

Abstract
Hearts utilize fatty acids as a primary source of energy. The sources of those lipids include free fatty acids and lipoprotein triglycerides. Deletion of the primary triglyceride-hydrolyzing enzyme lipoprotein lipase (LPL) leads to cardiac dysfunction. Whether heart LPL-knockout (hLPL0) mice are compromised due a deficiency in energetic substrates is unknown. To test whether alternative sources of energy will prevent cardiac dysfunction in hLPL0 mice, two different models were used to supply nonlipid energy. 1) hLPL0 mice were crossed with mice transgenically expressing GLUT1 in cardiomyocytes to increase glucose uptake into the heart; this cross-corrected cardiac dysfunction, reduced cardiac hypertrophy, and increased myocardial ATP. 2) Mice were randomly assigned to a sedentary or training group (swimming) at 3 mo of age, which leads to increased skeletal muscle production of lactate. hLPL0 mice had greater expression of the lactate transporter monocarboxylate transporter-1 (MCT-1) and increased cardiac lactate uptake. Compared with hearts from sedentary hLPL0 mice, hearts from trained hLPL0 mice had adaptive hypertrophy and improved cardiac function. We conclude that defective energy intake and not the reduced uptake of fat-soluble vitamins or cholesterol is responsible for cardiac dysfunction in hLPL0 mice. In addition, our studies suggest that adaptations in cardiac metabolism contribute to the beneficial effects of exercise on the myocardium of patients with heart failure.
Keywords: triglyceride, heart failure, glucose, lactate, energetics, glucose transporters
in the healthy adult human heart, most of the energy required for normal cellular processes is derived from the uptake and oxidation of fatty acids (FAs), with the remainder being contributed by glucose, lactate, and amino acids (36). FAs are delivered to the heart either as free fatty acids (FFAs) conjugated to albumin or via lipoprotein lipase (LPL)-mediated hydrolysis of lipoproteins, with subsequent uptake by FAT/CD36, other FA transporters, and nonreceptor mediated pathways (5).
Cardiomyocyte deletion of LPL (hLPL0) leading to reduced triglyceride (TG) lipolysis increased cardiac glucose uptake and reduced expression of several peroxisomal proliferator-activated receptor (PPAR) regulated genes (1). hLPL0 hearts developed reduced ejection fraction (2) and were unable to compensate when subjected to increased afterload due to aortic coarctation or hypertension (56). These data suggest that TG is an important source of lipid for cardiac energy.
In heart failure, traditional cardiac substrate usage of FAs is perturbed, and as a result the heart shifts away from FA oxidation to glucose utilization. These changes in heart energy use have been studied experimentally in isolated hearts using buffers containing FFAs (35). Whether this change in substrate preference is beneficial is unknown. It is estimated that to produce equivalent amounts of ATP, 10 molecules of glucose need to be oxidized for every molecule of FA (28). But if oxygen is limited, the heart performs 40% more efficiently with glucose (31). Thus, in the setting of ischemia and infarction, the heart's preference for glucose may be beneficial, but in heart failure this shift may be detrimental. In support of this, increased FA delivery to the heart through high-fat diets was found to be beneficial in rats with myocardial infarction and pressure overload-induced cardiac dysfunction (10, 41). In addition, a high-fructose diet rather than high-fat feeding increased mortality in hypertensive rats (42). Similarly, clinical investigators found that the antilipolytic drug acipimox, which reduces FA oxidation and enhances glucose utilization, worsened left ventricular function in patients with idiopathic dilated cardiomyopathy (51) or did not improve function in a more generalized group of heart failure subjects (20). This suggests that reduced lipid uptake and usage in the setting of heart failure may be a maladaptive response.
Treatment of heart failure has focused on pharmacological agents to attenuate ventricular remodeling and decrease cardiac workload. Mechanisms by which nonpharmacological interventions such as exercise bring about improvements in heart failure are not fully understood. Regular exercise can improve cardiac function by enhancing glycolysis and oxidative metabolism (9, 45). This may be attributed to the increase in cardiac mass that occurs with exercise and not changes in cardiac substrate usage (39). Other studies suggest that training improves FA utilization by the heart (8, 49). Whether FAs or glucose are needed for both hypertrophy and improved cardiac function is not known. If reduced FA utilization has deleterious effects on the failing heart, then promoting myocardial FA usage through exercise should help restore cardiac function, whereas the inability to use FAs should prevent any exercise-derived benefits.
To elucidate the importance of TG-derived FA uptake on heart function and whether the uptake of alternative energy substrates can compensate for their loss, we studied heart-specific LPL-knockout (denoted hLPL0) mice in which glucose uptake was augmented by introduction of a cardiomyocyte-specific GLUT1 transgene (MHC-GLUT1). Although this transgene leads to a marked increase in glucose uptake in isolated perfused hearts (>40-fold) (33), its effects on metabolism in vivo had not been determined. In addition, we studied whether training induces adaptive cardiac hypertrophy in hLPL0 mice and whether these animals have a defect in exercise-induced cardiac function. Our studies show that both exercise and the MHC-GLUT1 transgene improve heart function in hLPL0 mice and establish that defective cardiac TG metabolism induces heart dysfunction that can be corrected by the provision of alternative sources of fuel.
METHODS
Experimental animals.
hLPL0 and MHC-GLUT1 mice have been described previously (2, 33). MHC-GLUT1 mice were crossed with hLPL0 and then with Lplflox/flox mice to produce MHC-GLUT1/hLPL0 and littermate controls that were MHC-GLUT1/Lplflox/flox and Lplflox/flox. Hearts from these mice were assessed by echocardiography, gene expression, and lipids, as described below. Female mice were used to assess cardiac function in MHC-GLUT1-TG and GLUT1/hLPL0 mice.
