

Abstract
Proteostasis is the maintenance of the proper function of cellular proteins. Hypertonic stress disrupts proteostasis and causes rapid and widespread protein aggregation and misfolding in the nematode Caenorhabditis elegans. Optimal survival in hypertonic environments requires degradation of damaged proteins. Inhibition of protein synthesis occurs in response to diverse environmental stressors and may function in part to minimize stress-induced protein damage. We recently tested this idea directly and demonstrated that translation inhibition by acute exposure to cycloheximide suppresses hypertonicity-induced aggregation of polyglutamine::YFP (Q35::YFP) in body wall muscle cells. In this article, we further characterized the relationship between protein synthesis and hypertonic stress-induced protein damage. We demonstrate that inhibition of translation reduces hypertonic stress-induced formation and growth of Q35::YFP, Q44::YFP, and α-synuclein aggregates; misfolding of paramyosin and ras GTPase; and aggregation of multiple endogenous proteins expressed in diverse cell types. Activation of general control nonderepressible-2 (GCN-2) kinase signaling during hypertonic stress inhibits protein synthesis via phosphorylation of eukaryotic initiation factor-2α (eIF-2α). Inhibition of GCN-2 activation prevents the reduction in translation rate and greatly exacerbates the formation and growth of Q35::YFP aggregates and the aggregation of endogenous proteins. The current studies together with our previous work provide the first direct demonstration that hypertonic stress-induced reduction in protein synthesis minimizes protein aggregation and misfolding. Reduction in translation rate also serves as a signal that activates osmoprotective gene expression. The cellular proteostasis network thus plays a critical role in minimizing hypertonic stress-induced protein damage, in degrading stress-damaged proteins, and in cellular osmosensing and signaling.
Keywords: C. elegans, organic osmolytes, hypertonic stress, osmoregulation
proteostasis is defined as the homeostatic mechanisms that maintain the conformation, concentration, interactions, localization, and, hence, function of proteins. The proteostasis network is highly conserved across evolutionarily divergent species and comprises the tightly integrated activities of gene transcription, RNA metabolism and protein synthesis, folding, assembly, trafficking, disassembly, repair, and degradation (2, 10, 27).
Proteostasis is under constant challenge in both normal and pathophysiological states. For example, errors in protein translation occur at a rate of one in every 1,000–10,000 translated codons (12). If mistranslated proteins are misfolded and dysfunctional, they must be detected and repaired or destroyed. Gene mutations that disrupt protein structure and function, protein damaging stressors, and bacterial and viral infection that hijack protein synthesis and folding machinery further challenge proteostasis. Senescence causes a poorly understood decline in the capacity of cellular proteostasis networks that repair and degrade damaged proteins (19, 27). Because protein structure is inherently unstable and is under constant challenge, “cells live on the edge of a proteostasis catastrophe” (13).
Environmental stressors damage cellular proteins by inducing chemical modification, misfolding, denaturation, and/or aggregation. Reduction of protein synthesis is a widely observed cellular response to environmental stress and is thought to function to minimize stress-induced damage (15, 35). Inhibiting translation reduces energy consumption and the total number of cellular proteins that may be damaged by a stressor. This in turn is expected to free up energy resources and cellular machinery that can be used to minimize and reverse damage to existing proteins or remove damaged proteins from the cell.
When exposed to sublethal hypertonic stress the nematode Caenorhabditis elegans rapidly loses water. Water loss is followed by activation of systemic volume recovery mechanisms that are mediated first by salt and water uptake followed by accumulation of the organic osmolyte glycerol (8, 20, 22). Cellular water loss causes rapid and dramatic damage to both native and transgenic proteins (5, 6, 9). Survival of C. elegans in hypertonic environments requires the activity of genes that function to degrade damaged proteins (9). Hypertonic stress causes a rapid inhibition of protein synthesis and reduced translation in turn serves as a signal for activation of hypertonic stress response pathways including glycerol synthesis (6, 23).
The idea that inhibition of translation is protective against stress-induced protein damage is conceptually attractive and widely cited (15, 35). However, there is little experimental evidence to support this idea. Our recent studies in C. elegans provided to the best of our knowledge the first direct demonstration that reduced rates of translation minimize stress-induced protein damage. RNA interference (RNAi) silencing of essential translational components or acute exposure to cycloheximide suppresses hypertonicity-induced aggregation of a polyglutamine-YFP (Q35::YFP) reporter protein 50–80% in C. elegans. Dietary changes that increase protein production increase Q35::YFP aggregation 70–180% (6).
The relationship among cellular stress, protein synthesis, and protein damage has important implications for understanding disease and the pathophysiology associated with aging (2, 10, 19, 27, 27). In the current article, we further characterize the relationship between protein synthesis and hypertonic stress-induced protein damage in C. elegans. We demonstrate that inhibition of protein synthesis protects diverse proteins in diverse cell types from hypertonicity-induced aggregation and misfolding. We also show for the first time that hypertonic stress-induced reductions in translation rate mediated by activation of general control nonderepressible-2 (GCN-2) kinase signaling and eukaryotic initiation factor-2α (eIF-2α) phosphorylation function to limit protein damage. The current studies together with our previous work provide the foundation for development of a systems level understanding of the relationship among cellular stress, translation rate, protein damage, and activation of stress protective pathways.
