

Abstract
Adriamycin (ADR) administration in susceptible rodents such as the BALB/c mouse strain produces injury to the glomerulus mimicking human focal glomerular sclerosis. The goal of the present study was to use this model to investigate antiproteinuric action of nitro-oleic acid (OA-NO2), a nitric oxide-derived endogenous lipid product, which has exhibited multiple attractive signaling properties particularly in the kidney. BALB/c mice were pretreated for 2 days with OA-NO2 at 5 mg·kg−1·day−1 via an osmotic minipump, followed by a single injection of vehicle or adriamycin (10 mg/kg) via the tail vein. Albuminuria and renal function were analyzed at 1 wk post-ADR treatment. ADR mice developed prominent albuminuria, hypoalbuminemia, hyperlipidemia, and severe ascites. In contrast, the symptoms of nephrotic syndrome were greatly improved by OA-NO2 treatment. In parallel, plasma creatinine and plasma urea nitrogen were elevated in the ADR group, and the severity was less in the ADR+OA-NO2 group. OA-NO2 attenuates ADR-induced glomerulosclerosis, podocyte loss, and tubulointerstitial fibrosis. Indices of oxidative stress, including plasma and urinary thiobarbituric acid-reactive substances and renal expression of NAD(P)H oxidase p47phox and gp91phox, and inflammation, including renal expression of TNF-α, IL-1β, and MCP-1 in response to ADR, were all similarly suppressed. Together, these findings suggest that OA-NO2 exerts renoprotective action against ADR nephropathy likely via its anti-inflammatory and antioxidant properties.
Keywords: adriamycin, albuminuria, glomerulosclerosis, OA-NO2, oxidative stress
proteinuria is an early and predominant pathological feature of a wide variety of primary glomerular disease, and it is not only a marker for the progression and prognosis of kidney injury but also an important pathogenic mediator that causes subsequent oxidant stress, inflammatory, and fibrotic responses in the kidney, finally leading to end-stage renal disease. Podocytes are one of the principal components of the glomerular filtration barrier and play a crucial role in the regulation of glomerular function, and podocyte injury is one of the major causes leading to defective glomerular filtration, resulting in proteinuria (20, 25, 32). Therefore, protection of podocytes in proteinuric kidney disorders should be a major strategy to prevent the worsening of renal disease, but drugs clinically available for that goal are not very effective.
Adriamycin (ADR) is an anthracycline antibiotic used widely in solid and hematopoietic malignancy therapies as one of the simplest and most effective chemotherapeutic agents. It can cause podocyte foot process effacement and increase glomerular permeability, leading to proteinuria (8, 11, 17). The mechanism involved in ADR nephropathy is still incompletely understood. Among many possible pathogenic factors, oxygen free radicals and inflammation are thought to play a role (21, 28).
OA-NO2 is an electrophilic nitroalkenyl fatty acid with several attractive signaling properties. It is detected in healthy human blood, reaching ∼0.6 μM (3), and increased production is demonstrated during inflammatory and metabolic stress (4, 24). The signaling pathway transducing the action of OA-NO2 appears to be complex and remains to be defined particularly under pathophysiological condition in vivo. In vitro studies suggest that OA-NO2 may signal through activation of peroxisome proliferator-activated receptors (PPARs) (3), S-alkylation of critical thiols of Keap-1, and subsequent activation of Nrf2 (4, 16, 27), covalent modification of the p65 subunit of NF-κB, and suppression of its activity (9). A series of our previous studies demonstrates that OA-NO2 is protective against kidney injury induced by ischemia-reperfusion and endotoxin-induced sepsis, and in obese Zucker rats (18, 29–30). The present study was undertaken to examine the potential therapeutic action and mechanism of OA-NO2 in a murine model of ADR nephropathy.
MATERIALS AND METHODS
Materials.
OA-NO2 was synthesized and purified as described previously (3). ADR (doxorubicin HCl) was purchased from Bedford Laboratories. All other reagents were purchased from Sigma-Aldrich unless otherwise specified.
Animals and treatments.
