

Abstract
The cardioprotective effects of adiponectin (APN) against myocardial ischemia/reperfusion (MI/R) injury are well known. However, comprehension of the mechanisms mediating intracellular APN signaling remains incomplete. We recently demonstrate the antioxidant/antinitrative effects of APN are not dependent on AMPK. Protein kinase A (PKA) has been previously shown to be activated by APN, with uncertain relevance to APN cardiac protection. The current study determined whether the antioxidative/antinitrative effect of APN is mediated by PKA. Administration of APN (2 μg/g) 10 min before reperfusion significantly enhanced cardiac PKA activity, reduced oxidative stress, and decreased infarct size. Knockdown of cardiac PKA expression (PKA-KD) by intramyocardial injection of PKA-siRNAs (>70% suppression) significantly inhibited APN cardioprotection determined by cardiac apoptosis, infarct size, and cardiac function. Moreover, PKA-KD virtually abolished the suppressive effect of APN on MI/R-induced NADPH oxidase overexpression and superoxide overproduction and partially inhibited the effect of APN on nitrative protein modification in MI/R heart. Mechanistically, APN significantly inhibited MI/R-induced IKK/IκB phosphorylation and NF-κB activation, which were blocked in PKA-KD heart. Finally, the PKA-mediated antioxidant/antinitrative and cardioprotective effects of APN are intact in AMPK-deficient mice, suggesting that there is no cross talk between AMPK and PKA signaling in APN cardioprotection. Collectively, we demonstrate for the first time that APN inhibits oxidative/nitrative stress during MI/R via PKA-dependent NF-κB inhibition.
Keywords: protein kinase A, adiponectin, reperfusion injury, oxidative stress
the adipokine adiponectin (APN), of primarily adipose origin, regulates glucose and lipid metabolism (23). Prospective clinical observations and animal studies have demonstrated that reduced APN function may be a major contributor to increased ischemic heart disease morbidity in diabetic patients (21). Administration of APN attenuates myocardial ischemia and reperfusion (MI/R) injury in mice (20, 24). However, the detailed molecular mechanisms responsible for the cardioprotective effect of APN remain elusive.
It is well accepted that excess superoxide-mediated oxidative stress plays an important role in MI/R injury. Superoxide reacts with nitric oxide (NO) to form the highly cytotoxic molecule peroxynitrite (ONOO−), which induces nitrative stress and exacerbates MI/R injury. We demonstrated that APN-deficient mice exhibited much more severe I/R injury from overproduction of superoxide, peroxynitrite, and resultant nitrative stress, reversible by globular domain of APN (gAPN) administration (22). Therefore, APN is cardioprotective against ischemia/reperfusion injury by inhibiting oxidative/nitrative stress. We have recently demonstrated that the antioxidative/antinitrative property of APN is not mediated by AMP-activated protein kinase (AMPK), the most recognized APN intracellular signaling molecule (24). A comprehensive understanding of intracellular APN antioxidative signaling mechanisms remains lacking.
Protein kinase A (PKA) is an important mediator of signal transduction downstream of G protein-coupled receptors and plays a key role in the regulation of metabolism and triglyceride storage. In endothelial cells, APN inhibits palmitate-induced apoptosis by suppressing reactive oxygen species (ROS) generation via PKA pathways (27). However, whether PKA signaling mediates APN's antioxidative/antinitrative effect during MI/R injury has never been previously investigated.
Therefore, the aims of the present study were to investigate whether PKA mediates APN's antioxidative/antinitrative and cardioprotective effects against MI/R injury, and if so to identify the possible responsible intracellular signaling mechanisms.
MATERIALS AND METHODS
Animals.
Adult male wild-type (WT) mice, adult male AMPK-DN mice [with dominant negative α2-subunit (D157A) of AMPK], and their male littermate controls were utilized in this study. Generation, breeding, phenotype characteristics, and genotyping of AMPK-DN mice (>80% inhibition of cardiac AMPK activity) have been previously described in detail (28). The experiments were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the Thomas Jefferson University Committee on Animal Care.