Exercise protocol.
Three-month-old male hLPL0 mice and control littermates were subjected to swim training, as described previously (12). Each day consisted of two swimming sessions separated by at least a 4-h interval. Swimming sessions began with a 10-min duration on the first day. The swim duration was increased by 10 min/day until mice were swimming 90 min twice a day. Mice were trained for 32 additional days (42 days total).
Tissue analysis.
All mice were euthanized after a 5-h fast. Trained mice were euthanized 1 day after their last swimming session. Hearts were harvested, flash-frozen in liquid nitrogen, and stored at −80°C until further use. Heart weight (HW) and tibia length (TL) were also measured so that HW/TL ratios could be calculated.
Echocardiography.
Cardiac function was assessed by echocardiography 1 day prior to the animals being euthanized. Two-dimensional echocardiography was performed using a high-resolution imaging system with a 30-MHz imaging transducer (Vevo 770; VisualSonics) in unconscious mice. The mice were anesthetized with 1.5–2% isoflurane and maintained thereafter on 1–1.5% isoflurane throughout the procedure. Two-dimensional echocardiographic images were obtained and recorded in a digital format and analyzed offline by a researcher blinded to the murine genotype. Left ventricular end-diastolic dimension (LVDd) and left ventricular end-systolic dimension (LVDs) were measured. Percent fractional shortening, which quantifies contraction of the ventricular wall and is an indication of muscle function, was calculated as %FS = ([LVDd − LVDs]/LVDd) × 100.
RNA isolation and gene expression analysis.
Total RNA was extracted from the heart using the phenol guanidine isothiocyanate method (Trizol kit; Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Total RNA (0.7 μg) was reverse transcribed for 1 h at 50°C with the Thermoscript RT-PCR system (Invitrogen, Carlsbad, CA). The reverse-transcribed cDNA was then amplified using an iCycler (Bio-Rad) with the Brilliant SYBR Green QPCR master mix (Stratagene, Palo Alto, CA). Relative quantification of gene expression was performed using the ΔΔ method with 18s rRNA as the housekeeping gene. All primer sequences are listed in Table 1.
Table 1.
List of primers used for quantitative PCR
| Forward | Reverse | |
|---|---|---|
| 18S rRNA | CCATCCAATCGGTAGTAGCG | GTAACCCGTTGAACCCCATT |
| BNP | AGGTTTGCTATCTGGCA | ATGTCGAAGTTTAAGGCTCTGGA |
| ANP | GGAAATGGGATAGAGGTCAGGTG | GTGATTAAACCGGCAAGCAAGGC |
| MCP-1 | GTGACCATTGTGGAATGCT | CTCCGCTTTCTGTTCTTTGG |
| AOX | ATCACGGGCACTTATGC | TCTCACGGATAGGGACA |
| PDK4 | CCGCTGTCCATGAAGCA | GCAGAAAAGCAAAGGACGTT |
| MCAD | TGGCACGTTCTAACCC | ACTGTAGGTCTGGTTCTAT |
| PPARα | ACTACGGAGTTCACGCATGTG | TTGTCGTACACCAGCTTCAGC |
| CD36 | AAACGACTGCAGGTCA | TGTAGGCTCATCCACTAC |
| CPT-1 | TCTAGGCAATGCCGTTCAC | GAGCACATGGCACCATAC |
| DGAT1 | GAGTCTATCACTCCAGTGGG | GGCGGCACCACAGGTTGACA |
| DGAT2 | GCGCTACTTCCGAGAC | CCACGATGATGATAGCAT |
BNP, brain naturietic protein; ANF, atrial natriuretic protein; MCP-1, monocyte chemotactic protein-1; AOX, acyl-CoA oxidase; PDK4, pyruvate dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; PPARα, peroxisome proliferator-activated receptor-α; CPT-1, carnitine palmitoyltransferase; DGAT1 and -2, diaclyglycerol acyltransferase 1 and 2, respectively.
Heart lipid measurement.
To measure tissue lipids, hearts were perfused with PBS and homogenized in at 4°C in 1 M NaCl buffer containing lipase inhibitors to prevent TG hydrolysis. Lipids were extracted from heart tissues (50 mg) according to methods modified from that of Folch et al. (14). Heart TG and FFA were measured enzymatically using an Infinity kit (Thermo Fisher, Middletown, VA) and a NEFA kit (Wako).
Cardiac ATP measurement.
Frozen heart tissues from 5-h-fasted mice were used to measure ATP. Briefly, the hearts were homogenized with 0.1% trichloroacetic acid and centrifuged at 4°C. Subsequently, the supernatant was isolated and 50 mM TAE containing 2 mM EDTA (pH 7.8) added. Ten microliters of supernatant was used to measure ATP. An ATP bioluminescent assay kit (Invitrogen, Carlsbad, CA) was used for ATP measurement.
Cardiac lactate measurement.
Fresh heart tissues obtained after a 5-h fast were homogenized in lactate assay buffer (Sigma-Aldrich, St.Louis, MO), and the samples were centrifuged to remove insoluble material. Then samples were deproteinized with a 10-kDa MWCO spin filter to remove lactate dehydrogenase, and the soluble fraction was assayed.
In vivo 2-deoxy-d-[1-3H]glucose and [14C]VLDL-TG uptake assessment.
Basal glucose uptake was measured following an intravenous administration of 3 μCi of 2-deoxy-d-[1-3H]glucose (Perkin-Elmer, Waltham, MA) via tail vein in 5-h-fasted mice. Blood was collected 2, 15, 30, 45, and 60 min following injection. In some experiments, [14C]VLDL-TG was injected 45 min after labeled glucose was injected and blood collected at 45 and 60 min. At 60 min, tissues were excised after the body cavity was perfused with 10 ml of PBS by cardiac puncture. Radioactivity in the tissues was measured in a scintillation counter, and tissue uptake was normalized to 2-min plasma counts for 2-deoxy-d-[1-3H]glucose and 30 s for [14C]VLDL-TG.