Male BALB/c mice were purchased from the Jackson Laboratories (Bar Harbor, ME). Mice were maintained in a temperature-controlled barrier facility with a 12:12-h light-dark cycle and were given free access to standard laboratory chow and tap water. Mice were randomized into three groups: group 1: control, group 2: ADR, and group 3: ADR+OA-NO2. In group 3, OA-NO2 (dissolved in ethanol) was administered at 5 mg·kg−1·day−1 via a subcutaneously implanted osmotic minipump, and vehicle (ethanol) was given to the other two groups. This dose was chosen based a previous study (36). After 2 days of pretreatment with OA-NO2, groups 2 and 3 received a single tail vein injection of ADR at 10 mg/kg. Group 1 received a single tail vein injection of saline. Twenty-four-hour urine was collected using metabolic cages. Seven days after ADR treatment, all mice were euthanized and the kidneys were immediately harvested for gene expression or histological analyses. All protocols employing mice were conducted in accordance with the principles and guidance of the University of Utah Institutional Animal Care and Committee.
Measurement of biochemical parameters.
Urine samples were centrifuged for 5 min at 10,000 rpm. Blood samples from anesthetized mice were collected by puncturing the vena cava using a 1-ml insulin syringe containing 50 μl of 1 mM EDTA in the absence of protease inhibitors. Urine and plasma albumin were determined using a murine microalbuminuria enzyme-linked immunosorbent assay kit (1011, Exocell). The plasma triglyceride level was determined using a LabAssay Triglyceride ELISA Kit (290-63701, Wako). Urine and plasma levels of urea were measured with a Urea Nitrogen Direct Kit (0580-250, Stanbio Laboratory), and urine and plasma levels of creatinine were measured using a Creatinine Liquicolor Kit (0420-250, Stanbio Laboratory).
Morphological studies.
Under anesthesia, kidneys are removed and fixed with 4% paraformaldehyde. The tissues were subsequently embedded in paraffin, and 4-μm sections were cut and stained with periodic acid-Schiff (PAS). Glomerular sclerosis was assessed as follows using a semiquantitative score previously described (30–32): grade 0, normal appearance; grade I, involvement of up to 25% of the glomerulus; grade II, involvement of 25–50% of the glomerulus; grade III, involvement of 50–75% of the glomerulus; and grade IV, involvement of 75–100% of the glomerulus. A glomerulosclerosis index (GSI) was calculated by multiplying the number of glomeruli with a sclerosis score of I by one, the number with a score of II by two, III by three, and IV by four. These values were summed and divided by the number of glomeruli assessed, including those with a sclerosis score of zero. The GSI for each kidney specimen was a sum of the points from 30 glomeruli. Tubulointerstitial injury (defined as tubular atrophy, dilatation, thickening of the basement membrane, protein cast) was assessed by semiquantitative analysis (33, 34). Thirty cortical fields from each animal were examined at ×200 magnification and graded according to a scale of 0–4: 0, no tubulointerstitial injury; 1, <25% of the tubulointerstitium injured; 2, 25–50% of the tubulointerstitium injured; 3, 51–75% of the tubulointerstitium injured; and 4, 76–100% of the tubulointerstitium injured. All sections were examined in a blind manner.
Immunohistochemistry.
Immunohistochemical staining was performed as previously described (15). Anti-WT1 antibody was purchased from Dako (Mob437, Dako).
Quantitative RT-PCR.
