensional (3D) image cubes. Spatial locations along the optical axis are determined with a correction factor accounting for refractive index mismatch between the immersion media of the oil-coupled lens, refractive index = 1.518, and the refractive index of the platelets such that the axial increment is given by dz = (1.399/1.518) × 0.1 μm. Sets of 30 platelets from three separate donors per experimental condition were sectioned and analyzed. Contrast enhancement for edge detection was carried out using a combined Hilbert transform-Sobel edge detection algorithm (7) implemented in MATLAB (The MathWorks). The cross-sectional areas of distinct planes through the platelets were added together to yield platelet volume. Volumes are presented as means ± SE throughout.
Fluorescence microscopy.
Purified human platelets (5 × 106/ml) were treated as described before fixation in 4% paraformaldehyde and immobilization on poly-l-lysine-coated coverglass. For HDAC6 staining, immobilized platelets were washed in PBS and permeabilized with blocking buffer (PBS, 0.1% SDS, + 1% BSA) for 1 h. Platelets were stained with anti-HDAC6 (1:100) and anti-tubulin (1:100) in blocking buffer overnight at 4°C. Invitrogen Alexa Fluor secondary antibodies (1:200) were added in blocking buffer for 1 h. Coverslips were mounted with Fluoromount G (Southern Biotech) on glass slides and visualized with a Deltavision CoreDV Widefield Deconvolution System. For marginal band quantification analyses, three fields of tubulin-stained platelets from three separate experiments per experimental condition were visualized with a Zeiss ×63 oil immersion 1.40 NA plan-apochromat lens on a Zeiss Axiovert 200M microscope. For superresolution imaging studies, platelets were fixed in 4% paraformaldehyde before immobilization on poly-l-lysine coated MatTek 10 mm No. 1.5 coverglass dishes (Ashland, MA) and an additional fixation step with methanol (−20°C, 30 min). Platelets were stained with anti-tubulin (1:100) overnight at 4°C and Invitrogen Alexa Fluor secondary antibodies (1:200). Platelets were imaged on a Zeiss Elyra SR-SIM Superresolution Structured Illumination Microscope. A z-stack of sixty 0.14-μm image slices was used to reconstruct platelet cytoskeletal morphology in three dimensions using Imaris (Bitplane) software. In selected experiments, platelet marginal bands were visualized as a z-stack of three to four image slices summed together using ImageJ software (National Institutes of Health, Bethesda, MD).
Flow cytometry analysis.
Purified human platelets (2 × 107/ml) were incubated with vehicle (DMSO) or HDAC inhibitor for 10 min before stimulation with vehicle (PBS) or CRP (1 μg/ml) for 15 min in the presence of FITC-conjugated anti-human CD62E/CD62P (Acris Antibodies) or PAC-1 FITC (BD Biosciences). Samples were diluted into HEPES/Tyrode buffer and analyzed by flow cytometry using FACSCalibur (Becton Dickinson). Platelets were identified by logarithmic signal amplification for forward and side scatter as previously described (9).
Statistical analysis.
The Jarque-Bera test was used to evaluate normality of all parameters. One-way analysis of variance with Bonferonni post hoc correction was used to assess statistical significance among parameters across multiple normally distributed cell parameters. The Kruskal-Wallis test was used to assess significance among non-normally distributed parameters. P ≤ 0.05 was considered significant.
RESULTS
Platelet activation promotes a condensation of the tubulin cytoskeleton and the deacetylation of α-tubulin.
Upon engagement of the GPVI receptor by collagen or CRP, platelets undergo a dramatic change in shape during the processes of platelet activation, aggregation, and thrombus formation (40). To study the mechanisms that regulate changes in platelet morphology associated with activation, platelets were treated with CRP in solution before fixation with paraformaldehyde and plated onto poly-l-lysine-coated coverglass. Köhler-illuminated DIC microscopy revealed a change in platelet shape from a characteristic discoid morphology to a more condensed sphere upon stimulation with CRP (Fig. 1A). To better define the signaling pathways that regulate this change in platelet morphology upon activation with CRP, platelets were pretreated with inhibitors of signaling steps proximal to GPVI engagement, including the Src kinase inhibitor, PP2, or the Syk-specific inhibitor, BAY 61-3606, before stimulation with CRP, fixation and examination by DIC microscopy. As seen in Fig. 1A, both PP2 and BAY 61-3606 prevented characteristic changes in platelet morphology that occur upon activation with CRP, suggesting that these early mediators of platelet activation control signaling processes that regulate platelet shape changes. In contrast, platelet morphology was unaffected by the inhibition of the Rho GTPase, Rac, with EHT 1864 or inhibition of Rho kinase signaling with Y-27632 (Fig. 1A).
Fig. 1.