Myocardial ischemia/reperfusion.
Mice were anesthetized with 2% isoflurane. MI/R was induced by temporarily exteriorizing the heart via a left thoracic incision and placing a 6-0 silk suture slipknot around the left anterior descending coronary artery. Twenty minutes after MI, animals were randomized to receive either vehicle or gAPN (2 μg/g ip) (24). After 30 min of MI, the slipknot was released, and the myocardium was reperfused for 3 h (all assays except cardiac function and infarct size) or 24 h (cardiac function and infarct size only). All assays were performed utilizing tissue from the I/R area, the area at risk (AAR), identified by Evans blue negative staining. Sham-operated control mice (Sham MI/R) underwent the same surgical procedure except that the suture placed under the left coronary artery was not tied.
Inhibition of PKA with in vivo siRNA-mediated knockdown.
To specifically confirm the role of PKA in gAPN's protective effect on MI/R, a siRNA gene silencing technique was utilized to knock down mouse cardiac PKA expression (PKA-KD). In brief, two mouse-specific PKAα subunit-selective siRNAs targeting the nucleotide sequences 5′-CGTCCTGACCTTTGAGTATCT-3′ and 5′-CAGTGTGCTGTTGTAAACATA-3′ were employed. siRNA oligos of the same size possessing scrambled nucleotide sequences served as controls. All siRNAs were obtained from Integrated DNA Technologies. The siRNAs were diluted in 5% glucose and mixed with in vivo-jet PEI (Genesee Scientific). Adult mice were anesthetized with 2% isoflurane. The heart was exposed via left thoracotomy at the fifth intercostal space. PKA-specific siRNAs (20 μl; dose 0.8 μg/μl) or scrambled control was delivered via three separate intramyocardial injections (by 32G needle), temporarily blanching the left ventricular free wall. 48 h after siRNAs injection, the mice were subjected to MI/R.
Determination of myocardial apoptosis and myocardial infarct size.
Myocardial apoptosis was determined by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining and caspase-3 activity, as described in our previous study (24). Myocardial infarct size was assessed by Evans blue-2,3,5-triphenyl tetrazolium chloride double staining methods (24).
Determination of cardiac function.
Cardiac function was determined by echocardiography and left ventricular catheterization methods 24 h after reperfusion before thoracotomy, as described in our previous study (24).
Measurement of PKA activity.
PKA activity was measured utilizing nonradioactive PepTag assays (Promega, Southampton, Hampshire, UK) reliant on a change in charge of the PepTag A1 peptide from +1 to −1 following phosphorylation (14). Sample reaction mixtures were incubated at room temperature for 15 min. After incubation, samples were separated on a 0.5% (wt/vol) agarose gel at 100 V for 15–20 min. Purified PKA catalytic subunit served as a positive control, while the negative control contained only buffer. Bands were visualized under ultraviolet light.
Quantification of superoxide production.
Superoxide production in I/R heart tissue was measured by lucigenin-enhanced chemiluminescence as previously described (24). Superoxide production was expressed as relative light units (RLU) per second per milligram of heart weight.
Determination of total NOx content in cardiac tissue.
Cardiac tissue samples from AAR were rinsed, homogenized in deionized water (1:10, wt/vol), and centrifuged at 14,000 g for 10 min. Tissue NO and its in vivo metabolic products (NO2 and NO3, collectively known as NOx) in the supernatant were determined via chemiluminescence NO detector (SIEVER 280i NO Analyzer) as described in our previous study (24).
Quantitation of tissue nitrotyrosine content.
I/R cardiac tissue nitrotyrosine (NT) content (pmol NT/mg protein), an accepted footprint of in vivo peroxynitrite formation and a reliable index of nitrative stress, was determined via ELISA, described in our previous publications (22, 26).
