

Abstract
The transcription factor NF-κB p65 is a key regulator in the regulation of an inflammatory response and in the pathology of atherosclerosis. However, there is no direct evidence for the role of NF-κB in macrophages in the development of atherosclerosis. We investigated whether macrophage overexpression of p65 in apoE-knockout mice could improve atherosclerosis. Transgenic (Tg) mice overexpressing p65 in macrophages were generated by crossing fatty acid-binding protein 4 (aP2) promoter-controlled p65 mice with apoE-knockout (KO) mice. Tg mice exhibited functional activation of NF-κB signaling in macrophages and fat tissues. We observed that the atherosclerotic lesion was 40% less in the Tg mice compared with the apoE-KO controls fed a standard atherogenic diet for 16 wk (n = 12). The Tg mice were leaner from reduced fat mass by increased energy expenditure. Moreover, the overexpression of p65 in macrophages suppressed foam cell formation. Our results show that there is 1) an increased fatty acid oxidation in macrophages, 2) a reduced scavenger receptor CD36 expression and lipid accumulation in microphages, 3) reduced-inflammation cytokines in serum, and 4) enhanced energy expenditure in Tg mice. Our data suggest that activation of NF-κB in macrophages has atheroprotective effects in mice by enhancing lipid metabolism and energy expenditure.
Keywords: nuclear factor-κB, apolipoprotein E, inflammation, atherosclerosis, lipid metabolism, macrophages, energy expenditure
atherosclerosis is a progressive disease characterized by the accumulation of lipid in the arterial wall, which is known to be driven by inflammatory processes (1, 26, 33). The early atherosclerotic lesion develops from foam cells, which are derived from the accumulation of cholesterol-engorged macrophages (29, 31, 34). In response to excess unmetabolized lipoproteins deposited in the arterial wall, blood monocytes are recruited and developed into macrophages in the intima. The foam cells are formed when the macrophages do not have the capacity to remove the lipid. The excess of oxidized low-density lipoprotein (oxLDL) particles induces cell death, including the foam cells, which causes the formation of the necrotic extracellular lipid core in lesion progression (1, 26, 33).
Transcription factor nuclear factor-κB (NF-κB) integrates multiple processes in the formation of atherosclerotic plaques (6, 40). Activated NF-κB is present in the atherosclerotic lesion (3) and in the coronary vasculature in experimental hypercholesterolemia (26, 39). There was a colocalization of atherosclerosis and increased levels of components of the NF-κB system in mice (6, 16). All of these indicate that NF-κB is implicated in atherosclerosis. NF-κB has been considered as a proatherogenic factor mainly because of its regulation of many of the proinflammatory genes linked to atherosclerosis (6). NF-κB may also play an important role by possibly affecting the pro- and anti-inflammatory balance in atherosclerosis, as NF-κB regulates expression of a broad spectrum of genes (35). Its activity is found in many types of cells, including adipocytes and macrophages (17, 36). The common form of NF-κB contains two subunits: p65 (RelA) and p50 (NF-κB1). p65 contains the transactivation domain and mediates transcriptional activation of target genes. p50 usually inhibits the transcriptional activity of p65, and the inhibition disappears in the p50-knockout (KO) mice (11). However, there is no direct evidence that p65 in macrophages leads to atherosclerosis. In the current study, the NF-κB activity is enhanced in fat and macrophages with aP2 promoter-controlled p65 in apolipoprotein E (apoE)-KO transgenic (Tg) mice. The atherosclerosis was then evaluated in the Tg mice.
The differentiation of monocytes to macrophages is characterized by increased expression of two major scavenger receptors on the surface of cells: scavenger receptor class A (SR-A) and CD36 (2, 6, 9). They mediate the scavenging of modified lipids from the vessel wall, thereby facilitating the process of foam cell formation. The role of NF-κB in the scavenging of modified lipids or foam cell formation has not been studied extensively. It was shown that in the absence of p50, activated macrophages take up less modified LDL because of downregulation of SR-A (22). However, detailed analyses of the exact function of NF-κB p65 in foam cell formation are still lacking. We have reported that p50-KO and aP2-p65 overexpression mice have increased energy expenditure and prevented high-fat diet-induced obesity. In these two mouse strains inflammation was elevated, and the anti-inflammation genes were significantly activated (14, 37). We also reported that NF-κB inhibits PPARγ function. This inhibition may reduce CD36 expression because CD36 is a PPARγ target gene (12). NF-κB may prevent foam cell formation through regulation of lipid metabolism. To investigate the role of NF-κB activation in macrophages during atherogenesis, Kanters et al. (23) used LDL receptor-deficient mice with a macrophage-restricted deletion of IκB kinase-β (IKKβ), which is essential for NF-κB activation by proinflammatory signals. These mice showed increased atherosclerosis, as quantified by lesion area measurements. This suggests that NF-κB signal is required for the macrophage function. We investigated whether macrophage overexpression of p65 could improve atherosclerosis in apoE-KO mice.