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA), and first-strand cDNAs were synthesized from 2 μg of total RNA in a 20-ml reaction using SuperScript (Invitrogen). The first-strand cDNAs served as the template for quantitative (q) PCR performed in the Applied Biosystems 7900 Real Time PCR System using SYBR green PCR reagent (Applied Biosystems, Foster City, CA). The amplification was carried out for 40 cycles with conditions of 15-s denaturation at 95°C. The sequence of oligonucleotides used for qPCR (RT-PCR) is listed as follows: GAPDH sense: 5′-GTC TTCACTACCATGGAGAAGG-3′ and antisense: 5′-TCATGGATGACCTTGGCC AG-3′; fibronectin (FN) sense: 5′-CGTGGAGCAAGAAGGACAA-3′ and antisense: 5′-GTGAGTCTGCGGTTGGTAAA-3′; smooth muscle actin (SMA)-α sense: 5′-CCCTGAAGAGCATCC GACA-3′ and antisense: 5′-CCAGAGTCCAGCACAATACC-3′; transforming growth factor (TGF)-β sense: 5′-TAC GCCTGAGTGGCTGTCTT-3′ and antisense: 5′-CGTGGAGTTTGTTATCTTTGCT-3′; zonula occludens (ZO)-1 sense: 5′-GCGCGGAGAGAGACAAGA-3′ and antisense: 5′-CTGGCCCTC CTTTTAACACA-3′; p47phox sense: 5′-CACTCCCTTTGCTTCCATCT-3′ and antisense: 5′-ATGTTGCTATCCCAGCCAGT-3′; gp91phox sense: 5′-CCGTATTGT GGGAGACTGGA-3′ and antisense: 5′-CTTGAGAATGGAGGCAAAGG-3′; desmin sense: 5′- GTGGATGCAGCCACTCTAGC-3′ and antisense: 5′- TTAGCCGCGATG GTCCATAC-3; TNF-α sense: 5′-TCCCCAAAGGGATGAGAAG-3′ and antisense: 5′-CACTTGGTGGTTTGCTACGA-3′; IL-1β sense: 5′- ACTGTGAAATGCCACTTT TG-3′ and antisense: 5′-TGTTGATGTGCTGCTGTGAG-3′; MCP-1 sense: 5′-GCT CTCTCTTCCTCCACCAC-3′ and antisense: 5′-ACAGCTTCTTTGGGACACCT-3′; and collagen type III sense: 5′-AGGCAACAGTGGTTCTCCTG-3′ and reverse 5′-GAC CTCGTGCTCCAGTTAGC-3′.
Immunoblotting.
The kidney lysates were stored at −80°C until assayed. Protein concentrations were determined using Coomassie reagent. An equal amount of the whole tissue protein was denatured at 100°C for 10 min, separated by SDS-PAGE, and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS), followed by incubation for 1 h with a primary antibody. The blots were washed with TBS followed by incubation with a horseradish peroxidase-conjugated secondary antibody. Immune complexes were detected using ECL methods. The immunoreactive bands were quantified using the Gel and Graph Digitizing System (Silk Scientific, Tustin, CA).
Measurement of thiobarbituric acid-reactive substances.
The measurement of thiobarbituric acid-reactive substances (TBARS) in the mouse kidney was based on the formation of malondialdehyde (MDA) by using a commercially available TBARS Assay kit (10009055, Cayman Chemical) according to the manufacturer's instructions.
Statistical analysis.
All values are represented as means ± SE. Data were analyzed using an unpaired t-test or ANOVA followed by a Bonferroni posttest. Differences were considered to be significant when P < 0.05.
RESULTS
OA-NO2 attenuates albuminuria and renal dysfunction.
BALB/c mice were administered vehicle, ADR, or ADR in combination with OA-NO2; OA-NO2 was delivered via an osmotic minipump 2 days before ADR injection. At day 5 after ADR injection, albuminuria was most evident in the ADR group (508.89 ± 48.52 μg/24 h) compared with the control group (33.39 ± 3.50 μg/24 h) and was attenuated in the ADR+OA-NO2 group (342.40 ± 33.26 μg/24 h). (Fig. 1). These changes were observed at day 3 and maintained at day 7. At day 7, plasma albumin was significantly reduced in the ADR group (0.28 ± 0.08 g/dl) compared with the control group (1.01 ± 0.15 g/dl) and was significantly restored in the ADR+OA-NO2 group; the decrease in plasma albumin levels was significantly attenuated (0.58 ± 0.13 g/dl) (Fig. 2A). Ninety percent of ADR mice had severe ascites at death, in contrast to only 20% of ADR+OA-NO2 mice, who had mild ascites (Fig. 2B).
Fig. 1.

Nitro-oleic acid (OA-NO2) ameliorates albuminuria in adriamycin (ADR) nephropathy. ELISA analysis shows the levels of urinary albumin in different groups of mice at the indicated period of time after ADR injection. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
Fig. 2.