MATERIALS AND METHODS
C. elegans strains.
The following strains were obtained from the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN): wild-type N2 Bristol, ST60 [gcn-1(nc40)], AM140 rmIs132[Punc-54::Q35::YFP], NL5901 pkIs2386[Punc-54::α-synuclein::YFP], CB1402 [unc-15(e1402)], SD551 [let-60(ga89)], and GF80 dgEX80[pAMS66 vha-6p::Q44::YFP + rol-6(su1006) + pBluescriptII]. AM160 and ST60 strains were crossed to generate gcn-1(nc40);Q35::YFP worms. F2 progeny were selected for YFP expression, and the presence of the gcn-1(nc40) allele was verified by PCR. Unless stated otherwise, worms were cultured at 20°C using standard methods (3). Hypertonic agar plates were generated by adding additional NaCl to standard nematode growth medium.
Fluorescent protein aggregate measurements.
The number of body wall muscle cell Q35::YFP or intestine Q44::YFP aggregates were quantified manually using a Zeiss Stemi SV11 microscope (Chester, VA). Quantification of α-synuclein::YFP aggregates in body wall muscle cells located anterior to the nerve ring was performed using a Zeiss LSM510-Meta confocal microscope and Plan-Neofluar 40×/1.3 NA or Plan-Apochromat 63×/1.4 NA oil objective lenses. The volume of individual aggregates was estimated from optical sections obtained by confocal microscopy as described previously (5).
Temperature-sensitive mutant phenotype assays.
The effects of hypertonic stress and translation rate on expression of temperature-sensitive (ts) mutants phenotypes were characterized in unc-15(e1402) and let-60(ga89) mutant worms maintained at the permissive temperature of 16°C. A slow movement phenotype was determined for unc-15(e1402) mutants as described by Gidalevitz et al. (14). Briefly, worms were transferred to the center of a well in a 12-well growth plate that was seeded with a ring of bacteria on the outermost edge. Slow movement was defined as the number of animals that failed to reach the bacteria within 10 min. Defective egg hatching or larval arrest phenotypes were quantified by transferring 1-day-old gravid adults to 300 mM NaCl feeding plates and then removing them after 24 h. Eggs were scored for failure to hatch or develop past the L1 larval stage.
RNA interference.
RNAi was performed by feeding worms from the L1 larval stage with bacteria expressing a nonspecific scrambled dsRNA or dsRNA homologous to iftb-1. Adult RNAi worms were transferred to control or high NaCl growth plates seeded with dsRNA-expressing bacteria.
Survival and motility assays.
Synchronized late L4 wild-type and gcn-1(nc40) worms were transferred to control or high NaCl growth media at 20°C. Survival was determined after 24 h. Worms were considered to be dead if they did not respond to repeated prodding with a platinum wire.
Worm motility was measured as described previously (5). Briefly, wild-type and gcn-1(nc40) L4 worms were transferred to 300 mM NaCl growth medium for 24 h. Single worms were then placed on a fresh lawn of OP50 bacteria and removed after 30 s. Brightfield images of the lawns were obtained using a Zeiss Stemi 2000-CS microscope (Thornwood, NY) equipped with a CCD camera (DAGE-MTI, Michigan City, IN). The length of the tracks in bacterial lawn made by individual worms was measured with ImageJ software (National Institues of Health, Bethesda, MD).
Analysis of endogenous insoluble proteins.
Isolation of insoluble proteins was carried out as described previously (5). Briefly, late L4 stage larvae were exposed to 51 mM (control) or 500 mM NaCl growth media for 4 h. After removal, worms were washed; transferred to a buffer containing 100 mM MES, 1 mM EGTA, 0.1 mM EDTA, 0.5 mM MgSO4, and 20 mM NaF; drip frozen in liquid nitrogen; and ground to a powder with a mortar and pestle. Immediately upon thawing, 10 μl of the ground material were taken for analysis of total protein concentration using a bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL). Two additional 60-μl aliquots were placed in either a solubilization buffer (8 M urea, 2% SDS, 50 mM DTT, 50 mM Tris, and Roche complete protease inhibitor, pH 7.4) for total protein determination or RIPA buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% SDS, 0.5% SDO, 1% NP-40, and Roche complete protease inhibitor, pH 8). Insoluble proteins were isolated from samples in RIPA buffer by centrifugation at 16,100 g for 10 min. After supernatant removal, the insoluble protein pellet was resuspended in 100 μl of RIPA buffer, centrifuged a second time, and then solubilized in solubilization buffer.
Protein samples were analyzed by SDS-PAGE. Gels were stained with Bio-Safe Coomassie (Bio-Rad, Hercules, CA), and the amount of protein present in each lane was quantified with GeneTools software (Syngene, Frederick, MD).
Statistical analysis.