MATERIALS AND METHODS
Animals.
Male B6.129P2-apoE<tm1Unc>/J (4-wk-old) mice (apoE-KO) were purchased from The Jackson Laboratory (Bar Harbor, ME). aP2 (fatty acid-binding protein 4) promoter-controlled p65 overexpression mice (aP2-p65) were generated in our laboratory. In aP2-p65 mice, p65 is expressed in adipocytes and macrophages. ApoE-KO mice were crossed with aP2-p65, generating aP2-p65 ApoE KO mice (Tg). Twelve of the 8-wk-old Tg male mice, 12 of the 8-wk-old littermate apoE-KO male control mice, and six of the B6 wild-type controls were fed a standard atherosclerotic diet (TD 88137, 0.15% wt/wt cholesterol; Harlan-Teklad, Madison, WI) in which 42% of calories came from fat. All of the mice were housed in an animal facility with a 12:12-h light-dark cycle and constant temperature (22–24°C). The mice had free access to water and diet. All procedures were performed in accordance with the National Institutes of Health guidelines for the care and use of animals and were approved by the Institutional Animal Care and Use Committee at the Pennington Biomedical Research Center.
Nuclear magnetic resonance.
Body composition was measured for fat content using quantitative nuclear magnetic resonance, as described previously (13).
Metabolic chamber.
Energy expenditure, respiratory exchange ratio, spontaneous physical movement, and food intake were measured simultaneously for each mouse, using a comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, OH) as described previously (13).
Quantitative real-time RT-PCR.
Total RNA was extracted from frozen tissues (kept at −80°C) using Tri-Reagent (T9424; Sigma), as described previously (14, 19). TaqMan RT-PCR primer and probe were used to determine mRNA for cluster of differentiation 36 (CD36; Mm00432403_m1), 3-hydroxy-3-methyl-glutaryl-CoA reductase (Mm01282499_m1), 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2; Mm00550050_m1), hormone-sensitive lipase (HSL; Mm00495359_m1), lipoprotein lipase (LPL; Mm00434770_m1), liver X receptor (LXR; Mm00447996), sterol regulatory element-binding protein (SREBP; Mm00550338_m1), IL-1β (Mm00434228_m1), IL-6 (Mm00446190_m1), IL-10 (Mm01288386_ml), IL-1 receptor antagonist (IL-1Ra; Mm00446185_m1), TNFα (Mm00443258_m1), ATP-binding cassette, subfamily G member 1 (ABCG1; Mm00437390_m1), adiponectin (Mm00456425_m1), CD11β-type 1 macrophage marker (Mm00434455_m1), arginase 1 (Arg-1)-type 2 macrophage marker (Mm00475988-ml), leptin (Mm00434759_m1), monocyte chemoattractant protein-1 (Mm00441242_m1), F4/80 (Mm00802530_ml), and p65 (Mm00501346_m1). The reagents were purchased from Applied Biosystems (Foster City, CA). Mouse ribosomal 18S rRNA_s1 (without intron-exon junction) was used as an internal control. The reaction was conducted using a 7900HT Fast Real-Time PCR System (Applied Biosystems).
Peritoneal macrophages.
Primary peritoneal macrophages were isolated from apoE-KO, aP2-p65, and their wild-type control mice, as described elsewhere (42). The macrophages were induced intraperitoneally by injection of 2 ml of sterilized solution of 2% starch (85643; Sigma). The peritoneal macrophages were harvested 3 days later with 20 ml of cold PBS by lavage and then cultured in RPMI 1640 (supplemented with 10% FBS and 50 μg/ml gentamicin) in a 100-mm culture dish. Three days later, the cells were transferred to chamber plates (cat. no. PEZGS0416; Millipore) for the foam cell formation.
Lipid and cholesterol test.
Cells were homogenized in 500 μl of PBS. The lipids were extracted from the lysate using a chloroform-methanol (2:1) mixture (41). Triglyceride was determined using a serum triglyceride determination kit (TR0100; Sigma). Cholesterol was determined with Cholesterol Reagent (80015; Raichem) according to the instructions from the manufacturer. Serum LDL and HDL were measured with EnzyChrom AF HDL and LDL/VLDL Assay Kit (E2HL-100) from BioAssay Systems (Hayward, CA).
Immunohistostaining.
Formalin-fixed aortas were processed, embedded in paraffin, and sectioned at 5 μm. The slides were blotted with a monoclonal primary antibody to F4/80 (sc-71087; Santa Cruz Biotechnology) at a 1:200 dilution. A biotinylated secondary antibody (mouse IgG) in the ABC kit was used together with the AEC substrate kit (AEC101; Sigma) for a color reaction according to the manufacturer's instructions.
Immunoblot.