Measurement of nuclear translocation of NF-kB p65.
The relative increase of NF-kB p65 translocation into the nuclei of the I/R cardiomyoctyes was determined via ELISA per manufacturer's protocol (IMGENEX, San Diego, CA). In brief, cardiac tissue was cut into small pieces and homogenized. The mixture was centrifuged for 10 min at 10,000 rpm at 4°C. The supernatant represented the cytoplasmic fraction. The pellet was resuspended in nuclear lysis buffer and subsequently centrifuged for 10 min at 14,000 rpm at 4°C. This supernatant represented the nuclear fraction. A plate coated with anti-p65 antibody captured free p65 (both nuclear or cytoplasmic). Bound p65 was detected by adding a secondary antibody followed by alkaline phosphatase-conjugated secondary antibody. Each well's absorbance value was determined at 405 nm by a microplate reader (Molecular Devices). The relative ratio of nuclear to cytoplasmic p65 was calculated by the equation (absorbance value of nucleus)/(absorbance value of cytoplasm) (16).
Western blot analysis.
Protein from tissue homogenates was separated on SDS-PAGE gels, transferred to nitrocellulose membranes, and incubated with primary antibodies [against PKA C-α antibody, IKKβ, phospho-IKKβ, IκBα, phospho-IκBα (Cell Signaling Technology, Danvers, MA), iNOS (Upstate, Chicago, IL), gp91phox (Transduction Laboratories, San Jose, CA)] and horseradish peroxidase-conjugated secondary antibody. The blot was developed with a Supersignal Chemiluminescence detection kit (Pierce, Rockford, IL) and observed with a Kodak Image Station 400 (Rochester, NY).
Statistical analysis.
All values in the text and figures are presented as means ± SE of n independent experiments. All data (except Western blot density) were subjected to one-way ANOVA followed by Bonferoni correction for post hoc t-test. Western blot densities were analyzed with the Kruskal-Wallis test followed by Dunn's post hoc test. Probabilities of 0.05 or less were considered to be statistically significant.
RESULTS
PKA deficiency blocked gAPN inhibition of MI/R-induced myocardial infarct, apoptosis, and cardiac dysfunction.
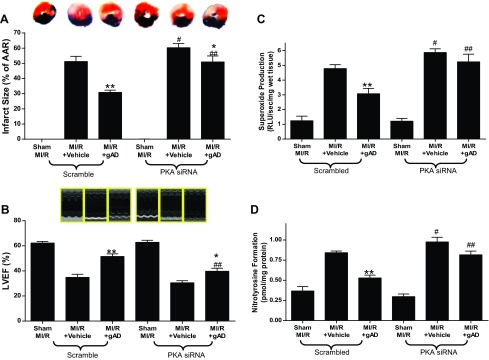
MI/R did not alter PKA expression and slightly reduced PKA activity. Administration of APN 10 min before reperfusion had no effect on PKA expression but significantly increased PKA activity (Fig. 1) and reduced myocardial infarct size (Fig. 2). To determine whether APN activation of PKA plays a causative role in APN cardioprotection, PKA expression was downregulated via intramyocardial siRNA injection. Pretreatment with the PKA-selective siRNAs reduced PKA expression by 70% (P < 0.01 relative to scramble group, n = 6; Fig. 2A). Cardiac-specific PKA knockdown blocked APN-induced PKA activation (Fig. 2B) and attenuated APN's infarct reduction effects (Fig. 2C).
Fig. 1.

Influence of adiponectin (APN) on PKA expression and activation. A: cardiac PKA expression in control (Sham MI/R) or myocardial ischemia/reperfusion (MI/R) treated with vehicle or globular domain of APN (gAPN, gAD). B: cardiac PKA activity in control or MI/R treated with vehicle or gAPN; n = 6–9 hearts/group. *P < 0.05 vs. vehicle.