OA-NO2 ameliorates hypoalbuminemia and ascites in ADR nephropathy. A: ELISA analysis of plasma albumin in different groups of mice at day 8 after ADR injection. B: photographs of ascites in different groups of mice at day 8 after ADR injection. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
ADR mice developed severe hyperlipidemia (plasma triglyceride: 396.18 ± 70.94 mg/dl) that was less in the ADR+OA-NO2 group (plasma triglyceride: 212.70 ± 39.22 mg/dl) (Fig. 3A). Plasma creatinine and blood urea nitrogen (BUN) were determined to reflect renal function. ADR mice had elevated plasma creatinine and BUN, both of which were significantly attenuated in the ADR+OA-NO2 group (Fig. 3, B and C).
Fig. 3.

OA-NO2 ameliorates hypertriglyceridemia and renal dysfunction in ADR nephropathy. A: plasma triglyceride. B: plasma creatinine. C: blood urea nitrogen (BUN). Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
OA-NO2 attenuates glomerular injury and renal fibrosis.
To correlate the reduction of albuminuria with glomerular structure, the effect of drug treatments on glomerulosclerosis was assessed by PAS staining. Being consistent with the data on albuminuria, results from the ADR mice showed marked glomerulosclerosis as evidenced by mesangial expansion and increased accumulation of extracellular matrix (ECM) in the mesangium (Fig. 4A). A semiquantitative glomerulosclerotic index of kidney sections confirmed the histological data. The ADR mice showed the highest score, and OA-NO2 treatment led to a marked reduction in the index (P < 0.05) (Fig. 4B).
Fig. 4.
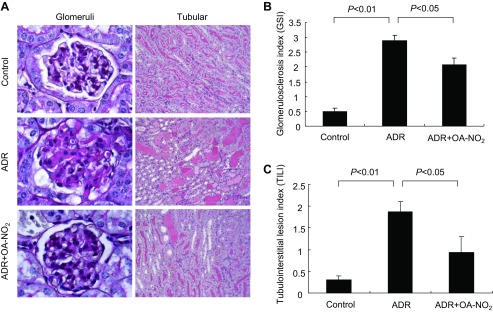
OA-NO2 ameliorates glomerulosclerosis and tubulointerstitial lesions in ADR nephropathy. A: representative micrographs showing kidney histology in different groups of mice at day 8 after ADR injection. Kidney sections were stained with periodic acid-Schiff reagent [magnification: ×200 (right), ×1,000 (left)]. B and C: glomerulosclerosis index (GSI) and tubulointerstitial lesion index (TILI) in different groups of mice. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
Because podocyte injury is an early and predominant pathological feature of the ADR model, we examined expression of a number of podocyte markers. WT1 is a pivotal transcription factor that is essential for the maintenance of the differentiated features of adult podocytes (35, 36). As illustrated in Fig. 5, A and B, immunoblotting revealed a marked reduction of WT1 after ADR injury compared with controls; OA-NO2 pretreatment prevented the downregulation of WT1 in the ADR mice (P < 0.05). The number of podocytes was semiquantitively analyzed by immunohistochemical analysis of WT-1. The number of WT1-positive cells was reduced in the ADR group and was partially restored in the ADR+OA-NO2 group (Fig. 5, C and D). qRT-PCR was performed to examine mRNA expression of ZO-1 and desmin. Renal ZO-1 mRNA exhibited a trend of reduction in the ADR group compared with the control group and a significant elevation in the ADR+OA-NO2 group (Fig. 5E). Desmin mRNA was upregulated in the ADR mice, and treatment with OA-NO2 prevented this increase (Fig. 5F).
Fig. 5.

OA-NO2 preserves podocyte markers in ADR nephropathy. A: immunoblot analysis of WT1 and β-actin in the kidney. B: densitometric analysis of WT1 protein. The densitometric value of WT1 protein was normalized by β-actin. C: immunohistochemical analysis of WT1 in the kidney. D: the number of WT1-positive cells per glomerulus. E: quantitative (q) RT-PCR analysis of zonula occludens (ZO)-1 in the kidney. F: qRT-PCR analysis of desmin. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
We further examined renal fibrosis by determining the expression of α-SMA and FN and TGF-β in the kidney. As shown in Fig. 6, ADR mice showed marked increases in α-SMA and FN expression at the mRNA level relative to the control by real-time PCR (Fig. 6, A and B), and Western blotting revealed marked upregulation of α-SMA and FN (Fig. 6, C and D). The densitometric values of these two proteins are shown in Fig. 6, E and F. OA-NO2 treatment prevented the upregulation of α-SMA and FN in the ADR mice (P < 0.05). In addition, the mRNA expression of several other fibrosis/sclerosis-related genes in the kidney was upregulated in the ADR mice, including TGF-β (Fig. 6G) and collagen III (Fig. 6H). OA-NO2 treatment induced a dramatic suppression of these genes in the kidney (P < 0.05). These data are consistent with the antisclerotic effect of OA-NO2 treatment.