Whole cell lysates were prepared by sonication in lysis buffer and used in Western blots, as described elsewhere (12). Akt (sc-8312), p65 (sc-8008), and phosphorylated (p)-IRS-1 Tyr632 (sc-17196) were purchased from Santa Cruz Biotechnology. p-Akt Ser473 (kp24001) was purchased from Calbiochem.
Fatty acid oxidation.
Fatty acid oxidation was measured as described elsewhere (19). Briefly, total [1-14C]palmitic acid (NEC-075H; Perkin-Elmer) oxidation was determined by measuring and summing 14CO2 production and 14C-labeled water-soluble metabolites. Gaseous 14CO2 produced from the oxidation of [1-14C]palmitic acid during the incubation was measured by transferring 1.0 ml of the incubation medium to a self-designed 50 wells (20 ml/well) of plastic plate trapper containing 1.0 ml of 70% perchloric acid and a 0.5-ml microcentrifuge tube containing 400 μl of 0.5 M sodium hydroxide. Liberated 14CO2 was trapped in the sodium hydroxide for 60 min at 37°C by shaking, and the microcentrifuge tube containing trapped 14CO2 was transferred into a scintillation vial with 4 ml of scintillation liquid and counted. 14C-labeled water-soluble metabolites were counted by sampling 0.5 ml of the aqueous phase of the lipid extraction in a scintillation vial.
Oxidative modification of LDL and foam cell formation.
To generate copper-oxidized LDL (oxLDL) (21), native LDLs (12-16-120412; Athens Research & Technology) diluted in PBS to a concentration of 2 mg/ml were incubated with copper sulphate (5 uM, final concentration) for 24 h at 37°C and then dialyzed against PBS for 48 h with a Slide-A-Lyzer Dialysis cassette (cat. no. 66380; Thermo Scientific). Primary macrophages were plated in four-well tissue culture plates (Millipore) at a density of 1 × 105 cells/ml. The macrophages were incubated 24 h later with oxLDL (25 μm/ml) for 24 h. The cells were fixed in 10% phosphate-buffered formalin. The foam cell formation was quantified with Oil Red O staining or 10 μg/μl Bodipy 493/503 (Invitrogen) for lipid staining. The nucleuses were stained with ProLong Gold antifade reagent with 4,6-diamidino-2-phenylindole (cat. no. P-36931, Invitrogen).
Quantitation of atherosclerosis.
The mice were anesthetized with a cocktail of ketamine-xylazine-acepromazine. After bleeding, the heart was perfused with Dulbecco's PBS through the left ventricle, whereas the right-side chamber was opened to allow flow. The aorta was fixed by perfusion with 4% paraformaldehyde for 20 min and was followed by 2 min of 60% isoprophenol. The lesion was stained by perfusion with 60% Oil Red O for 15 min. The flow was drowned from the right ventricle to avoid draining into the chamber. The nonspecific staining was removed by a perfusion of 60% isoprophenol until the flow was clear. The aorta was carefully removed while still attached to the heart. Fat and connective tissue were carefully removed and cleaned. The lesion in the aorta was quantified by scanning with an HP 7400c scanner and quantified again with NIH Image J software to get the digital signal. The upper half of the heart with the ascending aorta was dissected and paraffin-embedded. Sequential 5-μm-thick sections were cut from the heart toward the aortic root. From this point, one section was picked for every 15 cuts. The area evaluated is ∼2 mm of the proximal section of the aorta. A total of 15 sections were collected and photographed with Hamamatsu NanoZoomer Digital Slide Scanning System and quantified with NIH Image software. The fold change of Oil Red O staining was presented with eight samples for each group. For histological samples only, the vasculature was subsequently fixed with 4% paraformaldehyde. The hearts and entire aortas from all treatment groups were removed and immediately cleaned of fat and connective tissue. Aortas for biochemical analyses were frozen immediately (−70°C) without formalin fixation.
Serum cytokines.
Serum cytokines were measured using a magnetic multiplex kit (catalog no. MADPK-71k-05; Millipore). IL-1Ra ELISA kit was purchased from Alpco Diagnostics (Salem, NH).
Statistical analysis.
All data are represented as means ± SE. Statistical significance of differences between experimental groups was determined using Student's paired t-test. A value of P < 0.05 was considered statistically significant.
RESULTS
Reduced atherosclerotic lesion formation in Tg mice.