In vivo [14C]lactate and [3H]palmitate uptake.
[14C]lactate stock (Perkin-Elmer) was diluted with sterile saline such that each mouse received 105 counts intravenously. Following injection, blood was collected at 2, 10, 25, and 30 min. At 30 min, mice were perfused with PBS. [3H]palmitate was complexed with 6% BSA by warming at 40°C such that each mouse was injected with ∼106 counts intravenously. The labeled palmitate-BSA complex was injected at 25 min after labeled lactate injection and blood samples were collected at 30 s, 2 min, and 5 min postinjection.
At the conclusion of the kinetic studies, tissues were harvested, flash-frozen in liquid nitrogen, and stored at −80°C until further use. Radioactivity was determined in 10 μl of plasma and 200 μl of tissue homogenate on an LS 6500 multipurpose scintillation counter (Beckman-Coulter).
Statistical analysis.
All data are presented as means ± SD or means ± SE as indicated. Comparisons between two groups were performed using two-way ANOVA with Prism version 5.0. P value <0.05 was used to determine statistical significance.
RESULTS
MHC-GLUT1 prevents heart dysfunction.
At 5 mo of age, hLPL0 mice had systolic dysfunction with a reduction in fractional shortening compared with age-matched controls (Fig. 1A). This was associated with increased left ventricular end-diastolic and end-systolic diameter (Fig. 1, B and C). MHC-GLUT1 mice had normal echocardiographic parameters. Remarkably, MHC-GLUT1/hLPL0 mice had improved cardiac function that was not statistically different from control (echocardiographic images are shown in Fig. 1D). Thus, the dilated cardiomyopathy in hLPL0 mice was corrected by the MHC-GLUT1 transgene.
Fig. 1.

Cardiac overexpression of glucose transporter 1 (GLUT1) improved cardiac function. A–C: echocardiography showed increased fractional shortening (FS; %), reduced left ventricular end-diastolic dimension (LVDd), and left ventricular end-systolic diameter (LVDs) in GLUT1/hLPL0 mice compared with heart lipoprotein lipase (LPL)-knockout (hLPL0) mice. D: representative M-mode echocardiographic images in 5-mo-old female mice (n = 8–12). Data are shown as means ± SD. #P < 0.05 vs. control mice; *P < 0.05 vs. hLPL0 mice.
Gene expression in MHC-GLUT1/hLPL0 mice.
We assessed mRNA levels of genes involved in lipid and glucose metabolism in MHC-GLUT1 mice and how these mRNA levels were altered on the hLPL0 background. At this age (5 mo), the mice have early heart failure. As we had noted previously (1), hLPL0 mice had a reduction in PPARα mRNA levels, but differences in other mRNA levels were not significant, except for that of diaclyglycerol acyltransferase 2 (DGAT2; Fig. 2). The MHC-GLUT1 transgene on both the wild-type and hLPL0 backgrounds led to major changes in mRNA levels. PPARα remained reduced compared with control. Pyruvate dehydrogenase kinase-4, an indicator of glucose oxidation, was increased in both MHC-GLUT1 and MHC-GLUT1/hLPL0 hearts. There was a trend for reduced mRNA levels of several FA oxidation genes such as medium-chain acyl-CoA dehydrogenase, CD36, and acyl-CoA oxidase, but carnitine palmitoyltransferase-1βwas not altered.
Fig. 2.

Heart tissue mRNA expression. Quantitative reverse transcription-PCR analysis of mRNA expression using gene-specific primers. Data were normalized to 18S rRNA. Values represent fold change relative to control, which was set as 1; male mice, n = 5–7. Data are shown as means ± SD. #P < 0.05 and *P < 0.01 compared with control. PPARα, peroxisome proliferator-activated receptor-α; DGAT1 and -2, diaclyglycerol acyltransferase 1 and 2, respectively; PDK4, pyruvate dehydrogenase kinase-4; MCAD, medium-chain acyl-CoA dehydrogenase; AOX, acyl-CoA oxidase; CPT-1β, carnitine palmitoyltransferase-1β.
Lipid analysis.
DGAT1 and -2 were reduced by the MHC-GLUT1 transgene; this would suggest that these hearts have reduced TG synthesis from FFA. However, even on chow, MHC-GLUT1 mice had an increase in heart TG and FFA content (Fig. 3, A and B). It is possible that compensatory expression of other genes involved in TG synthesis or posttranslation modifications to DGAT1 and -2 may have occurred. Previous studies had shown that MHC-GLUT1 mice on high-fat diets develop heart failure associated with an increase in heart TG (57).
Fig. 3.

Heart lipid measurements. A: cardiac triglyceride (TG). B: cardiac free fatty acids (FFA). *P < 0.05 vs. littermate control mice; male mice, n = 5–9.
MHC-GLUT1/hLPL0 hearts have increased ATP.
We postulated that greater glucose uptake improved heart function by increasing energy production. Although hLPL0 mice had a reduced level of ATP after acute afterload (5) under nonstressed conditions, ATP levels were not reduced. Surprisingly, ATP was increased in MHC-GLUT1 hearts and also in the MHC-GLUT1/hLPL0 hearts (Fig. 4A). We expect that this improvement in energy stores is the reason that MHC-GLUT1 mice have improved function in the setting of ischemia-reperfusion (33). In addition, the ability to produce more ATP from glucose is likely to have restored heart function in the MHC-GLUT1/hLPL0 hearts.