Fig. 6.

OA-NO2 hampers renal fibrosis in ADR nephropathy. A and B: qRT-PCR analysis of renal mRNA levels of fibronectin (FN) and collage III. C and D: representative immunoblots of renal α-smooth muscle actin (SMA) and FN. β-Actin served as a loading control. E and F: densitometric analysis of immunoblots in C and D. G and H: qRT-PCR analysis of renal mRNA levels of transforming growth factor (TGF)-β and α-SMA. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
OA-NO2 hampers renal oxidative stress.
Among many possible pathogenic factors, oxidative stress has emerged as an important pathogenic factor in the development of ADR nephropathy. To investigate whether OA-NO2 had an antioxidative effect on ADR mice, we analyzed plasma and urinary levels of TBARS, a reliable product of lipid oxidation. As a result, the ADR group showed a marked increase in plasma (Fig. 7A), urinary (Fig. 7B), and kidney (Fig. 7C) TBARS compared with the control group. Treatment with OA-NO2 markedly attenuated the ADR-induced increase in plasma and urinary TBARS compared with ADR mice (Fig. 7, A and B). There was a trend of reduction of TBARS levels in response to OA-NO2 treatment, but this did not reach a statistical significance. NAD(P)H oxidase is an important source of reactive oxygen species (ROS) generation in various pathological conditions. We therefore examined renal expression of major subunits of NAD(P)H oxidase. As shown in Fig. 8, A and B, renal mRNA expression of p47phox and gp91phox was significantly increased in ADR mice compared with the control group, and the increase was less in the ADR+OA-NO2 group (P < 0.05). The change in gp91phox was further confirmed at the protein level (P < 0.01; Fig. 8, C and D).
Fig. 7.

Effect of OA-NO2 on thiobarbituric acid-reactive substances (TBARS) levels. A: measurement of plasma TBARS. B: measurement of urinary TBARS. C: measurement of kidney TBARS. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
Fig. 8.
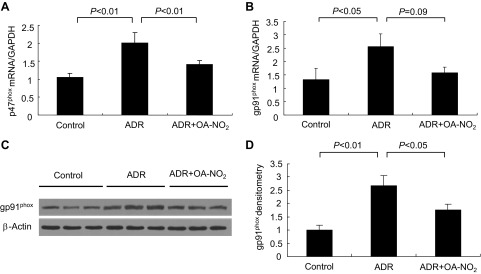
Effect of OA-NO2 on renal mRNA expression of NADPH oxidase subunits. A and B: qRT-PCR analysis of renal mRNA expression of p47phox and gp91phox. C and D: representative immunoblots and densitometric of gp91phox and β-actin in the kidneys. The densitometric value of gp91phox protein was normalized by β-actin. Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
OA-NO2 hampers renal inflammation.
ADR induces a proinflammatory response in the kidney, releasing cytokines and chemokines responsible for subsequent kidney injury. To examine whether OA-NO2 could reduce inflammation, we performed qRT-PCR analysis of TNF-α, IL-1β, and MCP-1. The renal expression of these proinflammatory mediators was in induced in parallel in ADR mice, and the inductions were all suppressed by OA-NO2 (Fig. 9, A–C).
Fig. 9.

OA-NO2 attenuates renal inflammation in ADR nephropathy. qRT-PCR was performed to determine renal mRNA expression of TNF-α (A), IL-β (B), and MCP-1 (C). Control: n = 8; ADR: n = 18; ADR+OA-NO2: n = 16. Values are means ± SE.