To compare the degree of atherosclerotic lesion formation in the three groups of Tg, apoE-KO, and B6 control mice, the standard atherogenic diet was fed to the mice for 16 wk to induce the severe atherosclerotic lesions in apoE- KO mice. The mice were euthanized, and blood was withdrawn from the right ventricle, followed by a perfusion with PBS from the left atrium until the flow was clear from the right ventricle. To fix the heart and aorta, 4% wt/wt saline-buffered formalin was perfused for 20 min and then replaced with Oil Red O solution, as described in materials and methods. This prevents lipid staining outside the aorta wall. The aorta and the heart were carefully isolated for analysis of lesion formation. We did not observe obvious atherosclerotic lesions in the B6 mice. We found that lesions in the aorta were reduced by 48% in Tg mice vs. the apoE-KO mice (Fig. 1A). The lesions in the ascending aorta were reduced by 36% in Tg mice (Fig. 1B). These results indicate that p65 overexpression in macrophages in apoE-KO mice reduces atherosclerotic lesion formation on the atherogenic diet.
Fig. 1.

Reduced atherosclerotic lesion formation in the transgenic (Tg) mice. A, top: plaque in aorta. A, bottom: quantification of the stain with National Institutes of Health (NIH) Image J software. B: plaque in proximal aorta and quantified with NIH image J software. Data were presented by 5 pairs of mice, *P < 0.001 by Student's paired t-test. WT, wild type; apoE-KO, apolipoprotein E knockout.
Reduced cholesterol in Tg mice/apoE-KO mice fed an atherogenic diet containing 1.25% wt/wt cholesterol developed severe hypercholesterolemia and atherosclerosis. To compare the extent of atherosclerotic lesion formation in the two groups, the mice were fed the atherogenic diet for 16 wk and then euthanized. Blood samples were analyzed for serum cholesterol and triglycerides. There were no significant changes in triglyceride in either group (Fig. 2A). However, serum cholesterol was reduced by 15% in Tg mice compared with apoE-KO controls (Fig. 2B). Serum LDL was reduced by 15% in Tg mice (Fig. 2C). Serum HDL was identical in both groups (data not shown). However, the HDL/LDL ratio increased from 0.26 ± 0.04 in apoE-KO to 0.34 ± 0.03 in Tg mice (Fig. 2D). In clinical studies, the HDL/LDL ratio is used to predict the chances of developing atherosclerosis and heart disease. A HDL/LDL ratio of >0.3 is considered healthy. The higher ratio of HDL/LDL in Tg mice suggests that Tg mice were protected from atherosclerosis on an apoE-KO background. The reduction of total cholesterol is a result of reduced LDL in Tg mice. The basal levels of triglyceride and cholesterol were identical at 8 wk of age before the feeding started (data not shown).
Fig. 2.
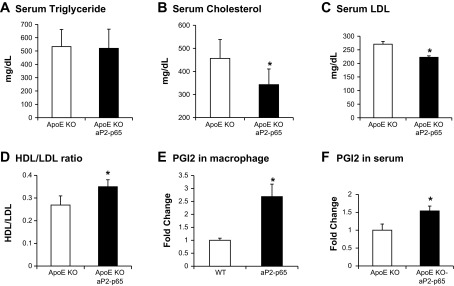
Reduced cholesterol in Tg mice. A: serum triglyceride. B: serum cholesterol. C: serum LDL. D: HDL/LDL ratio; n = 12. E: PGI2 in macrophages. F: PGI2 in serum. PGI2 was measured with 6-keto prostaglandin F1α EIA Kit (cat. no. 515211; Cayman Chemical); n = 6/group. Data presented by means ± SE. *P < 0.05 by Student's paired t-test.
PGI2 is an atheroprotective prostaglandin and a COX-2 target gene (5, 8). We measured PGI2 expression in primary peritoneal macrophages. The PGI2 expression was higher in aP2-p65 macrophages than in wild-type cells (Fig. 2E). We measured PGI2 in serum in apoE-KO mice and Tg mice. PGI2 was elevated in the Tg mice compared with the apoE-KO controls (Fig. 2F). This indicates that cholesterol is converted into an atheroprotective prostaglandin in the Tg mice. This may explain the reduced cholesterol in Tg mice.
Lipid metabolism in Tg mice.
We measured the cholesterol and triglyceride synthesis gene (HMG-CoA synthase 2, HMG-CoA reductase, HSL, LPL, LXR, SREBP, and ABCG1) expression of Tg mice in the liver using quantitative (q)RT-PCR, and there was no difference in the mRNA level between the Tg and apoE-KO control mice (Fig. 3A). p65 expression in liver was not changed (Fig. 3B). This suggests that the reduced cholesterol is not due to the changes of cholesterol synthesis in the Tg mice. However, HMGCS2, SREBP, and ABCG1 mRNAs were increased significantly in apoE-KO mouse livers compared with B6 wild-type controls. We quantified cholesterol and triglyceride levels in livers. B6 mice had fatty livers and had higher cholesterol and triglyceride in the liver (Fig. 3, C and D). These data may explain the phenotype difference of B6 and apoE-KO mice. Cholesterol contents were slightly lower in Tg mice compared with apoE-KO controls (Fig. 3C). We next quantified cholesterol and triglycerides in feces, and there was no difference between the Tg and apoE-KO mice (data not shown). This indicates that cholesterol absorption in the intestine is normal.