Fig. 4.

Cardiac ATP, glucose uptake, and lactate content. Cardiac ATP (A), lactate (B), and 2-deoxy-d-[3H]glucose uptake (C) of the mice. Data are showed as means ± SD. #P < 0.05 and §P < 0.01 vs. control mice, *P < 0.05 vs. hLPL0 mice; male mice, n = 5–7. DPM, disintegrations/min.
We assessed directly the reasons for greater ATP production by assessing glucose uptake. As noted previously, glucose uptake is increased in hLPL0 hearts; in this group of mice that increase was approximately threefold (Fig. 4B). MHC-GLUT1 increased 2-deoxyglucose (2-DG) uptake ∼4.2-fold and led to a significant increase above that in the hLPL0 mice (Fig. 4B). Lactate levels in the heart were increased in hLPL0 hearts (see below) but not with MHC-GLUT1 expression (Fig. 4C).
Exercise training improves cardiac function in mice with defective cardiac FA uptake.
Exercise-mediated improvements in cardiac function are associated with alterations in energy substrate utilization. To investigate the effect of exercise training on the cardiac structure and function of hLPL0 mice, a 6-wk swim training protocol was begun at 3 mo of age. Both control and hLPL0 mice had greater HW/TL ratios, which was consistent with the development of exercise-induced cardiac hypertrophy (Fig. 5A). Trained hLPL0 mice demonstrated significantly greater fractional shortening compared with their sedentary counterparts and led to a fractional shortening percentage identical to that seen in controls (Fig. 5B).There was no difference in cardiac contractility between trained hLPL0 and control mice, reflecting normalization of cardiac function (Fig. 5C).
Fig. 5.
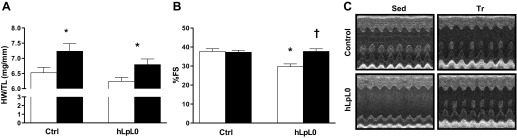
Effects of training on cardiac hypertrophy and function in 5-mo-old control (ctrl) and hLPL0 mice. Mice were randomly assigned to either a sedentary or training group that underwent swim training for 6 wk. Cardiac hypertrophy was determined by heart weight/tiba length ratio (HW/TL), whereas cardiac function was measured by 2-dimensional echocardiography. A: HW/TL ratio. B: fractional shortening (%). C: representative M-mode echocardiographic images. Open bars, sedentary mice; black bars, trained mice. Data are means ± SE. *P < 0.05 vs. controls, †P < 0.05 vs. sedentary mice; male mice, n = 8–18/group for echocardiographic analysis.
Exercise training does not further increase cardiac glucose uptake in hLPL0 mice.
We speculated that the improvements in cardiac function observed in trained hLPL0 mice might be due to an increase in the myocardial uptake of an alternate substrate. Since the hearts of these mice use more glucose than controls, we theorized that the improvements in cardiac function were due to the increased uptake of glucose by trained hearts, a process similar to that in MHC-GLUT1/hLPL0 mice. Although hLPL0 mice had increased cardiac 2-DG uptake compared with controls, exercise training did not increase glucose further (Fig. 6A). Plasma glucose did not significantly change immediately after exercise in any of the mice (Table 2).
Fig. 6.
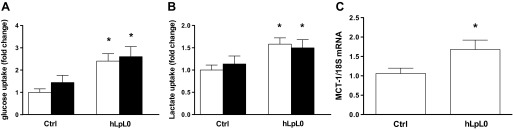
Cardiac glucose uptake, lactate uptake, and MCT-1 gene expression. A: control and hLPL0 mice fasted for 5 h; cardiac 2-deoxy-d-[1-3H]glucose uptake was determined by measuring cardiac radioactivity and normalized to 2-min plasma counts. B: [14C]lactate in mice was administered via tail vein injection and euthanized 60 min postinjection; radioactivity in the plasma was measured and normalized to 2-min plasma counts for [14C]lactate plasma clearance. C: gene expression of MCT-1, the primary cardiac lactate transporter, was analyzed by real-time PCR and normalized to 18S expression. Open bars, sedentary mice; black bars, trained mice. Data are means ± SE. *P < 0.05 vs. ctrl; male mice, n = 7–13/group.
Table 2.
Plasma lipids and glucose at rest and immediately after exercise
| Control |
hLPL0 |
|||
|---|---|---|---|---|
| Sed | Ex | Sed | Ex | |
| FFA, mEq | 1.38 ± 0.06 | 1.56 ± 0.15† | 1.27 ± 0.06 | 1.87 ± 0.22† |
| TG, mmol/l | 0.73 ± 0.17 | 0.75 ± 0.20 | 1.60 ± 0.35* | 1.65 ± 0.13 |
| Glucose, mmol/l | 1.58 ± 0.09 | 1.72 ± 0.07 | 1.44 ± 0.06 | 1.65 ± 0.09 |
Data are means ± SE. hLPL0, heart lipoprotein lipase knockout; Sed, sedentary group; Ex, exercise group; FFA, free fatty acids; TG, triglycerides.
P < 0.05 vs. control sed,
P < 0.05 vs. hLPL0 sed; male mice, n = 5–11/group.
Lactate uptake is increased in the hearts of hLPL0 mice.
Lactate becomes an important fuel for cardiac energy metabolism during exercise (24, 46) when plasma lactate concentrations rise. Using radiolabeled [14C]lactate, we specifically assessed myocardial lactate uptake. Plasma lactate levels and [14C]lactate clearance curves were not different between groups. Similar to monocarboxylate transporter-1 (MCT-1) mRNA expression, sedentary hLPL0 mice demonstrated an increase in radiolabeled lactate uptake compared with controls (Fig. 6B). Training did not further increase [14C]lactate uptake. MCT-1, the major lactate transporter in the heart, increases with training and heart failure (2). MCT-1 mRNA expression was significantly increased in sedentary hLPL0 hearts (Fig. 6C). Thus, hLPL0 hearts appeared to be programmed for greater uptake of lactate. Taken together, these data suggest that the increased capacity of hLPL0 hearts to consume lactate should benefit cardiac function most during exercise, when levels of plasma lactate rise drastically.