All data are presented as means ± SE. Statistical significance was determined using Student's two tailed t-test when two means were compared. P values of ≤0.05 were taken to indicate statistical significance.
RESULTS
Q35::YFP is a transgenic protein containing 35 contiguous glutamine (Q) residues fused to yellow fluorescent protein (YFP). When expressed in body wall muscle cells, the protein is soluble in young worms but undergoes rapid aggregation during water loss induced by hypertonic stress. Once formed, Q35::YFP aggregate volume increases rapidly and reaches a steady state ∼4 h after water loss is initiated (5). Inhibition of protein synthesis with cycloheximide reduces the number of Q35::YFP aggregates formed after a 1-h exposure to 500 or 700 mM NaCl (6). As shown in Fig. 1, cycloheximide also dramatically suppresses the rate of Q35::YFP aggregate growth. Worms were pretreated with 500 μg/ml cycloheximide on control growth medium for 15 min and then exposed to 500 mM NaCl medium containing the drug. Aggregates were detected within 1 h of exposure to 500 mM NaCl in control worms and grew rapidly reaching a maximum volume of ∼70 μm3 after 4 h of hypertonic stress (Fig. 1A). In contrast, no aggregates were detected in cycloheximide-treated worms at 1 h poststress. After formation, the aggregates grew more slowly reaching a maximum observed volume of ∼43 μm3 (Fig. 1A).
Fig. 1.
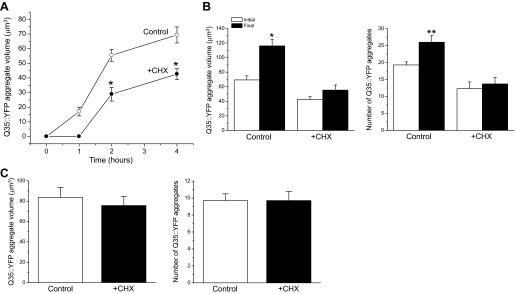
Characteristics of hypertonic stress-induced Q35::YFP aggregation during cycloheximide (CHX)-induced translation inhibition. A: effects of CHX on growth of Q35::YFP aggregates during hypertonic stress. Worms were exposed to 500 mM NaCl agar plates with or without 500 μg/ml CHX at time 0. Values are means ± SE (n = 8–30 aggregates measured in 7–9 worms). *P < 0.0003, compared with control worms. No Q35::YFP aggregates were detected at the 1-h time point in CHX-treated worms. B: effects of CHX on the growth and formation of Q35::YFP aggregates during recovery from hypertonic stress. Worms were exposed to 500 mM NaCl with or without CHX for 4 h and then returned to control medium without the drug for 3 h. Initial and final aggregate volumes and numbers were obtained after 4 h of hypertonic stress and 3 h of recovery, respectively. Both the size and number of Q35::YFP aggregates continued to increase in control worms even though the hypertonic stress was removed. CHX in contrast fully suppressed aggregation and aggregate growth. Values are means ± SE (n = 14–17). *P < 0.0001 and **P < 0.008, compared with the initial values. C: effects of CHX on posthypertonic stress growth and formation of Q35::YFP aggregates. Worms were exposed to 500 mM NaCl for 2 h and then transferred to control growth medium with or without CHX. Values are means ± SE (n = 10–21).
Hypertonicity-induced Q35::YFP aggregation continues even after cellular water balance is restored (5). This suggests that protein damage induced by hypertonic stress rapidly saturates cellular machinery that prevents aggregation and/or that protein aggregates represent a mechanism utilized by cells to isolate toxic intermediates including misfolded monomers and oligomeric structures (7, 17, 31). To further examine the impact of translation inhibition on Q35::YFP aggregation, we exposed worms to 500 mM NaCl for 4 h in the presence of cycloheximide and then returned them to control medium without the drug for 3 h. As shown in Fig. 1B, both the volume and number of Q35::YFP aggregates continued to increase significantly (P < 0.008) in control animals. In contrast, no significant (P > 0.1) change was observed in either aggregate number or volume during the recovery period in cycloheximide-treated worms (Fig. 1B). This indicates that inhibition of translation reduces initial hypertonic stress-induced protein damage and/or allows the cell to repair existing damage thus preventing further aggregation of Q35::YFP.
We also examined the effect of cycloheximide on Q35::YFP aggregation during the posthypertonic stress recovery period. Worms were exposed to 500 mM NaCl for 2 h and then returned for 2 h to control growth medium with or without cycloheximide. Inhibition of translation during the posthypertonic stress recovery period had no significant (P > 0.6) effect on aggregate volume or the number of aggregates formed (Fig. 1C). Thus the protective effect of translation inhibition occurs during hypertonic stress when protein unfolding and aggregation are being induced by increased cellular ionic strength and macromolecular crowding (5).