Fig. 3.
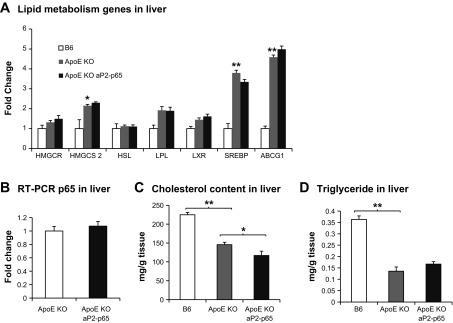
Lipid metabolism. A: lipid metabolism genes in livers. The liver samples were collected in mice that were fed the atherogenic diet for 16 wk. B: p65 expression in livers. C: cholesterol content in livers. D: triglyceride in livers; n = 5. Data presented by means ± SE. *P <0.05, **P < 0.001 by Student's paired t-test.
Reduced foam cell formation in the TG mice.
During the pathological examination of the lesions, we examined the foam cells in the aorta with a macrophage marker, F4/80, with histoimmunostaining. Surprisingly, the Tg mice had fewer foam cells in aortic walls compared with the apoE-KO controls (Fig. 4A). We did not observe macrophage staining in B6 controls. Because NF-κB has been shown to be involved in foam cell formation, we performed in vitro foam cell formation with primary peritoneal macrophages collected from aP2-p65 mice and their wild-type controls. In the aP2-p65 macrophages, lipid accumulation was reduced compared with the macrophages from wild-type controls of C57BL/6J, as indicated by a Bodipy lipid staining (Fig. 4B). We quantified triglyceride and cholesterol in cells with a triglyceride assay kit and a cholesterol assay kit. In the wild-type control cells, triglyceride accumulation was increased by 60% after 24 h of treatment with 25 μg/ml oxLDL. Conversely, the aP2-p65 macrophages reduced lipid accumulation after the treatment (Fig. 4C). There was a 28% increase in cholesterol in control cells but not in the aP2-p65 macrophages (Fig. 4D). To test the effect of NF-κB in lipid metabolism in macrophages, we measured CD36, a major scavenger receptor in lipid metabolism, using real-time RT-PCR. CD36 expression was inhibited by NF-κB, which indicated a reduced lipid uptake by the cells (Fig. 4E). We next measured fatty acid oxidation (FAO) in macrophages to test the capacity of the macrophages in removing the excess lipid. We found that the FAO was higher in the p65 overexpression macrophages (Fig. 4F). This suggested that the p65 overexpression macrophages have a better capacity to remove the lipid. The results indicate that overexpression of p65 in macrophages prevents foam cell formation.
Fig. 4.
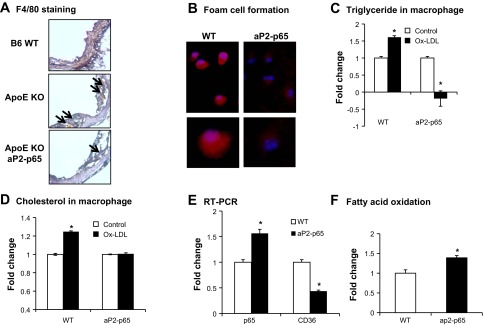
Reduced foam cell formation in aP2-p65 primary macrophages. A: immunohistostaining of macrophage marker of F4/80 of the proximal aorta with cryosections (5 μm); n = 5. B: primary macrophages were induced to foam cells, and the lipid was quantified with Bodipy staining (red). Nucleus was stained with 4,6-diamidino-2-phenylindole (blue). C and D: triglyceride and cholesterol were quantified in macrophages after incubation of oxidized LDL (oxLDL) for 24 h in triplicate. E: cluster of differentiation (CD36) expression in primary peritoneal macrophages. F: fatty acid oxidation in primary microphages, *P < 0.05 by Student's paired t-test.
Increased energy expenditure in aP2-p65 apoE-KO (Tg) mice.
The aP2 promoter-controlled p65 overexpression in fat and mice macrophages prevented high-fat diet-induced obesity by enhanced energy expenditure (37). To evaluate the role of NF-κB p65 in the protection against atherosclerosis, we measured the metabolic phenotype of the Tg mice fed a standard atherogenic diet. Body weight and body fat content were measured every 2 wk (Fig. 5, A and B). The lean mass was identical in both groups, which indicated that they developed normally (Fig. 5C). The body weight in both groups was identical at 21 days when the mice were weaned off the standard atherogenic diet. The body weight of the Tg mice was significantly lower than in the apoE-KO control mice due to the increased energy expenditure measured in the metabolic chamber (Fig. 5D). There was no significant difference in food intake for the two groups. The lower body weight in the Tg group is a result of reduced fat mass and enhanced energy expenditure. Reduced fat mass is beneficial for preventing metabolic syndrome.