Platelet activation results in a decrease in platelet volume and condensation of the tubulin cytoskeleton. A: replicate samples of purified human platelets were treated with the glycoprotein VI (GPVI)-agonist agonist collagen-related peptide (CRP; 1 μg/ml) in solution for 5 min in the presence of vehicle or the Src family kinase (SFK) inhibitor PP2 (10 μM), the Syk inhibitor BAY 61-3606 (BAY; 10 μM), the Rac inhibitor EHT 1864 (EHT; 50 μM), or the Rho kinase inhibitor Y-27632 (Y; 10 μM). Platelets were then fixed and visualized by differential interference contrast (DIC) microscopy. Wide field scale bar, 10 μm. Zoom field scale bar, 2 μm. Tubulin fluorescence of representative platelets was visualized by superresolution structured illumination microscopy (SR-SIM) and reconstructed into 3 dimensions using Imaris software. Scale bar, 500 nm. B: box plots of platelet volume determined by DIC optical sectioning microscopy as described in materials and methods. Central box marks indicate median values of platelet volumes; box edges represent 25th and 75th percentile values. Whiskers extend to the most extreme nonoutlier data points within ±2.7σ, where σ is the standard deviation of the data. *Inhibitor-treated samples with volumes significantly greater than that of vehicle-treated samples in the presence of CRP.
The changes in platelet morphology that follow platelet activation are coordinated by a number of signaling systems that regulate reorganization of the platelet cytoskeleton (23, 40). While the regulation of the platelet actin system by proteins such as the Rho GTPases has been well studied (5), the roles of other cytoskeletal elements, including microtubules, remain largely unexplored. To characterize the structure of the platelet tubulin cytoskeleton during activation, platelets were stained with primary antibodies against α-tubulin and fluorescent secondary antibodies before visualization by fluorescent superresolution structured illumination microscopy (SR-SIM) (22). SR-SIM sectioning and Imaris 3D reconstruction of the platelet tubulin cytoskeleton revealed characteristic marginal band structures at the discoid faces of resting platelets as well as finer interconnected tubulin structures (Fig. 1A). Upon treatment with CRP, tubulin marginal bands condensed towards the platelet center while supporting the extension of tubulin-containing filopodial structures (Fig. 1A). Pretreatment of platelets with PP2 or BAY 61-3606 blocked this centripetal condensation of the tubulin cytoskeleton upon stimulation with CRP (Fig. 1B). Platelet tubulin condensation was not affected by treatment with EHT 1864 or Y-27632 (Fig. 1A).
To determine whether changes in platelet morphology and tubulin structures are accompanied by changes in platelet volume, we employed a quantitative DIC-based optical technique (7) to analyze the volume of resting and activated platelets. As shown in Fig. 1B, resting platelets had a mean volume of 12.5 ± 0.5 fl, which condensed to 10.7 ± 0.3 fl upon activation with CRP. Pretreatment of platelets with the Src family kinase (SFK) inhibitor PP2 inhibited the ability of CRP to induce a change in volume (15.2 ± 0.5 fl). The Syk inhibitor BAY 61-3606 similarly prevented the ability of CRP treatment to promote a change in platelet volume (13.9 ± 0.6 fl). Similar to the results above demonstrating that Rac and ROCK inhibition do not prevent tubulin condensation, EHT 1864 and Y-27632 did not significantly prevent CRP from promoting a change in platelet volume (11.8 ± 0.6 and 9.9 ± 0.4 fl, respectively).
Advances in the studies of platelet formation from megakaryocytes have demonstrated the importance of microtubule dynamics to platelet production (11); however, the roles of microtubule dynamics in platelet physiological function remain less understood. Previous studies of the roles of tubulin dynamics in platelet formation have found that α-tubulin is heavily acetylated in platelets (31) and that tubulin acetylation has roles in platelet production (17). More recently, a report demonstrated that platelet α-tubulin lysine 40 is deacetylated following activation and that HDAC6-mediated deacetylation of α-tubulin plays a role in the kinetics of platelet spreading (33). To better understand the role of tubulin deacetylation in platelet activation, we next examined the agonist concentration and time dependence of platelet α-tubulin deacetylation by Western blotting for α-tubulin acetyl-lysine 40 (Fig. 2). To define the platelet signaling systems that mediate tubulin deacetylation upon activation, platelets were pretreated with pharmacological inhibitors targeting specific platelet signaling mediators prior to stimulation with the GPVI agonist CRP and analysis of tubulin acetylation by Western blot. The deacetylation of platelet α-tubulin following stimulation with CRP was abrogated by agents that block early signaling events in platelet activation, including the SFK- and Syk-specific inhibitors PP2 and BAY 61-3606, respectively (Fig. 3A). Inhibition of signaling by Rho GTPases, RhoA and Rac1, by pretreatment with the Rho kinase (ROCK) inhibitor Y-27632 and the Rac inhibitor EHT 1864, respectively, did not alter tubulin deacetylation following CRP stimulation (Fig. 3B). Together, these results suggest that signaling events downstream of Src kinases and upstream of Rho GTPases have a role in tubulin deacetylation that occurs over the time course of platelet cytoskeletal condensation.
Fig. 2.