FFA uptake is not increased in hLPL0 mice.
Since exercise via peripheral lipolysis generates FFAs that can fuel the heart, we assessed the effects of acute exercise on plasma FFA and TG levels (Table 2). HLPL0 had higher circulating TG, with no differences in circulating FFA levels. Acute exercise increased plasma FFA in controls and in hLPL0 mice, whereas plasma TG levels did not change. We then measured FFA and VLDL-derived TG uptake in controls and hLPL0 hearts. No differences in FFA or VLDL plasma tracer clearance were noted between groups. [3H]palmitate uptake in hLPL0 mice was twofold greater than in controls (Fig. 7A), and as expected, cardiac VLDL-TG uptake was reduced in hLPL0 mice (Fig. 7B). Endurance training increased palmitate uptake and reduced VLDL-derived TG uptake in controls but did not affect the uptake of either substrate in hLPL0 hearts.
Fig. 7.

Cardiac lipid uptake. A: control and hLPL0 mice, fasted for 5 h, were administered [3H]palmitate via femoral vein injection and euthanized 5 min postinjection. Radioactivity in the plasma was measured in a scintillation counter and normalized to 2-min plasma counts. B: in a separate group of mice, [14C]VLDL-TG was injected via femoral vein. Radioactivity was measured in the heart and normalized to 30-s plasma counts. Open bars, sedentary mice; black bars, trained mice. Data are means ± SE. *P < 0.05 vs. ctrl, †P < 0.05 vs. sedentary hLPL0 mice; male mice, n = 7–13/group.
DISCUSSION
The heart is a metabolic omnivore that is capable of responding to physiological and pathological stresses by modulating its intake of various energy substrates. At rest, the adult heart relies primarily on FA derived from either circulating albumin-bound FFA or lipoproteins. In contrast, ischemia shifts myocardial preference toward the more energetically efficient glucose. Whether this metabolic shift is beneficial in heart failure is unclear. Data from isolated perfused hearts that allow the quantification of FA and glucose oxidation by conversion of labeled substrates into CO2 suggest that glucose utilization may be beneficial (34). These studies are limited by nonphysiological levels of FA, albumin, and/or insulin in the perfusate, which likely affected the heart's preference for its fuel. In addition, the use of heparin to prevent the harvested heart from clotting has the unintended consequence of displacing LPL from its physiological location on the luminal endothelial surface (19). Although attempts have been made to make perfusate buffers physiological, the concentrations of circulating factors often vary between laboratories and likely affect cardiac function and substrate metabolism.
Previously, we created mice with a cardiomyocyte-specific knockout of LPL, leading to a defect in the myocardial utilization of TG-derived FAs and a compensatory reliance on glucose, a metabolic phenotype that mimics many forms of human heart failure. These mice, in response to age and increased afterload (2, 5, 56), also develop cardiac dysfunction. Aside from lipid uptake, hLPL0 hearts also have defects in the uptake of cholesterol and fat-soluble vitamins (5) such as vitamin A, whose deficiency has been implicated as a cause of heart failure. In this study, we demonstrate that the provision of alternative substrates, either by increasing glucose uptake or through the exercise-induced uptake of FFA and lactate, prevents the onset of heart failure in hLPL0 mice.
Overexpression of GLUT1 in cardiomyocytes led to a 4.2-fold increase in glucose uptake as assessed by the uptake of tracer 2-DG. This increase was greater than that found in hLPL0 hearts (2). By crossing the MHC-GLUT1 transgene onto the hLPL0 background, glucose uptake was increased, an amount that was sufficient to compensate for the defect in lipid uptake and to increase heart content of ATP. Although we assessed cardiac function only in female GLUT1/hLPL0 mice, we expect a similar preservation of cardiac function in males since increased cardiac glucose uptake was seen in male MHC-GLUT1 transgenic mice. It should be noted that MHC-GLUT1 transgenic hearts showed a 42-fold increase in glucose uptake when studied in isolated heart preparations (33). Therefore, these studies further illustrate the marked differences in heart metabolism seen when comparing in vivo and isolated heart measurements. Furthermore, because most glucose accumulated by the heart is used for oxidation, this discrepancy in uptake also illustrates the difficulty of correlating glucose oxidation data with in vivo metabolism.
Exercise training is beneficial in patients with heart failure (40) and improves exercise tolerance (27, 37), quality of life (16, 43), and cardiac structure (4, 46). Training-induced benefits in heart failure have been attributed predominantly to adaptations in the peripheral circulation and skeletal muscles (52, 53), whereas improvements in ventricular function have largely been ascribed to reverse remodeling secondary to afterload reduction and improved endothelial function (21, 22). Utilization of various energy substrates by the heart may be altered by exercise training and could directly improve heart function. Acute exercise has been shown to increase cardiac utilization of glucose and lactate (43a), with an increase in FFA utilization as exercise duration progresses (17). The effects of chronic exercise on substrate utilization patterns may differ. Responses to chronic training have been variable. Some studies have demonstrated increased FA utilization (7, 15, 38), whereas others showed either no change or an increase in carbohydrate uptake (30). These discrepancies likely reflect differences in species and the type of training protocol used.