Fig. 2.

Influence of PKA signaling blockade by PKA-selective siRNAs on myocardial PKA expression and activity and the resultant effect on myocardial infarct size. A: cardiac-specific PKA knockdown decreased PKA expression. B: PKA siRNAs significantly inhibited PKA expression. C: PKA-specific siRNAs significantly blocked gAPN inhibition of MI/R-induced myocardial infarction; n = 9–12 hearts/group. *P < 0.05, **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
MI/R resulted in significant cardiac injury including cardiac systolic and diastolic dysfunction (Fig. 3) and myocardial apoptotic cell death (Fig. 4). Consistent with our previous observations (22), gAPN administration significantly improved cardiac function after reperfusion (Fig. 3, Scramble) and reduced apoptosis (Fig. 4, Scramble). The knockdown of cardiac PKA expression (PKA siRNA) slightly worsened MI/R-induced cardiac dysfunction as measured by LVEF and LVEDP (Fig. 3, A and B) but significantly increased infarct size (Fig. 2C) and apoptosis (Fig. 4A). More importantly, the cardioprotective effect of APN was either completely abolished (antiapoptotic effect on Fig. 4) or significantly attenuated (Figs. 2C and 3) in the PKA-KD mice. Taken together, these results demonstrated that PKA activation plays a critical role in APN cardioprotection after MI/R.
Fig. 3.

PKA deficiency blocked gAPN inhibition of MI/R-induced myocardial dysfunction. Cardiac function determined by echocardiography (A, LVEF) and left ventricular catheterization hemodynamic assay (B, LVEDP; C, +dp/dtmax); n = 12–14 animals/group. *P < 0.05, **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
Fig. 4.

PKA deficiency blocked gAPN inhibition of MI/R-induced cardiomyocyte apoptosis. A: TUNEL staining. B: caspase-3 activity; n = 5–6 hearts/group. **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
PKA deficiency abolished antioxidative and inhibited antinitrative effects of gAPN post-I/R.
Having demonstrated that gAPN protects against reperfusion injury in a partially PKA-dependent manner, we investigated the underlying mechanisms potentially responsible. We reported previously that gAPN cardioprotection involves reduction of oxidative/nitrative stress (22). We performed two series of experiments assessing the relationship between PKA signaling and oxidative/nitrative stress in MI/R hearts. In the first experiment series, MI/R-induced oxidative stress was detected. Under basal conditions, no significant difference existed between superoxide production in scramble and PKA-KD mice. MI/R induced greater increases in superoxide production in PKA-KD animals (Fig. 5A). APN administration significantly inhibited MI/R-induced superoxide production in scramble animals (Fig. 5A). However, APN treatment did not reduce superoxide production in PKA-KD animals (Fig. 5A). We next identified potentially responsible molecular sources for the impaired antioxidant actions of gAPN in PKA-KD mice. Expression of gp91phox, the primary isoform of NADPH oxidase expressed in heart (1, 2, 29), was significantly increased after MI/R in both scramble and PKA-KD heart (Fig. 5B). gAPN treatment of Scramble animals subjected to MI/R significantly inhibited gp91phox expression. In contrast, the inhibitory effect of APN on MI/R-induced gp91phox overexpression was abolished in PKA-KD mice, in consistent fashion with superoxide generation trends in PKA-KD mice (Fig. 5B). Together, these results demonstrate that MI/R-induced oxidative stress was modestly increased in PKA-KD mice, and the antioxidant effect of gAPN was completely abolished by PKA signaling deficiency.
Fig. 5.