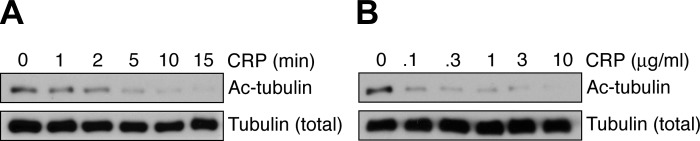
Time- and concentration-dependent deacetylation of tubulin upon platelet activation by CRP. A: purified human platelets were treated with CRP in solution (1 μg/ml) for 1–15 min before lysis into sample buffer and Western blot analysis for acetylated α-tubulin (Ac-tubulin) Lys 40. B: purified human platelets were treated with CRP (0.1–10 μg/ml) for 5 min before lysis into sample buffer and Western blot analysis. Total tubulin levels are shown as a control for protein loading. Results are representative of three separate experiments.
Fig. 3.

Platelet signaling inhibition blocks CRP-triggered tubulin deacetylation. Replicate samples of purified human platelets were pretreated with the SFK inhibitor PP2 (10 μM), the Syk inhibitor BAY 61-3606 (10 μM) (A), the Rho kinase (ROCK) inhibitor Y-27632 (10 μM), the Rac inhibitor EHT 1864 (50 μM) (B), or vehicle alone before stimulation with 1 μg/ml CRP (2–10 min). Platelets were lysed into sample buffer and analyzed for tubulin acetylation by Western blotting. Results are representative of three separate experiments.
HDAC6 deacetylates α-tubulin upon platelet stimulation.
In nucleated cells, the histone deacetylase HDAC6 serves as a primary cytosolic tubulin deacetylase in the control of diverse cellular functions (38). To better characterize the HDAC system in platelets, we analyzed the expression of HDAC proteins in human platelets compared with MDA-MB-231 breast cancer cells. Western blot analysis of total cell lysates revealed that platelets express HDAC6 and to a lesser extent HDAC4. HDAC1, HDAC2, and HDAC7 proteins, which were all present in MDA-MB-231 cells, were not detectable by Western blotting in human platelet lysates (Fig. 4A). Platelets also expressed small amounts of the sirtuin SIRT2, which belongs to a separate class of deacetylase enzymes also known to catalyze tubulin deacetylation (28). To first analyze the roles of deacetylase activity mediated by HDACs in platelets, platelets were lysed into assay buffer and analyzed for total HDAC activity using a histone deacetylase enzyme assay. Quantification of total HDAC enzyme activity from resting and CRP-treated platelets revealed that platelet lysates possess HDAC enzyme activity that does not change upon stimulation with CRP (Fig. 4B and data not shown). Under CRP-stimulated conditions, the pan-HDAC inhibitor TSA reduced platelet HDAC activity to 48.0 ± 8.1% of basal levels. The HDAC6-specific inhibitor tubacin (14) significantly reduced total HDAC activity to 59.1 ± 3.2% relative to control levels, suggesting that HDAC6 is a major contributor to total platelet HDAC activity. Inhibition of SIRT2 deacetylase activity had no significant effect on platelet deacetylase activity as treatment of platelets with 10 mM nicotinamide retained normal HDAC enzymatic activity levels relative to untreated controls (102.7 ± 1.8%, n = 3).
Fig. 4.

Histone deacetylase (HDAC) protein expression and activity in human platelets. A: purified human platelets and MDA-MB-231 cells were lysed directly into sample buffer and analyzed for HDAC 1, 2, 4, 6, and 7 and sirtuin 2 (SIRT2) protein expression by Western blotting. Tubulin serves as a loading control for total cellular protein. B: human platelets were treated with the pan-HDAC inhibitor trichostatin A (TSA; 10 μM), the HDAC6-specific inhibitor tubacin (10 μM), or vehicle alone before lysis into assay buffer and analysis of total HDAC activity (means ± SE; n = 4). *P < 0.05 compared with vehicle-treated platelets. C and D: replicate preparations of purified human platelets (n = 3) were fixed, immobilized on coverglass, stained for the presence of HDAC6 and α-tubulin protein, and examined by fluorescence deconvolution microscopy (C) or stained with α-tubulin and acetylated tubulin and visualized by SR-SIM (D). Scale bar, 1 μm.
To examine the localization of HDAC6 protein relative to the tubulin cytoskeletal system in platelets, human platelets were fixed with 4% paraformaldehyde, plated onto poly-l-lysine-coated coverglass, and stained for HDAC6 and α-tubulin. Deconvolution immunofluorescence microscopy demonstrated that HDAC6-positive punctae are found at the platelet tubulin marginal band as well as the platelet interior, suggesting that HDAC6 may localize to specific regions to mediate its roles in platelet function (Fig. 4C). To better determine the relative distribution of tubulin to acetylated tubulin, platelets were also stained with antibodies specific for α-tubulin as well as α-tubulin acetylated at Lys 40 and examined by fluorescence structured illumination microscopy (SIM). As seen in Fig. 4D, total α-tubulin and acetylated α-tubulin show a largely overlapping distribution, localizing to the platelet marginal band as well as the platelet interior.