The effect of exercise training on cardiac carbohydrate and FA metabolism in heart failure is largely unknown. One study in patients with idiopathic dilated cardiomyopathy demonstrated an increase in cardiac insulin-stimulated glucose uptake after training (44). There are no similar metabolic studies in other forms of human heart failure. Myocardial FA utilization had been reported to increase with training in rodents (49). Iemitsu et al. (26) demonstrated that regular exercise in aging rats prevented age-induced decreases in PPARα expression and subsequent age-induced heart dysfunction. Similarly, exercise training improved cardiac function in rabbits with myocardial infarction-induced heart failure; this was associated with greater FA utilization (8). Therefore, changes in the amount or type of substrates for cardiac energy production might prevent or improve heart failure.
Because we knew that hLPL0 mice had cardiac dysfunction and an increased but limited capacity to take up glucose under pathological stress (56), we speculated that exercise training would act as a physiological stressor on these hearts, leading to more cardiac dysfunction. To our surprise, left ventricular function in hLPL0 mice did not worsen after 6 wk of swim training but actually improved. Our studies with a model of heart failure due specifically to a genetic loss of lipid uptake by the heart are consistent with other studies in both animal models of heart failure (11, 32) and heart failure patients that found exercise-induced improvements in cardiac ejection fraction (18, 24, 55).
To explain the improvement in cardiac function in hLPL0 hearts, we explored changes in several energy substrates. We initially thought that training had increased glucose uptake, but our kinetic studies of glucose uptake did not support this. For this reason, we assessed the uptake of another cardiac energy substrate, lactate, whose utilization increases during exercise and heart failure (13, 23, 29, 43a). Cardiac lactate uptake occurs via a carrier-mediated system involving the lactate transporter MCT-1 (3, 50, 58), with uptake being proportional to arterial concentration and all lactate acquired by the heart used for oxidation (48). Since plasma lactate levels are highest during active exercise, this increased capacity to utilize lactate should be most advantageous during exercise.
Consistent with these studies, we found that hLPL0 hearts had an increased capacity to take up radiolabeled lactate. We also found increased cardiac mRNA expression of MCT-1. However, we did not find an increase in radiolabeled lactate uptake with training in control or hLPL0 hearts. Thus, it may be that levels of circulating lactate and not the transporters are rate-limiting in the cardiac acquisition of lactate. Moreover, because lactate utilization by the heart competes with FA oxidation (6), lactate use during exercise may mask any metabolic deficiencies in resting hLPL0 mice.
During exercise, peripheral lipolysis in adipose tissue raises plasma FFA levels, which can be utilized for energy by the heart. Since hLPL0 hearts have a defect in VLDL-derived FA and not FFA uptake, we also wondered whether these hearts had an increase in FFA uptake. hLPL0 hearts at baseline have an increase in radiolabeled palmitate uptake compared with controls; to some extent, this is likely due to a lack of competition between the tracer and unlabeled FFA generated by LPL-mediated hydrolysis of lipoprotein TG. Six weeks of training increased the uptake of labeled FFA in control hearts but not in hLPL0 hearts. Since hLPL0 hearts already have increased FFA uptake at baseline, the higher plasma FFA levels generated during exercise likely lead to greater cardiac uptake of this substrate. Taken together, we believe that in addition to other exercise-induced benefits on the failing heart such as improved endothelial function and oxidative stress, the uptake of energy substrates such as lactate and FFA can augment the function of the failing heart.
In conclusion, we describe two methods to compensate for reduced lipid uptake into the heart. hLPL0 hearts replicate the reduced FA uptake that is thought to be the “metabolic switch” seen in heart failure. In hLPL0 mice, this defect appears to cause energetic deficiency that leads to heart failure and can be ameliorated by increased uptake of glucose. Our findings also indicate that exercise is able to take advantage of the heart's increased capacity for lactate consumption by generating lactate peripherally, which then serves as an alternate energy fuel. Therefore, exercise training provides hLPL0 hearts with regular boluses of lactate and FFA, which serve to fuel the heart. Although controversial because of the greater oxygen requirements for lipid oxidation compared with glucose use, our data suggest that methods to prevent defective FA use might be a preventive strategy. However, once heart failure and reduced FA oxidation develop, another method to improve function may be to encourage the cardiac utilization of alterative substrates.
GRANTS
This work was supposrted by National Heart, Lung, and Blood Institute Grant HL-073029 (I. J. Goldberg). R. S. Khan was supported by postdoctoral training grant HL-007343. Y. Lin was supported by grant from the National Natural Science Foundation of China (81100170).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.S.K., P.C.S., R.T., and I.J.G. contributed to the conception and design of the research; R.S.K., Y.L., Y.H., N.-H.S., K.G.B., C.P., A.C., R.J., and S.Y. performed the experiments; R.S.K., Y.L., R.J., S.H., and I.J.G. analyzed the data; R.S.K., S.H., R.T., and I.J.G. interpreted the results of the experiments; R.S.K. and Y.L. prepared the figures; R.S.K. drafted the manuscript; R.S.K., P.C.S., R.T., and I.J.G. edited and revised the manuscript; R.S.K., Y.H., N.-H.S., C.P., A.C., R.J., S.Y., S.H., P.C.S., R.T., and I.J.G. approved the final version of the manuscript.