Fig. 5.

Enhanced energy expenditure in Tg mice. A: body weight was measured every 2 wk. B: fat percentage was presented by total fat mass normalized with body weight. C: muscle percentage was presented by total fat mass normalized with body weight. D: energy expenditure was measured in a metabolic chamber. V̇o2 was normalized with lean body mass; n = 12. Data presented by means ± SE. *P < 0.05, **P < 0.001 by Student's paired t-test.
Increased anti-inflammation response in aP2-p65 macrophages.
To study the inflammation response of p65 overexpression in macrophages, we quantified inflammation gene expression with real-time RT-PCR in peritoneal macrophages. In nonstimulated macrophages, expression of the major proinflammation genes such as TNFα, IL-1β, and IL-6 was higher in aP2-p65 macrophages compared with the wild-type control macrophages. In contrast, anti-inflammation genes IL-10 and IL-1Ra were overexpressed in aP2-p65 macrophages (Fig. 6A). TNFα and IL-1β are proatherosclerotic cytokines. IL-6 was reported to have proatherosclerotic or antiatherosclerotic properties. Its role in atherosclerosis is controversial (18, 27, 32). Although inflammation is potentially dangerous, it has a protective response that is controlled by numerous negative feedback mechanisms. Activation of anti-inflammation genes may overcome the effects of proinflammation in Tg mice. We quantified the production of cytokines in serum of the apoE-KO and Tg mice after a 16-wk cholesterol diet feeding. The production of inflammation cytokines IL-1β, TNFα, and IL-6 was lower in the Tg mice than in the apoE-KO control mice (Fig. 6B). NF-κB or IKK negative regulates IL-1β secretion (15, 28, 38). It is well established that IKKβ and NF-κB are involved in negative regulation of inflammasome activation (15, 28, 38). The anti-inflammation cytokines IL-10 and IL-1Ra were significantly elevated in Tg mice (Fig. 6C). IL-1Ra is an antiatherogenic cytokine in the suppression of lesion development. Reduced IL-1β and TNFα in circulation protected against lesion formation in the Tg mice. The reduced inflammation may be a result of enhanced energy expenditure in Tg mice.
Fig. 6.

Inflammation in macrophages and serum. A: inflammation genes in peritoneal macrophages. B and C: cytokines in serum. Serum cytokines were measured using multiplex kit; n = 8. Data presented by means ± SE. *P < 0.05 by Student's paired t-test. IL-1Ra, receptor antagonist.
NF-κB p65's effects in adipose tissues.
aP2 promoter is expressed in adipocytes and macrophages. We confirmed that p65 is specifically overexpressed in adipose tissues (Fig. 7A) and macrophages (Fig. 4E) but not in the livers (Fig. 3B). To test the role of p65 in adipocytes, we examined adipokine expression and insulin signaling in adipose tissues. We observed that IL-1β and TNFα expressions in adipose tissues were significantly increased in Tg mice. Leptin was reduced in Tg mice due to the small fat pad. Macrophage maker F4/80 in Tg was lower compared with apoE-KO controls due to the small fat pad in Tg mice (Fig. 5B). Type 1 macrophage maker gene CD11b was reduced in Tg mice, and the M2 macrophage Arg-1 was not changed (Fig. 7A). Insulin signaling was examined in fat tissues. Our result suggests that insulin signaling was improved in Tg mice compared with apoE-KO controls (Fig. 7B). This suggests that p65 overexpression in fat plays a partial role in the improvement of atherosclerosis.
Fig. 7.

NF-κB p65's effects in adipose tissues. A: adipokine and macrophage genes in epididymal fat (16 wk on a cholesterol diet) by quantitative RT-PCR. B: insulin signaling in epididymal fat (16 wk on a cholesterol diet) by Western blot. Fold changes of the signals that were quantified with NIH Image J software and animalized by Akt protein. Data presented by means ± SE. *P < 0.05, **P < 0.001 by Student's paired t-test; n = 5.
DISCUSSION
In this study, we analyzed the direct effect of NF-κB in apoE-KO mice, in which NF-κB p65 is overexpressed in macrophages. We quantified atherosclerotic lesions in those mice on a standard atherogenic diet. The lesion size was reduced by 48% compared with littermate apoE-KO controls. This indicated that activation of NF-κB in macrophages has a protective effect in atherosclerosis.