Next, to examine the role of HDAC6 activity in the regulation of platelet α-tubulin Lys 40 acetylation, we analyzed tubulin deacetylation following CRP treatment by Western blotting in HDAC-inhibited conditions. As seen in Fig. 5, pretreatment of platelets with the pan-HDAC inhibitor TSA or the HDAC6-specific inhibitor tubacin prevented the time-dependent deacetylation of platelet α-tubulin Lys 40 in response to CRP stimulation. Treatment of platelets with TC-H 106, an inhibitor of HDAC1 and HDAC3, had no effect on CRP-induced tubulin deacetylation (Fig. 5A). Inhibition of the sirtuins SIRT1 and SIRT2 with specific pharmacological agents EX 527 and AGK2, respectively, similarly did not affect tubulin deacetylation elicited by CRP (Fig. 5B). To test the hypothesis that α-tubulin deacetylation may also have roles in platelet agonist responses other than CRP, we next assayed tubulin deacetylation upon treatment with the PAR1 and PAR4-selective agonist peptides TRAP6 and GYPGKF, respectively. As seen in Fig. 5, C and D, PAR1 and PAR4 stimulation induced a time-dependent deacetylation of platelet α-tubulin Lys 40 that was inhibited by TSA as well as by tubacin.
Fig. 5.

HDAC6 inhibition prevents platelet tubulin deacetylation. A and B: purified human platelets were pretreated with the pan-HDAC inhibitor TSA (10 μM), the HDAC6-specific inhibitor tubacin (10 μM), the HDAC1/3 inhibitor TC-H 106 (10 μM), or vehicle alone (DMSO) (A) or the SIRT1 inhibitor EX 527 (10 μM), the SIRT2 inhibitor AGK2 (10 μM), or vehicle alone (DMSO) (B) before stimulation with 1 μg/ml CRP (2–10 min). Platelets were lysed into sample buffer and analyzed for tubulin acetylation by Western blot. C and D: purified human platelets were pretreated with the pan-HDAC inhibitor TSA (10 μM), the HDAC6-specific inhibitor tubacin (10 μM), or vehicle alone (DMSO) before stimulation (2–10 min) with the PAR1 agonist peptide TRAP6 (C) or the PAR4 agonist peptide GYPGKF (D). Western blot results are each representative of 4 experiments.
HDAC6 regulates platelet tubulin dynamics, aggregation, and signaling upon activation.
In nucleated cells, HDAC6 activity has been suggested to regulate the dynamics of tubulin organization (46); however, the mechanistic roles of HDAC6-mediated deacetylation in tubulin cell biological functions remain to be elucidated. To better characterize the role of HDAC6 in platelet tubulin dynamics, platelets were activated with CRP before fixation as described above and tubulin was stained under vehicle and HDAC6-inhibited conditions (Fig. 6A). In vehicle-treated platelets, the platelet marginal band was observed as a solid ring structure that collapses upon activation with CRP (Fig. 6A). Pretreatment of platelets with TSA or the HDAC6-specific inhibitor tubacin altered the distribution of tubulin in resting platelets, as evidenced by a punctate distribution of tubulin in tubacin- and TSA-treated platelets under basal conditions (Fig. 6A). Tubacin and TSA treatment resulted in 68.7 ± 3.7%- and 59.5 ± 5.3%-fold decrease in the numbers of platelets with nonpunctate marginal bands, respectively. Moreover, treatment of platelets with TSA or tubacin inhibited the degree of platelet tubulin condensation induced by platelet activation with CRP, suggesting that HDAC inhibition may alter cytoskeletal changes in platelets following stimulation (Fig. 6A). Platelets treated with tubacin showed a 47.0 ± 7.0%-fold decrease (31.2 ± 5.4%-fold decrease for TSA) in complete tubulin ring-to-sphere condensation following CRP stimulation. To further examine the effects of HDAC6-regulated tubulin dynamics on total platelet morphology, platelets treated with either TSA or tubacin were fixed before and after stimulation with CRP and subjected to volumetric analysis by DIC microscopy. As seen in Fig. 6, B and C, inhibition of HDAC activity had no significant effect on resting platelet or CRP-stimulated platelet morphology as determined my DIC microscopy. Volumetric analysis of TSA- and tubacin-treated platelets stimulated with CRP similarly demonstrated that HDAC inhibition does not significantly alter changes in platelet volume following CRP stimulation (volumes: 9.1 ± 0.3 fl and 10.8 ± 0.3 fl, respectively).
Fig. 6.

HDAC6 inhibition alters the structure of the platelet marginal band. A: purified human platelets were pretreated with TSA (10 μM), tubacin (10 μM), or vehicle alone before stimulation with 1 μg/ml CRP. After 5 min, platelets were fixed, immobilized on coverglass, stained with antibodies directed against α-tubulin, and examined by SR-SIM. Representative 0.56-μm slices from three separate experiments are shown. Wide field scale bar, 10 μm. Zoomed image scale bar, 1 μm. B–D: after fixation, platelets were also examined by DIC microscopy (B and C) and quantitative volumetric analysis (D) as described in materials and methods. Central box marks indicate median values of platelet volume; box edges represent 25th and 75th percentile values. Whiskers extend to the most extreme nonoutlier data points. *Values significantly lower than that of resting platelets (basal).