REFERENCES
- 1.Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R, Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem 279: 25050–25057, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Augustus AS, Buchanan J, Park TS, Hirata K, Noh HL, Sun J, Homma S, D'Armiento J, Abel ED, Goldberg IJ. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J Biol Chem 281: 8716–8723, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Baker SK, McCullagh KJ, Bonen A. Training intensity-dependent and tissue-specific increases in lactate uptake and MCT-1 in heart and muscle. J Appl Physiol 84: 987–994, 1998 [DOI] [PubMed] [Google Scholar]
- 4.Belardinelli RG, Cianci G, Berman N, Ginzton L, Purcaro A. Exercise training improves left ventricular diastolic filling in patients with dilated cardiomyopathy: clinical and prognostic implications. Circulation 91: 2775–2784, 1995 [DOI] [PubMed] [Google Scholar]
- 5.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS, Goldberg IJ. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem 285: 37976–37986, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bielefeld DR, Vary TC, Neely JR. Inhibition of carnitine palmitoyl-CoA transferase activity and fatty acid oxidation by lactate and oxfenicine in cardiac muscle. J Mol Cell Cardiol 17: 619–625, 1985 [DOI] [PubMed] [Google Scholar]
- 7.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, Allard MF. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol 287: H1055–H1063, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Chen CY, Hsu HC, Lee BC, Lin HJ, Chen YH, Huang HC, Ho YL, Chen MF. Exercise training improves cardiac function in infarcted rabbits: involvement of autophagic function and fatty acid utilization. Eur J Heart Fail 12: 323–330, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Coleman R, Weiss A, Finkelbrand S, Silbermann M. Age and exercise-related changes in myocardial mitochondria in mice. Acta Histochem 83: 81–90, 1988 [DOI] [PubMed] [Google Scholar]
- 10.Duda MK, O'Shea KM, Lei B, Barrows BR, Azimzadeh AM, McElfresh TE, Hoit BD, Kop WJ, Stanley WC. Low-carbohydrate/high-fat diet attenuates pressure overload-induced ventricular remodeling and dysfunction. J Card Fail 14: 327–335, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emter CA, Baines CP. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am J Physiol Heart Circ Physiol 299: H1348–H1356, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evangelista FS, Brum PC, Krieger JE. Duration-controlled swimming exercise training induces cardiac hypertrophy in mice. Braz J Med Biol Res 36: 1751–1759, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Evans RK, Schwartz DD, Gladden LB. Effect of myocardial volume overload and heart failure on lactate transport into isolated cardiac myocytes. J Appl Physiol 94: 1169–1176, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509, 1957 [PubMed] [Google Scholar]
- 15.Fröberg SO. Effects of training and of acute exercise in trained rats. Metabolism 20: 1044–1051, 1971 [DOI] [PubMed] [Google Scholar]
- 16.Gary RA, Sueta CA, Dougherty M, Rosenberg B, Cheek D, Preisser J, Neelon V, McMurray R. Home-based exercise improves functional performance and quality of life in women with diastolic heart failure. Heart Lung 33: 210–218, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest 82: 2017–2025, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giannuzzi P, Temporelli PL, Corrà U, Tavazzi L; ELVD-CHF Study Group Antiremodeling effect of long-term exercise training in patients with stable chronic heart failure: results of the Exercise in Left Ventricular Dysfunction and Chronic Heart Failure (ELVD-CHF) Trial. Circulation 108: 554–559, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res 50 Suppl: S86–S90, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halbirk M, Nørrelund H, Møller N, Schmitz O, Gøtzsche L, Nielsen R, Nielsen-Kudsk JE, Nielsen SS, Nielsen TT, Eiskjær H, Bøtker HE, Wiggers H. Suppression of circulating free fatty acids with acipimox in chronic heart failure patients changes whole body metabolism but does not affect cardiac function. Am J Physiol Heart Circ Physiol 299: H1220–H1225, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Hambrecht R, Fiehn E, Weigl C, Gielen S, Hamann C, Kaiser R, Yu J, Adams V, Niebauer J, Schuler G. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation 98: 2709–2715, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Hambrecht R, Hilbrich L, Erbs S, Gielen S, Fiehn E, Schoene N, Schuler G. Correction of endothelial dysfunction in chronic heart failure: additional effects of exercise training and oral l-arginine supplementation. J Am Coll Cardiol 35: 706–713, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto T, Kambara N, Nohara R, Yazawa M, Taguchi S. Expression of MHC-β and MCT1 in cardiac muscle after exercise training in myocardial-infarcted rats. J Appl Physiol 97: 843–851, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Haykowsky MJ, Liang Y, Pechter D, Jones LW, McAlister FA, Clark AM. A meta-analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: the benefit depends on the type of training performed. J Am Coll Cardiol 49: 2329–2336, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Iemitsu M, Miyauchi T, Maeda S, Tanabe T, Takanashi M, Irukayama-Tomobe Y, Sakai S, Ohmori H, Matsuda M, Yamaguchi I. Aging-induced decrease in the PPAR-α level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol 283: H1750–H1760, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Jetté M, Heller R, Landry F, Blümchen G. Randomized 4-week exercise program in patients with impaired left ventricular function. Circulation 84: 1561–1567, 1991 [DOI] [PubMed] [Google Scholar]
- 28.Ingwall JS. On substrate selection for ATP synthesis in the failing human myocardium. Am J Physiol Heart Circ Physiol 293: H3225–H3226, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Kaijser L, Berglund B. Myocardial lactate extraction and release at rest and during heavy exercise in healthy men. Acta Physiol Scand 144: 39–45, 1992 [DOI] [PubMed] [Google Scholar]
- 30.Kainulainen H, Komulainen J, Takala T, Vihko V. Effect of chronic exercise on glucose uptake and activities of glycolytic enzymes measured regionally in rat heart. Basic Res Cardiol 84: 174–190, 1989 [DOI] [PubMed] [Google Scholar]