DISCUSSION
OA-NO2 is a recently identified as an endogenous signaling molecule capable of activating PPARs, inhibiting NF-κB, and releasing NO. A series of studies from our group demonstrated that OA-NO2 exerts renoprotective action in animal models of kidney injury induced by ischemia-reperfusion, endotoxemia, and metabolic syndrome. Of particular note, OA-NO2 effectively lowered proteinuria in obese Zucker rats, suggesting a possible protective action of OA-NO2 against glomerular disease (29). ADR-induced kidney damage is one of the few exciting murine models of acquired glomerulonephropathy, in which progressive renal damage leads to terminal renal failure (10, 12, 19, 31, 35). Despite the incomplete understanding of the underlying mechanism, ADR in susceptible genetic backgrounds induces pathological glomerular changes similar to human focal glomerular sclerosis (10). In the present paper, we use this model to explore the therapeutic potential and mechanism of OA-NO2 in the management of glomerular disease.
In the BALB/c background, ADR administration induced the typical nephritic syndrome, characterized by severe albuminuria, hypoalbuminemia, hyperlipidemia, and ascites. As a major mechanism of these symptoms, ADR can directly injure the podocytes, resulting in proteinuria and glomerular sclerosis. Greater proteinuria leads to the lowering of plasma albumin concentration and plasma osmolality, which sequentially reduce GFR via a prerenal mechanism. Increasing evidence suggests that proteinuria alone can cause kidney injury. Hyperlipidemia is one of the major features of ADR nephropathy (6, 19). Increasing evidence suggests that hyperlipidemia and renal lipotoxicity are important pathogenic factors in kidney damage (5, 23) and may contribute to the development and the progression of ADR nephropathy.
Administration of OA-NO2 significantly improved all the symptoms mentioned above as well as renal function. The major mechanism of the protective action of OA-NO2 in this model is likely through the protection of podocytes. Podocytes play a crucial role in the regulation of glomerular function. Injury to podocytes can disrupt the structural and functional integrity of the slit diaphragm, leading to proteinuria. In this model, podocyte injury was an early-occurring event. After 7 days of ADR injection, WT1 protein was markedly downregulated, and OA-NO2 was able to significantly preserve its expression. We also examined the mRNA levels of the epithelial maker ZO-1 and the mesenchymal marker desmin. OA-NO2 reversed the reduction of ZO-1 and increase in desmin. Consistent with these results, histologically OA-NO2 treatment ameliorated ADR-induced glomerulosclerosis, alleviated the accumulation of mesangial matrix, attenuated the prominent tubular dilation, reduced the intraluminal protein casts, and improved the narrow Bowman's capsule. It is somewhat puzzling that on day 7 of OA-NO2 treatment, despite the marked improvement of kidney injury, the attenuation of albuminuria was relatively modest (the P value is still <0.05) compared with the two other early time points. The reason for this phenomenon is unclear. One possibility is that severe hypoabuminemia after 7 days of ADR treatment had reduced the further leakage of albumin from the damaged glomeruli and therefore had masked the therapeutic effect of OA-NO2.
Oxidative stress has emerged as an important pathogenic factor in the development of ADR nephropathy (11, 17, 33–34). In the present study, we showed that pretreatment with OA-NO2 ameliorated albuminuria concomitantly with a reduction of TBARS levels in both urine and plasma. The NADPH oxidase system is a major superoxide-generating system contributing to ROS generation in chronic kidney disease, including ADR nephropathy (11). We found that OA-NO2 significantly attenuated ADR-induced upregulation of NADPH oxidase subunits gp91phox and p47phox at both the mRNA and protein levels; gp91phox and p47phox are of particular importance as the former contains the catalytic domain and the latter is necessary for cytosolic subunit translocation and for initiation of NADPH oxidase assembly in the kidney. These results suggest that the antioxidant property of OA-NO2 via suppressing NADPH oxidase expression may in part account for the renoprotective action of this compound.