To examine the consequences of HDAC6 inhibition on platelet function, replicate samples of washed human platelets were pretreated with tubacin or vehicle alone for 10 min prior to stimulation with CRP in a Born aggregometer. As seen in Fig. 7A, tubacin inhibited the aggregation of platelets in response to CRP. Platelet aggregation responses to CRP were similarly inhibited by the pan-HDAC inhibitor TSA (Fig. 7B) but not affected by EX 527 or AGK2, specific inhibitors of SIRT1 or SIRT2 deacetylases, respectively (Fig. 7C). The inhibition of platelet aggregation by TSA and tubacin treatment was likely not due to alterations in platelet secretion processes, as HDAC inhibition did not significantly decrease platelet P-selectin surface exposure upon stimulation with CRP (Fig. 7D). Moreover, neither TSA nor tubacin affected the activation of platelet GPIIbIIIa integrins by CRP, as measured by the binding of active-site specific mAb, PAC-1, to platelets (Fig. 7E). Together, with previous studies showing a role for HDAC6 in microtubule dynamics (46) as well as the regulation of platelet activation on glass surfaces (33), these results suggest that, upon platelet activation, HDAC6 deacetylates tubulin to regulate tubulin dynamics and that this process contributes to the regulation of platelet aggregation in response to the GPVI agonist CRP.
Fig. 7.

HDAC inhibition blocks platelet aggregation in solution. A: washed human platelets (2 × 108/ml) were incubated with vehicle (DMSO) or the HDAC6-specific inhibitor tubacin (10 μM) prior to stimulation with increasing concentrations of CRP, and the change in optical density indicative of platelet aggregation was recorded. Aggregation traces representative of three separate experiments are shown. B and C: washed human platelets (2 × 108/ml) were incubated with vehicle (DMSO) or the pan-HDAC inhibitor TSA (10 μM) (B) or the SIRT1 inhibitor EX 527 (10 μM) or the SIRT2 inhibitor AGK2 (10 μM) (C) prior to stimulation with CRP (10 μg/ml) in the presence of 2 U/ml apyrase, and the change in optical density indicative of platelet aggregation was recorded. D and E: platelet surface P-selectin levels (D) and integrin αIIbβ3 activation (PAC-1) (E) analyzed by flow cytometry following vehicle (DMSO), TSA, and tubacin treatment and incubation with vehicle (white bars) or CRP (1 μg/ml; black bars) stimulation. MFI, mean fluorescence intensity. Data are represented as means ± SE.
The partial disruption of platelet aggregation by HDAC6 inhibition supports the hypothesis that specific steps in platelet signaling cascades triggered by GPVI engagement may be regulated by the acetylation state of platelet tubulin. Furthermore, the data above suggest that HDAC6 functions downstream of Src kinases and upstream of Rho GTPases to drive platelet function (Fig. 3). To test the hypothesis that HDAC6 coordinates specific platelet signaling processes upon activation by CRP, total platelet lysates from resting or CRP-stimulated platelets pretreated with vehicle alone or the HDAC6-inhibitor tubacin were subjected to Western blot analysis for the activation and phosphorylation of specific platelet signaling mediators (Fig. 8). Lysates were separated by SDS-PAGE, transferred to nitrocellulose, and blotted with a monoclonal antibody directed against phosphotyrosine moieties (4G10), as well as phosphorylated FAK and Syk. As seen in Fig. 8A, CRP stimulation elicited a robust increase in total protein tyrosine phosphorylation that was not inhibited by pretreatment with tubacin. Tubacin similarly did not prevent the CRP-induced phosphorylation of FAK and Syk (data not shown). Together with the experiments above (Fig. 3A), these data suggest that HDAC6 does not regulate early tyrosine kinase signaling events in platelets, but rather regulates signaling upstream of Rho GTPase signaling (Fig. 3B). To analyze the activation of the Rho GTPase Rac under HDAC6-inhibited conditions, platelet lysates were probed for the phosphorylation of the p21-activated kinase PAK (1), a Rho GTPase effector with roles in platelet activation downstream of GPVI and PAR engagement (2, 4). Tubacin treatment prevented the activation of PAK following stimulation with CRP (Fig. 8B). Upon activation, platelet PAK phosphorylates the LIM domain kinase LIMK1 (2, 4). To demonstrate that a loss of PAK autophosphorylation resulted in a loss of PAK activity, we next examined the phosphorylation state of the PKA substrate LIMK1 under vehicle and tubacin-treated conditions. Tubacin prevented the PAK-mediated phosphorylation of LIMK1 in platelets following stimulation with CRP (Fig. 8B). In addition to LIMK1, PAK also has a role in MAPK activation through phosphorylation and activation of MEK proteins (12). Consistent with a role for HDAC6 upstream of PAK activation, tubacin prevented PAK-mediated phosphorylation of MEK phosphorylation and the downstream MEK-driven phosphorylation of ERK following stimulation with CRP. PAK has also been shown to play roles in organizing Akt signaling processes (16, 24). Our data show that phosphorylation of both Akt Thr 308 and Ser 473 by CRP treatment was markedly reduced by tubacin. Together, these results suggest that HDAC6 works upstream of Rac1 and PAK activation to coordinate platelet signaling events.