The cycloheximide treatment protocol used in our studies functions to acutely inhibit protein synthesis. We therefore also examined the effects of chronic reductions in the rate of translation on protein damage by knockdown of iftb-1, which encodes the eukaryotic translation initiation factor eIF-2β. RNAi silencing of iftb-1 inhibits protein synthesis in C. elegans ∼80% and activates gpdh-1 expression (23). Because constitutive activation of gpdh-1 usually results in elevation of whole animal glycerol levels (22), it is conceivable that any reductions in protein damage observed in iftb-1(RNAi) worms reflect reductions in water loss and shrinkage rather than a direct effect of reduced translation. However, hypertonicity-induced water loss and shrinkage are similar in control and iftb-1(RNAi) worms (6).
RNAi silencing of iftb-1 reduces the number of Q35::YFP aggregates formed during hypertonic stress (6). As shown in Fig. 2A, iftb-1 knockdown also greatly reduced the rate of aggregate growth during continuous exposure to 500 mM NaCl.
Fig. 2.
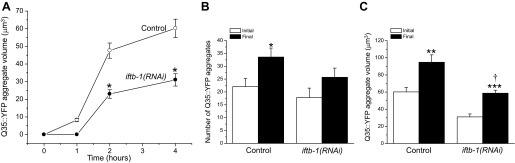
Characteristics of hypertonic stress-induced Q35::YFP aggregation in iftb-1(RNAi) worms. A: effects of RNA interference (RNAi) silencing of iftb-1 on growth of Q35::YFP aggregates during hypertonic stress. L1 larval worms were fed bacteria producing a scrambled dsRNA (control) or dsRNA homologous to iftb-1. L4 worms were exposed to 500 mM NaCl agar plates at time 0. Values are means ± SE (n = 26–50 aggregates measured in 7–9 worms). *P < 0.001, compared with control worms. No Q35::YFP aggregates were detected in iftb-1(RNAi) worms at the 1-h time point. B and C: effects of RNAi silencing of iftb-1 on the formation and growth of Q35::YFP aggregates during recovery from hypertonic stress. Worms were exposed to 500 mM NaCl for 4 h and then returned to control medium for 3 h. Initial and final aggregate volumes and numbers were obtained after 4 h of hypertonic stress and 3 h of recovery, respectively. Values are means ± SE (n = 6–49). *P < 0.05, **P < 0.01, and ***P < 0.001, compared with the initial values. †P < 0.0003, compared with final value in control worms.
During acute hypertonic stress (500 mM NaCl for 4 h) followed by a 3-h recovery in control medium, the increase in Q35::YFP aggregate number was arrested in iftb-1(RNAi) worms (Fig. 2B). Unlike cycloheximide-treated worms (Fig. 1B), the Q35::YFP aggregate volume continued to increase in iftb-1(RNAi) animals (Fig. 2C). However, the increase was significantly (P < 0.0003) less than observed in control worms. The reason for the difference in the growth in aggregate volume with cycloheximide treatment vs. iftb-1 knockdown is unclear. Chronic reductions in protein synthesis induce changes in gene expression (15, 35). Such changes may alter the cellular proteostasis environment, and this in turn may alter the mechanisms by which damaged proteins are handled. For example, misfolded proteins are toxic to cells and aggregation represents one mechanism by which that toxicity is reduced (7, 17, 31). The continued growth of existing aggregates in iftb-1(RNAi) worms may reflect a detoxification mechanism activated by prolonged translation inhibition.
The Q35::YFP used in these studies is expressed in body wall muscle cells. To determine whether the effect of translation inhibition is cell specific and/or protein specific, we examined additional reporters of protein damage. Figure 3A shows the effect of cycloheximide and RNAi silencing of iftb-1 on hypertonic stress-induced aggregation of Q44::YFP expressed in intestinal cells. As noted previously (28), aging-induced Q44::YFP aggregation is highly variable between animals. We therefore expressed the data as the percentage of animals exhibiting at least 10 Q44::YFP aggregates (28). Under control conditions, Q44::YFP aggregates were not detected in any of the 60 worms examined. Following a 2-h exposure to 500 mM NaCl, we observed that ≥10 aggregates formed in 17.5% of the animals. Treatment with cycloheximide or knockdown of iftb-1 significantly (P < 0.01) reduced the percentage of worms with ≥10 hypertonic stress-induced aggregates to ∼1.5–2.5%. Both experimental maneuvers also significantly (P < 0.002) reduced Q44::YFP aggregate volume by ∼50% (Fig. 3B).
Fig. 3.