- 31.Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol 278: H1345–H1351, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Lachance D, Plante E, Bouchard-Thomassin AA, Champetier S, Roussel E, Drolet MC, Arsenault M, Couet J. Moderate exercise training improves survival and ventricular remodeling in an animal model of left ventricular volume overload. Circ Heart Fail 2: 437–445, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 106: 2125–2131, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schönekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta 1213: 263–276, 1994 [DOI] [PubMed] [Google Scholar]
- 35.Lopaschuk GD, Saddik M, Barr R, Huang L, Barker CC, Muzyka RA. Effects of high levels of fatty acids on functional recovery of ischemic hearts from diabetic rats. Am J Physiol Endocrinol Metab 263: E1046–E1053, 1992 [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Maiorana A, O'Driscoll G, Cheetham C, Collis J, Goodman C, Rankin S, Taylor R, Green D. Combined aerobic and resistance exercise training improves functional capacity and strength in CHF. J Appl Physiol 88: 1565–1570, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Moreau D, Guilland JC, Athias P, Dumas JP, Klepping J, Didier JP. [Utilization of free fatty acids and triglycerides by the perfused isolated rat heart after a prolonged swimming training program]. C R Seances Soc Biol Fil 172: 465–469, 1978 [PubMed] [Google Scholar]
- 39.Murakami T, Shimomura Y, Fujitsuka N, Sugiyama S. Differential adaptation to endurance training between heart and gastrocnemius muscle mitochondria in rats. Biochem Mol Biol Int 36: 285–290, 1995 [PubMed] [Google Scholar]
- 40.Piña IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, Duscha BD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ; American Heart Association Committee on exercise, rehabilitation, and prevention Exercise and heart failure: A statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation 107: 1210–1225, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Rennison JH, McElfresh TA, Okere IC, Vazquez EJ, Patel HV, Foster AB, Patel KK, Chen Q, Hoit BD, Tserng KY, Hassan MO, Hoppel CL, Chandler MP. High-fat diet postinfarction enhances mitochondrial function and does not exacerbate left ventricular dysfunction. Am J Physiol Heart Circ Physiol 292: H1498–H1506, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Sharma N, Okere IC, Duda MK, Johnson J, Yuan CL, Chandler MP, Ernsberger P, Hoit BD, Stanley WC. High fructose diet increases mortality in hypertensive rats compared to a complex carbohydrate or high fat diet. Am J Hypertens 20: 403–409, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Smart N, Haluska B, Jeffriess L, Marwick TH. Exercise training in systolic and diastolic dysfunction: effects on cardiac function, functional capacity, and quality of life. Am Heart J 153: 530–536, 2007 [DOI] [PubMed] [Google Scholar]
- 43a.Stanley WC. Myocardial lactate metabolism during exercise. Med Sci Sports Exerc 23: 920–924, 1991 [PubMed] [Google Scholar]
- 44.Stolen KQ, Kemppainen J, Kalliokoski KK, Luotolahti M, Viljanen T, Nuutila P, Knuuti J. Exercise training improves insulin-stimulated myocardial glucose uptake in patients with dilated cardiomyopathy. J Nucl Cardiol 10: 447–455, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Stuewe SR, Gwirtz PA, Agarwal N, Mallet RT. Exercise training enhances glycolytic and oxidative enzymes in canine ventricular myocardium. J Mol Cell Cardiol 32: 903–913, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Sullivan MJ, Higginbotham MB, Cobb FR. Exercise training in patients with severe left ventricular dysfunction. Hemodynamic and metabolic effects. Circulation 78: 506–515, 1988 [DOI] [PubMed] [Google Scholar]
- 48.Taegtmeyer H. Tracing cardiac metabolism in vivo: one substrate at a time. J Nucl Med 51: 80S–87S, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terblanche SE, Gohil K, Packer L, Henderson S, Brooks GA. The effects of endurance training and exhaustive exercise on mitochondrial enzymes in tissues of the rat. Comp Biochem Physiol A Mol Integr Physiol 128: 889–896, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Thomas C, Bishop D, Moore-Morris T, Mercier J. Effects of high-intensity training on MCT1, MCT4, and NBC expressions in rat skeletal muscles: influence of chronic metabolic alkalosis. Am J Physiol Endocrinol Metab 293: E916–E922, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Tuunanen H, Engblom E, Naum A, Någren K, Hesse B, Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, Knuuti J. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114: 2130–2137, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Tyni-Lenné R, Gordon A, Europe E, Jansson E, Sylvén C. Exercise-based rehabilitation improves skeletal muscle capacity, exercise tolerance, and quality of life in both women and men with chronic heart failure. J Card Fail 4: 9–17, 1998 [DOI] [PubMed] [Google Scholar]
- 53.Tyni-Lenné R, Gordon A, Jansson E, Bermann G, Sylvén C. Skeletal muscle endurance training improves peripheral oxidative capacity, exercise tolerance, and health-related quality of life in women with chronic congestive heart failure secondary to either ischemic cardiomyopathy or idiopathic dilated cardiomyopathy. Am J Cardiol 80: 1025–1029, 1997 [DOI] [PubMed] [Google Scholar]
- 55.Wisløff U, Støylen A, Loennechen JP, Bruvold M, Rognmo Ø, Haram PM, Tjønna AE, Helgerud J, Slørdahl SA, Lee SJ, Videm V, Bye A, Smith GL, Najjar SM, Ellingsen Ø, Skjaerpe T. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation 115: 3086–3094, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Yamashita H, Bharadwaj KG, Ikeda S, Park TS, Goldberg IJ. Cardiac metabolic compensation to hypertension requires lipoprotein lipase. Am J Physiol Endocrinol Metab 295: E705–E713, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zonderland ML, Bär PR, Reijneveld JC, Spruijt BM, Keizer HA, Glatz JF. Different metabolic adaptation of heart and skeletal muscles to moderate-intensity treadmill training in the rat. Eur J Appl Physiol Occup Physiol 79: 391–396, 1999 [DOI] [PubMed] [Google Scholar]