Fig. 8.

HDAC6 mediates p21-activated kinase (PAK), MAPK, and Akt signaling in platelets. Purified human platelets were pretreated with tubacin (10 μM) or vehicle prior to stimulation with CRP (1 μg/ml). After 5 min, platelets were lysed in sample buffer and analyzed by Western blotting for acetyl tubulin, total phosphotyrosine (pTyr) moieties (A), phosphorylated PAK Ser 141, phosphorylated LIM domain kinase (LIMK1) Thr 508 (B), phospho-MEK, phospho-ERK (C), and phospho-Akt Thr 308 and Ser 473 (D). Western blotting results are representative of six separate experiments.
DISCUSSION
Here, we demonstrate that HDAC6-mediated deacetylation of α-tubulin lysine 40 in platelets is associated with changes in platelet cytoskeletal morphology and the signaling events that accompany platelet activation. Upon stimulation with the GPVI-specific agonist CRP, platelets condensed in volume in concert with a centripetal collapse of the platelet tubulin marginal band. This collapse of the marginal band was associated with an agonist concentration- and time-dependent deacetylation of α-tubulin, which was blocked by pharmacological inhibitors of early signaling mediators and HDACs, but not by inhibitors of Rho GTPase signaling. The HDAC6-specific inhibitor tubacin and the pan-HDAC inhibitor TSA prevented CRP-induced deacetylation of tubulin and inhibited platelet aggregation. TSA and tubacin treatment also altered the structure of the marginal band in resting platelets yet did not significantly change platelet morphology or platelet volume decreases in response to stimulation with CRP. Inhibition of HDAC6 also blocked key platelet signaling events downstream of Rho GTPase activation. Together, these results suggest that HDAC6 regulates tubulin morphology in resting and activated platelets and may have roles in organizing signaling processes following tyrosine kinase activation as well as regulating changes in platelet volume, platelet aggregation, and Rho GTPase family signaling events.
Microtubules have long been recognized to play a role in the morphological changes that underlie platelet activation (26, 37); however, the specific mechanisms by which microtubules contribute to platelet activation remain elusive. Early electron microscopy studies of platelets noted the presence of a circumferential marginal band that disappears or condenses upon platelet activation (36, 42, 43). It was proposed that as platelets activate, microtubules condense towards the platelet center, trapping granules and organelles (36). Inhibitors of microtubule polymerization prevent this condensation process as well as platelet aggregation and secretion, suggesting that microtubule dynamics play roles in aggregation and activation (26, 36, 37). Early immunofluorescence studies showed a centripetal redistribution of myosin and microfilaments upon platelet activation (29). Together, these studies led to the hypothesis that this centripetal relocalization towards the phase dense organellar or glomerular zone brings granules to the platelet open canalicular system upon activation (10, 36, 41). More recently, it was shown that actin filaments and microtubules as well as motor proteins have a specific role in this centripetal trafficking of organelles and granules in platelets (10).
Studies of platelet populations have shown a decrease in mean platelet volume upon aggregation (20). Using an optical sectioning DIC-based method of volume analysis (7), we find that tubulin condensation is associated with a volume change (Fig. 1). Signaling events transmitted from receptors through Src kinases have a role in platelet condensation, as the Src inhibitor PP2 inhibited platelet shrinkage (Fig. 1). While previous studies have taken advantage of electron microscopy (EM) to study tubulin before and after platelet activation in solution, our study provides the first 3D superresolution visualization of the tubulin cytoskeleton. Similar to earlier EM studies, superresolution structured illumination microscopy visualized the tubulin marginal band at the periphery of resting platelets (Fig. 1). Notably, in addition to the marginal band, finer web- or netlike structures were also observed at the discoid faces and interior of resting platelets, perhaps representing an association of tubulin filament structures with the open canalicular system (39, 41) (Fig. 1). Like earlier EM studies (36), we also observed a centripetal collapse of the tubulin cytoskeleton upon platelet activation with the GPVI agonist CRP (Fig. 1).