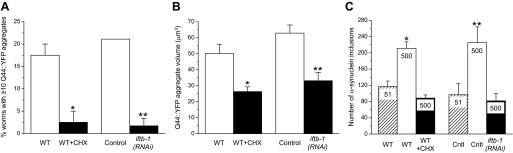
Characteristics of hypertonic stress-induced Q44::YFP and α-synuclein::YFP aggregation during translation inhibition by CHX or RNAi silencing of iftb-1. A: percentage of worms exhibiting ≥10 Q44::YFP aggregates in intestinal cells following a 2 h exposure to 500 mM NaCl. Wild-type (WT) worms were exposed to 500 mM NaCl in the presence or absence of 500 μg/ml CHX. Control worms were fed bacteria producing a scrambled dsRNA and compared with iftb-1(RNAi) worms. Values are means ± SE (n = 3 independent experiments with 20 worms each). *P < 0.01, compared with 500 mM NaCl alone; and **P < 001, compared with control worms. B: effects of translation inhibition on Q44::YFP aggregate volumes. Values are means ± SE (n = 9–19). *P < 0.002, compared with 500 mM NaCl alone; and **P < 0.001 compared with control worms. C: effects of translation inhibition on the number of α-synuclein::YFP inclusions in body wall muscle cells following a 2 h exposure to 500 mM NaCl. Concentration of NaCl is shown inside each bar. Values are means ± SE (n = 5–11). *P < 0.003, compared with WT worms on 51 mM NaCl; and **P < 0.02, compared with unstressed control (Cntl) worms maintained on 51 mM NaCl and fed bacteria producing scrambled dsRNA. Number of inclusions in CHX-treated and iftb-1(RNAi) worms was not significantly (P > 0.07) different from that of WT or control worms maintained on 51 mM NaCl.
α-Synuclein is an inherently disordered protein expressed abundantly in the brain and is found in protein inclusions associated with various neurodegenerative disorders (32). Human α-synuclein::YFP fusion protein expressed in C. elegans body wall muscle cells accumulates in inclusion bodies in an age-dependent manner (34) and in response to hypertonic stress (5). When worms were exposed to 500 mM NaCl for 2 h, α-synuclein::YFP inclusions increased significantly (P < 0.02) by 1.8- to 2.3-fold. In cycloheximide-treated or iftb-1(RNAi) worms, hypertonic stress did not significantly (P > 0.07) increase the number of inclusions above that observed in animals maintained at 51 mM NaCl (Fig. 3C).
Hypertonic stress induces rapid aggregation of endogenous proteins in C. elegans (5, 6, 9). To further assess the impact of translation rate on protein damage, we isolated total protein and a detergent insoluble fraction, which contains protein aggregates (5), from cycloheximide treated wild-type worms and iftb-1(RNAi) worms exposed for 4 h to 500 mM NaCl. Consistent with our previous findings (5), we observed that hypertonic stress induced significant (P < 0.04) increases in insoluble protein (Fig. 4). However, inhibition of translation fully suppressed this increase. The levels of insoluble protein in cycloheximide-treated and iftb-1(RNAi) worms were not significantly (P > 0.3) different from those in unstressed wild-type and control worms (Fig. 4).
Fig. 4.

Effect of inhibition of protein synthesis by CHX or RNAi silencing of iftb-1 on aggregation of endogenous proteins during hypertonic stress. CHX-treated WT (A) and iftb-1(RNAi) (B) worms were exposed for 4 h to 500 mM NaCl. Insoluble protein was quantified as a fraction of total protein and is plotted relative to unstressed (maintained on 51 mM NaCl) WT or control worms fed bacteria expressing a scrambled dsRNA. Values are means ± SE (n = 3–5). *P < 0.04, compared with WT 51 NaCl; **P < 0.04, compared with WT 500 mM NaCl; ***P < 0.004 compared with Control 51 NaCl; and †P < 0.01, compared with Control 500 NaCl.
It is widely accepted that ts mutations give rise to proteins that fold and function correctly at low or “permissive” temperatures. However, at elevated temperatures, temperature-sensitive (ts) mutant proteins misfold giving rise to mutant phenotypes (4, 14, 33). We have shown recently that hypertonic stress causes apparent misfolding of ts mutant proteins under permissive temperature conditions (5).
The let-60(ga89) and unc-15(el402) encode ts mutant ras GTPase and paramyosin, respectively. At elevated temperatures or under hypertonic conditions, these mutations give rise to egg-hatching defects and larval arrest (5). Mutations in unc-15 also give rise to motility defects (26). Figure 5A shows the effects of cycloheximide on the motility of L4/young adult wild-type worms and unc-15(e1402) mutants exposed for 2 h to 500 mM NaCl with or without cycloheximide and then allowed to recover for 3 h on normal growth medium without the drug. Slow movement is defined as the inability to reach a bacterial food source within 10 min (14) (see materials and methods). Under control conditions, most wild-type worms moved to food within the 10-min time frame. Hypertonic stress slowed movement in ∼50% of wild-type worms, but this was unaffected by cycloheximide. In unc-15(e1402) mutants, hypertonic stress significantly increased (P < 0.001) the number of slow moving worms from ∼43 to ∼93% (Fig. 5A). Cycloheximide treatment significantly (P < 0.05) reduced the fraction of worms exhibiting the mutant phenotype by ∼20% (Fig. 5A).
Fig. 5.