In the study of NF-κB in atherosclerosis, Kanters et al. (23) generated macrophage-specific deletion of NF-κB-activating kinase IKKβ in LDL receptor-KO mice. IKKβ deletion in macrophages inhibits NF-κB activation and leads to an increase in lesion size in the aortic root. The macrophages undergo more apoptosis and necrosis in the absence of IKKβ. The deletion of IKKβ inhibits both proinflammation and anti-inflammation gene expression. IL-10, an antiatherogenic cytokine, was strongly reduced in macrophage IKKβ knockouts. The effect of IL-10 on atherogenesis has been studied extensively using different mouse models (4, 7, 30). Inhibition of NF-κB impaired antiatherosclerotic inflammation cytokines. IL-1Ra is another anti-inflammation cytokine and an atheroprotective cytokine (20, 24). IL-10 and IL-1Ra are antiatherogenic factors in preventing atherogenic lesion formation in mice models. In our study, we measured the expression of inflammation cytokines in overexpression of NF-κB macrophages. We found that there was a strong activation of IL-10 and IL-1Ra in the aP2-p65 macrophages compared with the wild-type controls. The serum proinflammation cytokines TNFα, IL-1β, and IL-6 were reduced in Tg mice. IL-1Ra and IL-10 were increased in Tg mice. The anti-inflammation signal may overturn the effects of proinflammation for atheroprotection.
CD36 is a major scavenger receptor and drives macrophage foam cell development by uptaking oxidized lipoproteins (10). Absence of CD36 in apoE-KO resulted in a 70% decrease in total lesion area in Western diet-fed mice. PPARγ is a transcription factor that plays a key role in inducing membrane expression of CD36 (a PPARγ target gene) on human monocytes (9). In our previous study, PPARγ is inhibited by the activation of NF-κB (12). In this study, we observed that CD36 was inhibited in the aP2-p65 macrophages compared with the wild-type controls. Reduced CD36 represents a partial effect in the prevention of foam cell formation. In the liver, lipid metabolic genes are not altered (Fig. 2E). This suggests that lipid synthesis is not the cause of the improved atherosclerotic lesion in Tg mice.
Other than lipid uptake and synthesis in cells, lipid consumption may also prevent foam cell formation and atherosclerotic lesion formation in mice. In this study, we measured mitochondrial function in primary macrophages. We observed a significant increase in fatty acid oxidation in the aP2-p65 macrophages (Fig. 4F). This suggests that the aP2-p65 macrophages have a strong capacity to remove the lipid and prevent foam cell formation. The Tg mice, similarly to the aP2-p65 mice, exhibit markedly reduced total body and visceral fat weight that results from increased energy expenditure. Because p65 was overexpressed also in adipocytes, we observed an increased uncoupling in brown fat in aP2-p65 mice (14, 37) and in Tg mice (data not shown). The enhanced energy expenditure is beneficial for the prevention of metabolic syndrome. This study indicates that activation of NF-κB in macrophages prevents atherosclerosis by eliminating lipid accumulation on the aortic walls.
In summary, the overexpression of NF-κB in macrophages ameliorates atherosclerosis in apoE-KO mice. The mechanisms are involved in increased lipid conversion and oxidation in macrophages, suggesting that macrophages in Tg mice have a strong capacity to remove the lipid for improving atherosclerosis. Our data suggest that activation of NF-κB in macrophages has atheroprotective effects in mice by enhancing lipid metabolism and energy expenditure. The mechanism provides an explanation about the discrepancy of nonsteroidal anti-inflammatory agents in effectively combating inflammation but augmenting atherosclerotic complications (25).
GRANTS
This work was supported by American Diabetes Association Junior Faculty Award 1-09-JF-17 (to Z. Gao) and an National Institutes of Health (NIH) COBRE grant (P20-RR-021945). The qRT-PCR test and metabolic phenotyping and imaging studies were conducted in the genomic core, phenotyping core, and imaging core of the Pennington Biomedical Research Center, which are supported by the NIH Grants 2P30-DK-072476-06 and P20-RR-021945.
DISCLOSURES
The authors declare that they have no conflicts of interest, financial or otherwise, concerning this article.
AUTHOR CONTRIBUTIONS
X.Y., X.J., and K.C. performed the experiments; X.Y., W.G., and Z.G. analyzed the data; X.J. prepared the figures; Z.G. contributed to the conception and design of the research; Z.G. interpreted the results of the experiments; Z.G. drafted the manuscript; Z.G. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Roya Khosravi for proofreading the manuscript.