PKA deficiency blocked the anti-oxidative effects of gAPN post MI/R. Production of superoxide (A; n = 6–7 hearts/group); gp91phox expression (B), representative Western blots shown (n = 5–6 hearts/group). **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
In the second experiment series, we assessed MI/R-induced myocardial nitrative stress. There was no significant NOx content difference between PKA-KD and Scramble animals during basal conditions or after MI/R (Fig. 6A). Administration of gAPN significantly inhibited MI/R-induced NOx overproduction in both Scramble and PKA-KD mice (Fig. 6A). Inducible nitric oxide synthase (iNOS) expression was significantly increased after MI/R was induced in Scramble and PKA-KD animals (Fig. 6B). Treatment with gAPN significantly inhibited iNOS expression, an effect not inhibited by PKA-KD (Fig. 6B). These results demonstrate that, although the antioxidant effect of gAPN is mediated by PKA, the iNOS-inhibitory effect of gAPN is not mediated by PKA.
Fig. 6.

PKA deficiency partially blocked the anti-nitrative effects of gAPN post MI/R. NOx content (A; n = 5–6 hearts/group), inducible nitric oxide synthase (iNOS) expression (B), representative Western blots shown (n = 5–6 hearts/group); nitrotyrosine content, determined by ELISA (C; n = 6–8 hearts/group). *P < 0.05, **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
Superoxide rapidly reacts with NO to form the cytotoxic molecule peroxynitrite. NT content, the accepted footprint of peroxynitrite production, was increased by MI/R injury both in Scramble and in PKA-KD mice (Fig. 6C). Administration of gAPN significantly inhibited MI/R-induced NT generation in Scramble animals, an effect that was partially blocked in PKA-KD animals (Fig. 6C).
gAPN decreased IKK-IκB-NF-κB signaling activation in I/R myocardium, which was blocked by PKA deficiency.
Decreased IκB phosphorylation preventing nuclear NF-κB translocation attenuates myocardial reperfusion injury (17) and preserves cardiac function post-MI. PKA signaling is involved in gAPN-suppressed IKK-IκB-NF-κB activation in endothelial cells subjected to tumor necrosis factor-α or high-glucose treatment (27). We investigated the possible mechanism underlying the PKA-dependent cardioprotective effects of gAPN and determined activation of IKK-IκB-NF-κB by MI/R. Consistent with others' previous results (15, 30), MI/R induced significant IKK phosphorylation (Fig. 7A), resulting in IκBa phosphorylation (Fig. 7B) and NF-κB activation (Fig. 7C) in both Scramble and PKA-KD mice. MI/R-induced NF-κB activation by IKK/IκB phosphorylation was significantly inhibited by gAPN in Scramble animals (Fig. 7); however, gAPN did not inhibit NF-κB activity during PKA knockdown (Fig. 7). These data suggest that the cardioprotective effect of gAPN was partially due to inhibition of IKK-IκB-NF-κB activation and that such an effect was dependent on PKA signaling.
Fig. 7.

gAPN decreased IKK-IκB-NF-κB signaling activation in MI/R myocardium, which was blocked by PKA deficiency. A: phosphorylated IKK expression, representative Western blots shown (n = −6 hearts/group). B: phosphorylated IκB expression, representative Western blots shown (n = 5–6 hearts/group). C: activated NF-κB, determined by ELISA (n = 6–8 hearts/group). *P < 0.05, **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
PKA-mediated cardioprotective effect of APN against MI/R injury is AMPK independent.
Despite considerable evidence supporting the essential role of AMPK in APN intracellular signaling (4, 5, 7, 8, 11), we recently provided direct evidence of AMPK-independent gAPN-mediated cardioprotection in the intact animal (24, 25). The current study suggests that gAPN is cardioprotective in mice subjected to MI/R, with involvement of PKA signaling. We next investigated whether there is cross talk between PKA and AMPK pathways. Adult male mice overexpressing a dominant negative α2-subunit (D157A) of AMPK (AMPK-DN) mice were pretreated with PKA siRNAs, and the effects of gAPN administration on MI/R injury were determined. Consistent with our previous data (24), gAPN inhibited MI/R-induced infarction (Fig. 8A) and improved cardiac function (Fig. 8B) in AMPK-DN animals. Importantly, PKA knockdown significantly attenuated this protective effect, consistent with WT mice results presented in Figs. 1–3. Moreover, PKA deficiency blocked the antioxidative/antinitrative effects of gAPN, evidenced by augmented superoxide (Fig. 8C) and peroxynitrite (Fig. 8D) generation. Together, these results support that the PKA-mediated gAPN cardioprotective effect against MI/R injury is independent of AMPK signaling.