Effect of inhibition of protein synthesis on hypertonic stress-induced protein misfolding in unc-15(e1402) and let-60(ga89) temperature-sensitive (ts) mutant worms. unc-15(e1402) and let-60(ga89) are ts mutant alleles encoding paramyosin and ras GTPase, respectively. A: effect of CHX on slow movement phenotype-induced by hypertonic stress in WT and unc-15(e1402) mutant worms. Animals were exposed for 2 h to 500 mM NaCl with or without CHX and then allowed to recover for 3 h on normal growth medium without the drug. Slow movement is defined as the inability to reach a bacterial food source within 10 min. Hypertonic stress increases the slow movement phenotype in both WT and mutant worms. In unc-15(e1402) mutant worms, this phenotype is partially reversed by CHX-induced inhibition of translation. Values are means ± SE (n = 3 independent experiments with 50 worms in each). *P < 0.001, compared with control unc-15(e1402) worms; and **P < 0.05, compared with unc-15(e1402) worms exposed to 500 mM NaCl without CHX. B: effect of RNAi-induced inhibition of translation on egg hatching/larval arrest phenotype induced by hypertonic stress in WT and let-60(ga89) worms. Worms were grown from the L1 larval stage on 51 mM NaCl growth medium with bacteria expressing scrambled dsRNA or dsRNA homologous to iftb-1, which encodes the eukaryotic translation initiation factor eukaryotic initiation factor-2β (eIF-2β). Gravid adults were transferred to 300 mM NaCl RNAi plates and allowed to lay 40–50 eggs over an ∼24 h period. The percentage of eggs failing to hatch or that hatched L1 larvae that arrested was scored. Values are means ± SE (n = 5 independent experiments with 40–50 eggs in each). *P < 0.002, compared with WT; and **P < 0.03, compared with let-60(ga89) worms fed bacteria expressing scrambled dsRNA. WT, unc-15(e1402), and let-60(ga89) worms were maintained at 16°C throughout the experiments.
We were unable to induce the let-60(ga89) mutant phenotype with short-term hypertonic stress. Because worms cannot tolerate prolonged treatment with cycloheximide, we utilized RNAi to inhibit protein synthesis. Wild-type and let-60(ga89) L1 larvae were fed bacteria producing a scrambled dsRNA or dsRNA homologous to iftb-1. Gravid adults were transferred to 300 mM NaCl and allowed to lay 40–50 eggs over an ∼24-h period. The percentage of eggs failing to hatch or that hatched L1 larvae that arrested was scored. As shown in Fig. 5B, only ∼10% of the eggs from wild-type worms fed scrambled dsRNA failed to hatch or arrested at L1. In contrast, nearly 60% of the eggs from let-60(ga89) worms fed scrambled dsRNA showed the hatching/arrest phenotype. Inhibition of protein synthesis by iftb-1 RNAi significantly (P < 0.03) reduced the hatching/arrest phenotype to ∼23%. Taken together, the data in Fig. 5, A and B, demonstrate that inhibition of protein synthesis also suppresses hypertonic stress-induced protein misfolding.
The results shown in Figs. 1–5 as well as our previous findings (6) clearly demonstrate that inhibition of protein synthesis by cycloheximide or iftb-1 knockdown minimizes diverse types of hypertonic stress-induced protein damage in diverse cells types. Hypertonic stress inhibits proteins synthesis rapidly in C. elegans via activation of the GCN kinase GCN-2 with subsequent phosphorylation of the translation initiation factor eIF-2α (23). An important question is whether this GCN-2-mediated reduction in protein synthesis also functions to minimize hypertonic stress-induced protein damage. To test this possibility, we examined the effect of water loss on protein damage in a mutant worm strain expressing a loss-of-function allele of gcn-1 [gcn-1(nc40)]. GCN-2 activation requires interaction with the accessory protein encoded by gcn-1 (29). Hypertonic stress-induced phosphorylation of eIF-2α and inhibition of protein synthesis are completely suppressed in gcn-1(nc40) mutant worms (23).
We first examined the effect of the gcn-1(nc40) allele on 24-h survival during hypertonic stress. As shown in Fig. 6A, wild-type worms survived significantly (P < 0.03) better on growth media containing 400–600 mM NaCl compared with gcn-1(nc40) loss-of-function mutants.
Fig. 6.

Effect of general control nonderepressible (GCN) kinase signaling on survival and formation of Q35::YFP aggregates. Activation of GCN kinase signaling inhibits protein synthesis in C. elegans during hypertonic stress. Loss-of-function mutations in either gcn-1 or gcn-2 prevent translation inhibition (23). A: effect of the gcn-1 loss-of-function allele, gcn-1(nc40) on survival during hypertonic stress. Survival was measured 24 h after exposure to high NaCl growth media. Values are means ± SE (n = 3–4 independent experiments with 85–140 worms in each). *P < 0.03, **P < 0.02, and †P < 0.001, compared with WT worms. B: effect of gcn-1(nc40) loss-of-function allele on motility. Worms were exposed to 300 mM NaCl for 24 h and motility was quantified as the distance moved in 30 s. Values are means ± SE (n = 12). *P < 0.0001 compared with WT worms. C: rate of Q35::YFP aggregate formation in WT worms and gcn-1(nc40) mutants. Values are means ± SE (n = 8–17). *P < 0.0001, compared with WT worms. D: rate of Q35::YFP aggregate growth in gcn-1(nc40) mutants. Values are means ± SE (n = 18–26). *P < 0.01 and **P < 0.001, compared with WT worms (see Fig. 1A).