Inflammation is an important component of the pathophysiology of ADR nephropathy (7, 22, 26, 33). Histological assessment of the kidneys of ADR-treated mice shows tubulointerstitial inflammation with infiltration of T and B lymphocytes and macrophages. While studies in severe combined immunodeficient (SCID) mice shows that ADR nephropathy does not require lymphocytes, deletion and reconstitution studies demonstrate a pivotal role of macrophages in the disease process (1, 8, 14). Our results revealed excessive renal production of proinflammatory cytokines TNF-α, IL-1β, and MCP-1 in the early stage of ADR nephropathy. Administration of OA-NO2 significantly inhibited the induction of the proinflammatory cytokines. In support of this finding is the observation that in cultured RAW264.7 cells, a murine macrophage cell line, OA-NO2 attenuates the endotoxin-elicited inflammatory response via diverse mechanisms involving activation of mitogen-activated protein kinase phosphatase 1 (13) and nitroalkylation of NF-κB p65 (9, 11). Moreover, our previous study demonstrated anti-inflammatory and renoprotective actions of OA-NO2 in endotoxin-induced endotoxemia in mice (30).
In summary, the present study demonstrates that OA-NO2 protects against ADR nephropathy possibly through the inhibition of oxidative stress and inflammation. These findings suggest the therapeutic potential of OA-NO2 for management of human glomerular disease, such as focal segmental glomerulosclerosis. Our study will provide a basis for the rational design of such clinical trials.
GRANTS
This work was supported by the National Basic Research Program of China 973 Program 2012CB517600 (2012CB517602), a Merit Review from the Department of Veterans Affairs, and Established Investigator Award from the American Heart Association, and National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-079162. T. Yang is a Research Career Scientist in the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.L., Z.J., L.Z., and Y.L. performed experiments; S.L., Z.J., Y.L., H.L., S.-F.Z., G.G., and T.Y. analyzed data; S.L. and L.Z. prepared figures; S.L., Y.D., and T.Y. drafted manuscript; H.L., Y.D., and T.Y. provided conception and design of research; H.L., S.-F.Z., A.Z., Y.D., G.G., and T.Y. interpreted results of experiments; A.Z., Y.D., and T.Y. edited and revised manuscript; T.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Bruce A. Freeman (University of Pittsburgh) for providing OA-NO2.
REFERENCES
- 1.Adeagbo AS, Zhang X, Patel D, Joshua IG, Wang Y, Sun X, Igbo IN, Oriowo MA. Cyclo-oxygenase-2, endothelium and aortic reactivity during deoxycorticosterone acetate salt-induced hypertension. J Hypertens 23: 1025–1036, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Albrecht FE, Xu J, Moe OW, Hopfer U, Simonds WF, Orlowski J, Jose PA. Regulation of NHE3 activity by G protein subunits in renal brush-border membranes. Am J Physiol Regul Integr Comp Physiol 278: R1064–R1073, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem 280: 42464–42475, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker PR, Schopfer FJ, O'Donnell VB, Freeman BA. Convergence of nitric oxide and lipid signaling: anti-inflammatory nitro-fatty acids. Free Radic Biol Med 46: 989–1003, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens 19: 393–402, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boonsanit D, Kanchanapangka S, Buranakarl C. l-Carnitine ameliorates doxorubicin-induced nephrotic syndrome in rats. Nephrology (Carlton) 11: 313–320, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Cao Q, Wang Y, Zheng D, Sun Y, Lee VW, Zheng G, Tan TK, Ince J, Alexander SI, Harris DC. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol 21: 933–942, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng H, Wang S, Jo YI, Hao CM, Zhang M, Fan X, Kennedy C, Breyer MD, Moeckel GW, Harris RC. Overexpression of cyclooxygenase-2 predisposes to podocyte injury. J Am Soc Nephrol 18: 551–559, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem 281: 35686–35698, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fogo AB. Animal models of FSGS: lessons for pathogenesis and treatment. Semin Nephrol 23: 161–171, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Guo J, Ananthakrishnan R, Qu W, Lu Y, Reiniger N, Zeng S, Ma W, Rosario R, Yan SF, Ramasamy R, D'Agati V, Schmidt AM. RAGE mediates podocyte injury in adriamycin-induced glomerulosclerosis. J Am Soc Nephrol 19: 961–972, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He W, Kang YS, Dai C, Liu Y. Blockade of Wnt/beta-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol 22: 90–103, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichikawa T, Zhang J, Chen K, Liu Y, Schopfer FJ, Baker PR, Freeman BA, Chen YE, Cui T. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology 149: 4086–4094, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jania LA, Chandrasekharan S, Backlund MG, Foley NA, Snouwaert J, Wang IM, Clark P, Audoly LP, Koller BH. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E(2) biosynthesis. Prostaglandins Other Lipid Mediat 88: 73–81, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia Z, Wang H, Yang T. Microsomal prostaglandin E synthase 1 deletion retards renal disease progression but exacerbates anemia in mice with renal mass reduction. Hypertension 59: 122–128, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kansanen E, Jyrkkanen HK, Volger OL, Leinonen H, Kivela AM, Hakkinen SK, Woodcock SR, Schopfer FJ, Horrevoets AJ, Yla-Herttuala S, Freeman BA, Levonen AL. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J Biol Chem 284: 33233–33241, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koshikawa M, Mukoyama M, Mori K, Suganami T, Sawai K, Yoshioka T, Nagae T, Yokoi H, Kawachi H, Shimizu F, Sugawara A, Nakao K. Role of p38 mitogen-activated protein kinase activation in podocyte injury and proteinuria in experimental nephrotic syndrome. J Am Soc Nephrol 16: 2690–2701, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Liu H, Jia Z, Soodvilai S, Guan G, Wang MH, Dong Z, Symons JD, Yang T. Nitro-oleic acid protects the mouse kidney from ischemia and reperfusion injury. Am J Physiol Renal Physiol 295: F942–F949, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okuda S, Oh Y, Tsuruda H, Onoyama K, Fujimi S, Fujishima M. Adriamycin-induced nephropathy as a model of chronic progressive glomerular disease. Kidney Int 29: 502–510, 1986 [DOI] [PubMed] [Google Scholar]
- 20.Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol 5: 463–468, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol 296: F213–F229, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Polhill T, Zhang GY, Hu M, Sawyer A, Zhou JJ, Saito M, Webster KE, Wang Y, Grey ST, Sprent J, Harris DC, Alexander SI, Wang YM. IL-2/IL-2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J Am Soc Nephrol 23: 1303–1308, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruan X, Guan Y. Metabolic syndrome and chronic kidney disease. J Diabetes 1: 236–245, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, Baker PR, Ramani R, Freeman BA. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc Res 85: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 69: 2131–2147, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol 20: 593–603, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villacorta L, Zhang J, Garcia-Barrio MT, Chen XL, Freeman BA, Chen YE, Cui T. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol 293: H770–H776, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogtlander NP, Tamboer WP, Bakker MA, Campbell KP, van der Vlag J, Berden JH. Reactive oxygen species deglycosilate glomerular alpha-dystroglycan. Kidney Int 69: 1526–1534, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Liu H, Jia Z, Guan G, Yang T. Effects of endogenous PPAR agonist nitro-oleic acid on metabolic syndrome in obese Zucker rats. PPAR Res 2010: 601562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Liu H, Jia Z, Olsen C, Litwin S, Guan G, Yang T. Nitro-oleic acid protects against endotoxin-induced endotoxemia and multiorgan injury in mice. Am J Physiol Renal Physiol 298: F754–F762, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Pang W, Cui Z, Shi J, Liu Y, Liu B, Zhou Y, Guan Y, Hammock BD, Wang Y, Zhu Y. Upregulation of soluble epoxide hydrolase in proximal tubular cells mediated proteinuria-induced renal damage. Am J Physiol Renal Physiol 304: F168–F176, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Wu H, Wang YM, Wang Y, Hu M, Zhang GY, Knight JF, Harris DC, Alexander SI. Depletion of gammadelta T cells exacerbates murine adriamycin nephropathy. J Am Soc Nephrol 18: 1180–1189, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Zheng S, Epstein PN. Metallothionein over-expression in podocytes reduces adriamycin nephrotoxicity. Free Radic Res 43: 174–182, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Yang W, Wang J, Shi L, Yu L, Qian Y, Liu Y, Wang W, Cheng S. Podocyte injury and overexpression of vascular endothelial growth factor and transforming growth factor-beta 1 in adriamycin-induced nephropathy in rats. Cytokine 59: 370–376, 2012 [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Villacorta L, Chang L, Fan Z, Hamblin M, Zhu T, Chen CS, Cole MP, Schopfer FJ, Deng CX, Garcia-Barrio MT, Feng YH, Freeman BA, Chen YE. Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ Res 107: 540–548, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]