Fig. 8.
PKA-mediated cardioprotective effect of APN against MI/R injury is AMPK-independent. A: myocardial infarction, determined by Evans blue/TTC double staining (n = 9–12 hearts/group). B: cardiac function, determined by echocardiography assay (n = 12–15 animals/group). C: superoxide production (n = 6–8 hearts/group). D: nitrotyrosine content, determined by ELISA (n = 6–8 hearts/group). *P < 0.05, **P < 0.01 vs. vehicle; #P < 0.05, ##P < 0.01 vs. Scramble with the same treatment.
DISCUSSION
Several important observations have been made in the present study. First, we demonstrate that gAPN could not inhibit postischemic myocardial apoptosis, infarction, or cardiac dysfunction in cardiac-specific PKA knockdown animals. This is the first evidence that PKA signaling mediates the cardioprotective effects of APN in ischemic heart disease in vivo. Second, we provide the first direct evidence that the antioxidative and antinitrative effects of gAPN in I/R hearts are mediated by PKA. Third, we demonstrate that PKA signaling is involved in adiponectin-mediated inhibition of IKK-IκB-NF-κB activation after MI/R. Finally, we provide evidence that PKA-mediated gAPN's cardioprotective effect against MI/R injury is independent of AMPK signaling.
APN is an abundant circulating adipocytokine secreted primarily from adipose tissue, exerting at least three major functions, including an insulin sensitization/metabolic regulatory function (in the liver and muscle), an anti-inflammatory/vasculoprotective function, and an anti-ischemic/cardioprotective function. Numerous epidemiological studies underline the correlation between hypoadiponectinemic states and increased morbidity/mortality of cardiovascular ischemic diseases (6, 12, 13, 18). Consistent with our and others' previous results, the present study confirms that exogenous APN supplementation can significantly mitigate myocardial apoptosis, infarct size, and cardiac dysfunction.
The signal transduction pathway mediating the antioxidant effect of APN remains intensely investigated. PKA is involved in APN-mediated suppression of ROS and apoptosis in endothelial cells (9, 19). Therefore, the current study sought to determine the role of PKA in gAPN's cardioprotective effect against MI/R injury, utilizing in vivo gene silencing techniques. In WT mice, we demonstrate that gAPN reversed MI/R-induced myocardial infarction, cardiomyocytes apoptosis, and cardiac dysfunction. However, in mice subjected to intramyocardial PKA knockdown, such protective effects of gAPN were either completely abolished or significantly attenuated. This is direct evidence that a significant portion of gAPN-mediated cardioprotection is PKA dependent in intact animals.
Simultaneous overproduction of superoxide and NO not only causes inactivation of the cytoprotective NO but generates the highly cytotoxic molecule peroxynitrite. Therefore, those therapeutic interventions preventing concomitant NO/superoxide overproduction effectively block peroxynitrite formation and offer great tissue protection. The current study's results demonstrate that gAPN administration reduced synchronized NO/superoxide overproduction from NADPH oxidase and iNOS and effectively block peroxynitrite formation in I/R hearts. Here, we demonstrate for the first time that gAPN inhibits I/R-induced oxidative in a PKA-dependent fashion, which may be the mechanism responsible for reduced cardioprotection by gAPN observed in PKA-deficient animals.