The regulation of the tubulin cytoskeleton in platelets as well as nucleated cells is not well understood. A number of mechanisms, notably specific posttranslational modifications of tubulin, regulate microtubule polymerization, depolymerization, stability, and traffic along tubule structures (19). Acetylation of tubulin has roles in microtubule organization and stability (15, 19). Previous studies noted that the marginal band is heavily acetylated (31) and that platelet tubulin is deacetylated by HDAC6 following stimulation (33). Here, we find that platelets deacetylate tubulin in an agonist concentration- and time-dependent manner following the time course of platelet centripetal condensation and aggregation (Fig. 2). Tubulin deacetylation was blocked by inhibitors of early platelet signaling events including the SFKs, Src, and Syk. This provides evidence that signals from platelet receptors such as GPVI may regulate acetylation signaling processes through a Src kinase-based pathway (40) or may prevent changes in platelet cytoskeletal signaling events that could drive signaling processes upstream of tubulin deacetylation. HDAC6 has a role downstream of Src activation but apparently upstream of Rho GTPase activity, as Rho kinase and Rac inhibitors had no effect on agonist-induced tubulin deacetylation (Fig. 3). Furthermore, inhibition of HDAC6 activity in stimulated platelets failed to block Src activation but did inhibit the phosphorylation of effector molecules downstream of Rho GTPase activation in platelets, including PAK, LIMK1, MAPKs, and Akt. This suggests that deacetylation is required for signaling downstream of immunoreceptor tyrosine activation motif (ITAM)-coupled receptors, such as GPVI. Interestingly, previous studies in nucleated cells have suggested that HDACs play a role in organizing signaling systems to regulate cell migration processes related to platelet aggregation (8, 38). The role of acetylation in regulating similar processes in platelets is an important topic that warrants further investigation.
Using SR-SIM, we examined the structure of the tubulin cytoskeleton under HDAC6-inhibited conditions in resting and activated platelets (Fig. 6). Quiescent platelets treated with tubacin or TSA showed a less structured, more punctate, and thicker marginal band than that of control platelets (Fig. 6). This apparent disorganization of the resting platelet marginal band may have reduced the ability of the marginal band to condense upon platelet activation, as HDAC6-inhibited platelets showed a reduced ability to condense their marginal band (Fig. 6). Together, these results demonstrate a role for HDAC6-mediated deacetylation of tubulin in the maintenance and regulation of platelet microtubule morphology and provide insights into potential mechanisms used by platelets to regulate microtubule dynamics. Future studies of platelet tubulin acetylation will clarify the roles of this process in platelet activation while advancing the understanding of HDAC6 in cytosolic signaling events.
GRANTS
Superresolution microscopy studies were supported in part by the M. J. Murdock Charitable Trust. This work was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute Grant R01 HL-101972 to O. J. T. McCarty) and grants from the American Heart Association (13POST13730003 to J. E. Aslan and 13EIA12630000 to O. J. T. McCarty). O. J. T. McCarty is an American Heart Association Established Investigator. K. G. Phillips is supported by a Medical Research Foundation Early Clinical Investigator Award. A. Itakura is a Vertex Scholar. J. E. Aslan is a 2012-2013 Fulbright Scholar.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.E.A. and A.I. conception and design of research; J.E.A., K.G.P., A.I., and J.P. performed experiments; J.E.A., K.G.P., L.D.H., and A.I. analyzed data; J.E.A. and K.G.P. interpreted results of experiments; J.E.A. prepared figures; J.E.A. drafted manuscript; J.E.A. and O.J.T.M. edited and revised manuscript; J.E.A., K.G.P., L.D.H., A.I., J.P., and O.J.T.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank S. Kaech and A. Snyder (OHSU) and B. Chhun (Zeiss) for assistance with superresolution microscopy and B. Kusanto, S. Baker-Groberg, C. Loren, and L. Replogle for technical assistance.
Footnotes
This article is the topic of an Editorial Focus by Karin Sadoul (33a).
REFERENCES
- 1.Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell 100: 97–108, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Aslan JE, Baker SM, Loren CP, Haley KM, Itakura A, Pang J, Greenberg DL, David LL, Manser E, Chernoff J, McCarty OJ. The PAK system links Rho GTPase signaling to thrombin-mediated platelet activation. Am J Physiol Cell Physiol 305: C519–C528, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aslan JE, Itakura A, Gertz JM, McCarty OJ. Platelet shape change and spreading. Methods Mol Biol 788: 91–100, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Aslan JE, Itakura A, Haley KM, Tormoen GW, Loren CP, Baker SM, Pang J, Chernoff J, McCarty OJ. p21 activated kinase signaling coordinates glycoprotein receptor VI-mediated platelet aggregation, lamellipodia formation, and aggregate stability under shear. Arterioscler Thromb Vasc Biol 33: 1544–1551, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost 11: 35–46, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood 118: 3129–3136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker SM, Phillips KG, McCarty OJ. Development of a label-free imaging technique for the quantification of thrombus formation. Cell Mol Bioeng 5: 488–492, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26: 5468–5476, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Calaminus SD, McCarty OJ, Auger JM, Pearce AC, Insall RH, Watson SP, Machesky LM. A major role for Scar/WAVE-1 downstream of GPVI in platelets. J Thromb Haemost 5: 535–541, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerecedo D, Cisneros B, Mondragon R, Gonzalez S, Galvan IJ. Actin filaments and microtubule dual-granule transport in human adhered platelets: the role of alpha-dystrobrevins. Br J Haematol 149: 124–136, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Cramer EM, Norol F, Guichard J, Breton-Gorius J, Vainchenker W, Masse JM, Debili N. Ultrastructure of platelet formation by human megakaryocytes cultured with the Mpl ligand. Blood 89: 2336–2346, 1997 [PubMed] [Google Scholar]