Even though 100% of wild-type and gcn-1(nc40) worms survived for 24 h on 300 mM NaCl medium (Fig. 6A), the mutants appeared to be considerably less healthy. To characterize this apparent difference, we quantified motility by measuring the distance worms moved in 30 s after a 24-h exposure to 300 mM NaCl, which is sufficient time for full volume recovery and glycerol accumulation in wild-type animals (20). Wild-type worms moved nearly 80% faster than the gcn-1(nc40) mutants (Fig. 6B). These studies and our previous work demonstrate that GCN-1/GCN-2 signaling not only downregulates protein synthesis during hypertonic stress (23) but is required for optimal survival of C. elegans under hypertonic conditions.
We next crossed gcn-1(nc40) mutant worms with worms expressing transgenic Q35::YFP in their body wall muscle cells and quantified Q35::YFP aggregation during exposure to 500 mM NaCl. The rate of aggregate formation was significantly (P < 0.0001) faster in the gcn-1(nc40) mutants (Fig. 6C). In addition, the aggregates grew at a significantly (P < 0.01) greater rate (Fig. 6D) than observed in wild-type worms (compare to Fig. 1A).
As final test of the role of GCN-2 signaling in modulating protein damage, we quantified endogenous protein aggregation in wild-type and gcn-1(nc40) mutant worms. Hypertonic stress-induced protein aggregation was ∼1.6-fold greater (P < 0.02) in gcn-1(nc40) mutant compared with wild-type worms (Fig. 7). Taken together, the data in Figs. 6 and 7 demonstrate that inhibition of protein synthesis during hypertonic stress reduces protein damage and improves survival.
Fig. 7.

Effect of GCN kinase signaling on aggregation of endogenous proteins during hypertonic stress. A: examples of SDS-PAGE gels of total and detergent insoluble proteins isolated from WT and gcn-1(nc40) worms exposed to 500 mM NaCl for 4 h. B: quantification of insoluble protein. Insoluble protein was quantified as a fraction of total protein and is plotted relative to unstressed WT control animals maintained at 51 mM NaCl. Values are means ± SE (n = 3). *P < 0.02, compared with WT at 500 mM NaCl.
DISCUSSION
Reductions in protein translation occur in response to a wide variety of environmental stressors and are widely thought to play a role in minimizing stress-induced protein damage (15, 35). Our studies in C. elegans have tested this idea directly for the first time using the aggregation prone reporter Q35::YFP expressed in body wall muscle cells. Cycloheximide treatment during hypertonic stress strikingly reduces the number of Q35::YFP aggregate formed (6), reduces the rate of growth of single Q35::YFP aggregates, and completely blocks the formation and growth of aggregates that occurs after hypertonic stress is removed (Fig. 1, A and B). Inhibition of translation by RNAi silencing of iftb-1 has similar effects (Fig. 2).
The effect of translation inhibition is not restricted to a specific protein or cell type. Cycloheximide or RNAi silencing of iftb-1 reduces formation and growth of Q44::YFP aggregates in intestinal cells and the formation of α-synuclein inclusions in muscle cells (Fig. 3). Translation inhibition also reduces hypertonic stress-induced aggregation of multiple unidentified endogenous proteins (Fig. 4) and misfolding of UNC-15 and LET-60 (Fig. 5). unc-15 encodes paramyosin, which is expressed in all C. elegans muscle cells. LET-60 is a ras GTPase present in numerous diverse cell types. The larval lethality phenotype induced by hypertonic stress (Fig. 5B) is likely due to failure of the excretory system to form properly in mutant animals (36).
Our previous work (6) and data in Figs. 1–5 clearly demonstrate that exogenous inhibition of translation protects diverse proteins in diverse cell types from hypertonic stress-induced aggregation and misfolding. What roles do endogenous translation regulatory mechanisms play in modulating protein damage? In response to water loss, GCN-2 kinase signaling is activated in C. elegans resulting in the phosphorylation of eIF-2α and an ∼70% inhibition of protein synthesis (23). Phosphorylated eIF-2α inhibits translation by preventing the formation of elongation competent ribosomes (16). Disruption of the GCN-2 signaling cascade completely blocks hypertonic stress-induced translation inhibition (23), reduces worm survival (Fig. 6, A and B), dramatically increases the number and rate of growth of Q35::YFP aggregates (Fig. 6, C and D), and increases the aggregation of endogenous proteins (Fig. 7). Consistent with these results, Baker et al. (1) have recently shown that C. elegans clk-1 mutants, which have elevated levels of reactive oxygen species (24), exhibit increased oxidative protein damage that is exacerbated by loss of GCN-2 kinase function.