NF-κB is a crucial transcription factor in the induction of genes involved in various physiological processes, including response to injury and inflammation. Activation of NF-κB requires phosphorylation of the inhibitor of NF-κB (IκB) by IκB kinase (IKK), resulting in 26S proteasome-mediated degradation of IκB. This allows the translocation of NF-κB from the cytosol to the nucleus, where the heterodimer binds the promotor region of specific target genes. Activation of NF-κB leads to the transcription of factors promoting inflammation (i.e., adhesion molecules, cytokines, and chemokines), but may also contribute to tissue remodeling, inflammation resolution, and the transcription of genes resulting in anti-inflammatory effects. Notably, NF-κB activation is implicated in the pathophysiology underlying acute myocardial infarction, heart failure, endothelial dysfunction, and unstable angina pectoris (3, 10, 30). Furthermore, IKK inhibition attenuates myocardial injury and dysfunction following acute I/R (30), and inhibition of IκB phosphorylation and resultant NF-κB activation attenuates MI/R injury in cardiomyocytes. We currently demonstrate that gAPN inhibits IKK-IκB-NF-κB activation in the Scramble heart. However, gAPN could not block MI/R-induced IKK and IκB phosphorylation and resultant NF-κB activation in PKA-deficient mice. Together, these results indicate that gAPN confers cardioprotection via inhibition of IKK-IκB-NF-κB-mediated inflammation, and intact PKA signaling is necessary for such gAPN-mediated anti-inflammatory protection against I/R injury.
Substantial evidence supports the essential role AMPK plays in APN intracellular signaling. Pharmacological or genetic inhibition of AMPK activity virtually abolishes the metabolic, anti-inflammatory and vasculoprotective effects of APN. However, we previously demonstrated that APN cardioprotection against MI/R injury is retained in AMPK-DN mice. APN administration to AMPK-DN mice reduces MI/R-induced oxidative and nitrative stress, indicating that AMPK-independent mechanisms mediate gAPN's cardioprotective effect (24). Our present study data iare consistent with our previous findings. In AMPK-DN mice, the antioxidative/antinitrative effects (and consequent cardioprotective effects) of gAPN were dependent on whether PKA was silenced or not. Our data support the responsibility of PKA signaling, in part, for the protective actions of APN against MI/R injury.
In summary, we demonstrate for the first time that APN inhibits oxidative/nitrative stress during MI/R via PKA signaling in an AMPK-independent fashion. This represents new understanding in the intracellular signaling mechanisms of APN, as we continue to identify novel therapeutic targets in the treatment of diabetic I/R injury.
GRANTS
This research was supported by the National Natural Science Foundation of China Grant 81170144 (to X.-L. Wang), 81170199 (to Y.-J. Wang) 81270185 (to J. Zhao), and the National Heart, Lung, and Blood Institute Grants HL-63828 and HL-096686, the American Diabetes Association Research Award 7-08-RA-98 (to X.-L. Ma).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z., X.-L.W., J.Z., Y.-J.W., Y.-X.Y., and E.-H.G. performed experiments; Y.Z. and J.Z. prepared figures; X.-L.W., Y.-J.W., Y.-X.Y., and E.-H.G. analyzed data; X.-L.W. and W.B.L. drafted manuscript; W.B.L., W.J.K., and X.-L.M. edited and revised manuscript; W.J.K. and X.-L.M. approved final version of manuscript; X.-L.M. conception and design of research.