- 12.Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol 22: 6023–6033, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest 115: 3355–3362, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA 100: 4389–4394, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol 20: 71–76, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higuchi M, Onishi K, Kikuchi C, Gotoh Y. Scaffolding function of PAK in the PDK1-Akt pathway. Nat Cell Biol 10: 1356–1364, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Iancu-Rubin C, Gajzer D, Mosoyan G, Feller F, Mascarenhas J, Hoffman R. Panobinostat (LBH589)-induced acetylation of tubulin impairs megakaryocyte maturation and platelet formation. Exp Hematol 40: 564–574, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson SP, Schoenwaelder SM. Antiplatelet therapy: in search of the ‘magic bullet’. Nat Rev Drug Discov 2: 775–789, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12: 773–786, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Karpatkin S. Heterogeneity of human platelets. VI. Correlation of platelet function with platelet volume. Blood 51: 307–316, 1978 [PubMed] [Google Scholar]
- 21.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J 19: 1176–1179, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung BO, Chou KC. Review of super-resolution fluorescence microscopy for biology. Appl Spectrosc 65: 967–980, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 30: 2341–2349, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J, Liang Q. Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol 44: 429–434, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VL, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem 280: 39474–39484, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menche D, Israel A, Karpatkin S. Platelets and microtubules. Effect of colchicine and D2O on platelet aggregation and release induced by calcium ionophore A23187. J Clin Invest 66: 284–291, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer I, Kunert S, Schwiebert S, Hagedorn I, Italiano JE, Jr, Dutting S, Nieswandt B, Bachmann S, Schulze H. Altered microtubule equilibrium and impaired thrombus stability in mice lacking RanBP10. Blood 120: 3594–3602, 2012 [DOI] [PubMed] [Google Scholar]
- 28.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11: 437–444, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Painter RG, Ginsberg MH. Centripetal myosin redistribution in thrombin-stimulated platelets. Relationship to platelet Factor 4 secretion. Exp Cell Res 155: 198–212, 1984 [DOI] [PubMed] [Google Scholar]
- 30.Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani RA, Hartwig JH, Italiano JE., Jr Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood 106: 4076–4085, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel-Hett S, Richardson JL, Schulze H, Drabek K, Isaac NA, Hoffmeister K, Shivdasani RA, Bulinski JC, Galjart N, Hartwig JH, Italiano JE., Jr Visualization of microtubule growth in living platelets reveals a dynamic marginal band with multiple microtubules. Blood 111: 4605–4616, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruggeri ZM. Platelets in atherothrombosis. Nat Med 8: 1227–1234, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Sadoul K, Wang J, Diagouraga B, Vitte AL, Buchou T, Rossini T, Polack B, Xi X, Matthias P, Khochbin S. HDAC6 controls the kinetics of platelet activation. Blood 120: 4215–4218, 2012 [DOI] [PubMed] [Google Scholar]
- 33a.Sadoul K. Tubulin acetylation a valuable accessory of the platelet cytoskeleton. Focus on “Histone deacetylase 6-mediated deacetylation of α-tubulin coordinates cytoskeletal and signaling events during platelet activation.” Am J Physiol Cell Physiol (October 9, 2013). 10.1152/ajpcell.00309.2013 [DOI] [PubMed] [Google Scholar]
- 34.Schwer HD, Lecine P, Tiwari S, Italiano JE, Jr, Hartwig JH, Shivdasani RA. A lineage-restricted and divergent beta-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr Biol 11: 579–586, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Shepro D, Belamarich FA, Chao FC. Retardation of clot retraction after incubation of platelets with colchicine and heavy water. Nature 221: 563–565, 1969 [DOI] [PubMed] [Google Scholar]
- 36.Sneddon JM. Effect of mitosis inhibitors on blood platelet microtubules and aggregation. J Physiol 214: 145–158, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steiner M, Ikeda Y. Quantitative assessment of polymerized and depolymerized platelet microtubules. Changes caused by aggregating agents. J Clin Invest 63: 443–448, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 18: 291–297, 2008 [DOI] [PubMed] [Google Scholar]
- 39.van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HF. The platelet interior revisited: electron tomography reveals tubular alpha-granule subtypes. Blood 116: 1147–1156, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost 3: 1752–1762, 2005 [DOI] [PubMed] [Google Scholar]
- 41.White JG. Effects of colchicine and vinca alkaloids on human platelets. II. Changes in the dense tubular system and formation of an unusual inclusion in incubated cells. Am J Pathol 53: 447–461, 1968 [PMC free article] [PubMed] [Google Scholar]
- 42.White JG. Fine structural alterations induced in platelets by adenosine diphosphate. Blood 31: 604–622, 1968 [PubMed] [Google Scholar]
- 43.White JG, Krivit W. An ultrastructural basis for the shape changes induced in platelets by chilling. Blood 30: 625–635, 1967 [PubMed] [Google Scholar]
- 44.White JG, Rao GH. Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. Am J Pathol 152: 597–609, 1998 [PMC free article] [PubMed] [Google Scholar]
- 45.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26: 5310–5318, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci 122: 3531–3541, 2009 [DOI] [PubMed] [Google Scholar]