Stress-induced activation of GCN-2 kinase signaling and subsequent reduction in protein synthesis occur in response to diverse stressors in organisms ranging from yeast to mammals (11, 15, 35). Our studies together with those of Baker et al. (1) demonstrate that GCN-2 kinase signaling also plays an important role in minimizing protein damage under stress conditions. In C. elegans, inhibition of protein synthesis, either by hypertonic stress, knockdown of genes required for translation, or protein synthesis inhibitors activates gpdh-1 transcription (20, 22, 23). Increased gpdh-1 expression is required for synthesis of the organic osmolyte glycerol and optimal survival of worms under hypertonic conditions (22). GCN-2-induced inhibition of protein synthesis during hypertonic stress thus serves two critical roles, minimization of protein damage brought about by water loss and as a signal that activates genes required for survival.
Reductions in translation extend lifespan in numerous diverse species (18, 25), and extended lifespan is associated with increased resistance to multiple stressors (18, 30) including hypertonic stress (21). The relationship between reductions in protein synthesis and improved stress resistance is a problem of considerable biological importance. In C. elegans, the highly conserved WNK and Ste20 kinases, WNK-1 and GCK-3, function downstream from GCN-2-mediated inhibition of translation to activate gpdh-1 transcription (23). Defining the signaling mechanisms by which these kinases detect changes in translation and activate osmotic stress resistance pathways will provide important insights into how protein synthesis, proteostasis networks, and stress response mechanisms are coordinated and how this impacts numerous diseases and the pathophysiology of aging.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-61168 (to K. Strange) and National Institute of General Medical Sciences Institutional Development Award (IDeA) P20-GM-103423. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.K. and K.S. conception and design of research; H.K. performed experiments; H.K. and K.S. analyzed data; H.K. and K.S. interpreted results of experiments; H.K. and K.S. prepared figures; H.K. and K.S. drafted manuscript; H.K. and K.S. edited and revised manuscript; H.K. and K.S. approved final version of manuscript.
REFERENCES
- 1.Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet 8: e1002760, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 319: 916–919, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Brenner S. The genetics of Caenorhabditis elegans. Genetics 77: 71–94, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest 99: 1432–1444, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burkewitz K, Choe K, Strange K. Hypertonic stress induces rapid and widespread protein damage in C. elegans. Am J Physiol Cell Physiol 301: C566–C576, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkewitz K, Choe KP, Choung-Hee LE, Deonarine A, Strange K. Characterization of the proteostasis roles of glycerol accumulation, protein degradation and protein synthesis during osmotic stress in C. elegans. PLoS One 7: e34153, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 6: 140–147, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Choe KP, Strange K. Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am J Physiol Cell Physiol 293: C915–C927, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Choe KP, Strange K. Genome-wide RNAi screen and in vivo protein aggregation reporters identify degradation of damaged proteins as an essential hypertonic stress response. Am J Physiol Cell Physiol 295: C1488–C1498, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci 9: 759–767, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci 70: 3493–3511, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10: 715–724, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh K, Dill K. Cellular proteomes have broad distributions of protein stability. Biophys J 99: 3996–4002, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gidalevitz T, Ben Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311: 1471–1474, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6: 318–327, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486–489, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Kenyon CJ. The genetics of ageing. Nature 464: 504–512, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev 10: 205–215, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamitina ST, Morrison R, Moeckel GW, Strange K. Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am J Physiol Cell Physiol 286: C785–C791, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Lamitina ST, Strange K. Transcriptional targets of the DAF-16 insulin signaling pathwayprotect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol 288: C467–C474, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Lamitina T, Huang CG, Strange K. Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci USA 103: 12173–12178, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee EC, Strange K. GCN-2 dependent inhibition of protein synthesis activates osmosensitive gene transcription via WNK and Ste20 kinase signaling. Am J Physiol Cell Physiol 303: C1269–C1277, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20: 2131–2136, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta R, Chandler-Brown D, Ramos FJ, Shamieh LS, Kaeberlein M. Regulation of mRNA translation as a conserved mechanism of longevity control. Adv Exp Med Biol 694: 14–29, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Miller RK, Qadota H, Mercer KB, Gernert KM, Benian GM. UNC-98 and UNC-96 interact with paramyosin to promote its incorporation into thick filaments of Caenorhabditis elegans. Mol Biol Cell 19: 1529–1539, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78: 959–991, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Prahlad V, Morimoto RI. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci USA 108: 14204–14209, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sattlegger E, Hinnebusch AG. Separate domains in GCN1 for binding protein kinase GCN2 and ribosomes are required for GCN2 activation in amino acid-starved cells. EMBO J 19: 6622–6633, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shore DE, Ruvkun G. A cytoprotective perspective on longevity regulation. Trends Cell Biol S0962–S8924, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 437: 257–261, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem 103: 17–37, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Van Dyk TK, Gatenby AA, LaRossa RA. Demonstration by genetic suppression of interaction of GroE products with many proteins. Nature 342: 451–453, 1989 [DOI] [PubMed] [Google Scholar]
- 34.van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet 4: e1000027, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamasaki S, Anderson P. Reprogramming mRNA translation during stress. Curr Opin Cell Biol 20: 222–226, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yochem J, Sundaram M, Han M. Ras is required for a limited number of cell fates and not for general proliferation in Caenorhabditis elegans. Mol Cell Biol 17: 2716–2722, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]