REFERENCES
- 1.Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA, Watson AD, Lusis AJ. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation 91: 2488–2496, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, Hörkkö S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V, Witztum JL. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann NY Acad Sci 947: 214–222; discussion 222–223, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest 97: 1715–1722, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, Nicoletti A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med 9: 10–17, 2003 [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 296: 539–541, 2002 [DOI] [PubMed] [Google Scholar]
- 6.de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol 25: 904–914, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Eefting D, Schepers A, De Vries MR, Pires NM, Grimbergen JM, Lagerweij T, Nagelkerken LM, Monraats PS, Jukema JW, van Bockel JH, Quax PH. The effect of interleukin-10 knock-out and overexpression on neointima formation in hypercholesterolemic APOE*3-Leiden mice. Atherosclerosis 193: 335–342, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, Fitzgerald GA. COX-2-derived prostacyclin confers atheroprotection on female mice. Science 306: 1954–1957, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 108: 785–791, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman MW. Scavenger receptors in atherosclerosis. Curr Opin Hematol 4: 41–47, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Gadjeva M, Tomczak MF, Zhang M, Wang YY, Dull K, Rogers AB, Erdman SE, Fox JG, Carroll M, Horwitz BH. A role for NF-kappa B subunits p50 and p65 in the inhibition of lipopolysaccharide-induced shock. J Immunol 173: 5786–5793, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Gao Z, He Q, Peng B, Chiao PJ, Ye J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J Biol Chem 281: 4540–4547, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Z, Wang Z, Zhang X, Butler AA, Zuberi A, Gawronska-Kozak B, Lefevre M, York D, Ravussin E, Berthoud HR, McGuinness O, Cefalu WT, Ye J. Inactivation of PKCθ leads to increased susceptibility to obesity and dietary insulin resistance in mice. Am J Physiol Endocrinol Metab 292: E84–E91, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Gao Z, Yin J, Zhang J, He Q, McGuinness OP, Ye J. Inactivation of NF-kappaB p50 leads to insulin sensitization in liver through post-translational inhibition of p70S6K. J Biol Chem 284: 18368–18376, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O'Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130: 918–931, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 97: 9052–9057, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 132: 344–362, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol 19: 2364–2367, 1999 [DOI] [PubMed] [Google Scholar]
- 19.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab 284: E741–E747, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, Iwakura Y, Ohsuzu F. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 24: 1068–1073, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K, Zhang Z, Nozaki S, Matsuzawa Y. Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol 20: 1953–1960, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Kanters E, Gijbels MJ, van der Made I, Vergouwe MN, Heeringa P, Kraal G, Hofker MH, de Winther MPJ. Hematopoietic NF-kappaB1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood 103: 934–940, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest 112: 1176–1185, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 23: 656–660, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Libby P, Crea F. Clinical implications of inflammation for cardiovascular primary prevention. Eur Heart J 31: 777–783, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Lusis AJ. Atherosclerosis. Nature 407: 233–241, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madan M, Bishayi B, Hoge M, Amar S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis 197: 504–514, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mankan AK, Canli O, Schwitalla S, Ziegler P, Tschopp J, Korn T, Greten FR. TNF-alpha-dependent loss of IKKbeta-deficient myeloid progenitors triggers a cytokine loop culminating in granulocytosis. Proc Natl Acad Sci USA 108: 6567–6572, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Namiki M, Kawashima S, Yamashita T, Ozaki M, Sakoda T, Inoue N, Hirata K, Morishita R, Kaneda Y, Yokoyama M. Intramuscular gene transfer of interleukin-10 cDNA reduces atherosclerosis in apolipoprotein E-knockout mice. Atherosclerosis 172: 21–29, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Plenz G, Robenek H. Monocytes/macrophages in atherosclerosis. Eur Cytokine Netw 9: 701–703, 1998 [PubMed] [Google Scholar]
- 32.Schieffer B, Selle T, Hilfiker A, Hilfiker-Kleiner D, Grote K, Tietge UJ, Trautwein C, Luchtefeld M, Schmittkamp C, Heeneman S, Daemen MJ, Drexler H. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 110: 3493–3500, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Nerem RM. The pathogenesis of atherosclerosis: an overview. Clin Cardiol 14, 2 Suppl 1: I1–I16, 1991 [DOI] [PubMed] [Google Scholar]
- 34.Schwartz CJ, Valente AJ, Sprague EA, Kelley JL, Suenram CA, Rozek MM. Atherosclerosis as an inflammatory process. The roles of the monocyte-macrophage. Ann NY Acad Sci 454: 115–120, 1985 [DOI] [PubMed] [Google Scholar]
- 35.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47: 921–928, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 116: 1793–1801, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, Ko HJ, Ong H, Kim JK, Mynatt R, Martin RJ, Keenan M, Gao Z, Ye J. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem 285: 4637–4644, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693–733, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Wilson SH, Caplice NM, Simari RD, Holmes DR, Jr, Carlson PJ, Lerman A. Activated nuclear factor-kappaB is present in the coronary vasculature in experimental hypercholesterolemia. Atherosclerosis 148: 23–30, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Xanthoulea S, Curfs DM, Hofker MH, de Winther MP. Nuclear factor kappa B signaling in macrophage function and atherogenesis. Curr Opin Lipidol 16: 536–542, 2005 [DOI] [PubMed] [Google Scholar]
- 41.Xu F, Gao Z, Zhang J, Rivera CA, Yin J, Weng J, Ye J. Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: a role of lipid mobilization and inflammation. Endocrinology 151: 2504–2514, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293: E1118–E1128, 2007 [DOI] [PubMed] [Google Scholar]