REFERENCES
- 1.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105: 293–296, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Borchi E, Parri M, Papucci L, Becatti M, Nassi N, Nassi P, Nediani C. Role of NADPH oxidase in H9c2 cardiac muscle cells exposed to simulated ischaemia-reperfusion. J Cell Mol Med 13: 2724–2735, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-kappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation 103: 2296–2302, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJG, Prabhu SD, Valente AJ. Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-(kappa)B/PTEN suppression. J Biol Chem 283: 24889–24898, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng KK, Lam KS, Wang Y, Huang Y, Carling D, Wu D, Wong C, Xu A. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 56: 1387–1394, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, Ding YY, Russell RG, Lindemann D, Hartley A, Baker GR, Obici S, Deshaies Y, Ludgate M, Rossetti L, Scherer PE. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 145: 367–383, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Dyck JRB. The ischemic heart: starving to stimulate the adiponectin-AMPK signaling axis. Circulation 116: 2779–2781, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Fang X, Palanivel R, Cresser J, Schram K, Ganguly R, Thong FS, Tuinei J, Xu A, Abel ED, Sweeney G. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am J Physiol Endocrinol Metab 299: E721–E729, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 6: 27–35, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grabellus F, Levkau B, Sokoll A, Welp H, Schmid C, Deng MC, Takeda A, Breithardt G, Baba HA. Reversible activation of nuclear factor-kappaB in human end-stage heart failure after left ventricular mechanical support. Cardiovasc Res 53: 124–130, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Hattori Y, Nakano Y, Hattori S, Tomizawa A, Inukai K, Kasai K. High molecular weight adiponectin activates AMPK and suppresses cytokine-induced NF-kappaB activation in vascular endothelial cells. FEBS Lett 582: 1719–1724, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Hui X, Lam KS, Vanhoutte PM, Xu A. Adiponectin and cardiovascular health: an update. Br J Pharmacol 165: 574–590, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawano T, Saito T, Yasu T, Saito T, Nakamura T, Namai K, Tamemoto H, Kawakami M, Saito M, Ishikawa SE. Close association of hypoadiponectinemia with arteriosclerosis obliterans and ischemic heart disease. Metabolism 54: 653–656, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Khaliulin I, Parker JE, Halestrap AP. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc Res 88: 324–333, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C, Kao RL, Ha T, Kelley J, Browder IW, Williams DL. Early activation of IKKβ during in vivo myocardial ischemia. Am J Physiol Heart Circ Physiol 280: H1264–H1271, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Ohsugi T, Horie R, Kumasaka T, Ishida A, Ishida T, Yamaguchi K, Watanabe T, Umezawa K, Urano T. In vivo antitumor activity of the NF-kappaB inhibitor dehydroxymethylepoxyquinomicin in a mouse model of adult T-cell leukemia. Carcinogenesis 26: 1382–1388, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, Muto S, Itai A, Isobe M. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res 63: 51–59, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Ouchi N, Shibata R, Walsh K. Cardioprotection by adiponectin. Trends Cardiovasc Med 16: 141–146, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, Hough K, Scalia R, Goldstein BJ. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes 55: 1840–1846, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 11: 1096–1103, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T, Chronos NA. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation 105: 788–793, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115: 1408–1416, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Tishinsky JM, Robinson LE, Dyck DJ. Insulin-sensitizing properties of adiponectin. Biochimie 94: 2131–2136, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, Ma XL. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 119: 835–844, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Tao L, Yuan Y, Lau WB, Li R, Lopez BL, Christopher TA, Tian R, Ma XL. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am J Physiol Endocrinol Metab 297: E384–E391, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol 32: 934–942, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Mahadev K, Fuchsel L, Ouedraogo R, Xu SQ, Goldstein BJ. Adiponectin suppresses IκB kinase activation induced by tumor necrosis factor-α or high glucose in endothelial cells: role of cAMP and AMP kinase signaling. Am J Physiol Endocrinol Metab 293: E1836–E1844, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative (alpha)2 subunit of AMP-activated protein kinase. J Biol Chem 278: 28372–28377, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng Q, Wang Y, Yuan Y, Wang X, Tao L, Li R, Koch W, Ma XL. Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid Redox Signal 15: 1779–1788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zingarelli BF, Hake PW, FAUYang Z, Yang ZF, O'Connor MF, Denenberg AF, Wong HR. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-kappaB and AP-1 activation and enhances myocardial damage. FASEB J 16: 327–342, 2002 [DOI] [PubMed] [Google Scholar]