

Abstract
Nuclear factor-κB (NF-κB) is a master regulator of genes that control a large number of cellular processes, including angiogenesis and inflammation. We recently demonstrated that cytochrome P-450 1B1 (Cyp1B1) deficiency in endothelial cells (EC) and pericytes (PC) results in increased oxidative stress, alterations in migration, attenuation of capillary morphogenesis, sustained activation of NF-κB, and increased expression of thrombospondin-2 (TSP2), an endogenous inhibitor of angiogenesis. On the basis of a growing body of evidence that phenethyl isothiocyanate (PEITC) and pyrrolidine dithiocarbamate (PDTC) function as antioxidants and suppressors of NF-κB activation, we investigated their potential ability to restore a normal phenotype in Cyp1B1-deficient (cyp1b1−/−) vascular cells. PEITC and PDTC inhibited NF-κB activity and expression in cyp1b1−/− EC and PC. We also observed restoration of migration and capillary morphogenesis of cyp1b1−/− EC and decreased cellular oxidative stress in cyp1b1−/− EC and PC without restoration to normal TSP2 levels. In addition, expression of a dominant-negative inhibitor κBα, a suppressor of NF-κB activation, decreased NF-κB activity without affecting TSP2 expression in these cells. In contrast, knockdown of TSP2 expression resulted in attenuation of NF-κB activity in cyp1b1−/− vascular cells. Furthermore, expression of TSP2 in wild-type (cyp1b1+/+) cells resulted in increased NF-κB activity. Together, our results demonstrate an important role for TSP2 in modulation of NF-κB activity and attenuation of angiogenesis. Thus Cyp1B1 expression in vascular cells plays an important role in the regulation of vascular homeostasis through modulation of the cellular reductive state, TSP2 expression, and NF-κB activation.
Keywords: isothiocyanates, NF-κB, angiogenesis, cell migration, thrombospondins, oxidative stress
nuclear factor-κB (NF-κB) is a transcription factor involved in cell proliferation, angiogenesis, inflammation, and survival. Rel family members (p65/RelA, RelB, c-Rel, p50, and p52) form homo- or heterodimeric complexes that comprise the NF-κB complex. In resting cells, NF-κB is inactive because of its sequestration by inhibitor κB (IκB) proteins, which mask the nuclear localization sequence of NF-κB, thereby retaining it in the cytoplasm and preventing DNA binding and, subsequently, gene transcription (1). Upon stimulation, IκB is phosphorylated, ubiquitinated, and degraded by the proteosome. The NF-κB complexes are then translocated to the nucleus and bind DNA and activate transcription of target genes. Dysregulation of NF-κB activity is involved in a number of disease states, including cancer, chronic inflammation, glaucoma, retinal diseases, and diabetes (10). Recent studies from our laboratory have shown that a cytochrome P-450 1B1 (Cyp1B1) deficiency in endothelial cells (EC) or pericytes (PC) leads to increased oxidative stress, increased levels of the matricellular protein thrombospondin-2 (TSP2), and sustained p65 NF-κB activation (31, 39). The mechanisms underlying NF-κB activation, as well as its further role in the dysregulation of angiogenesis in Cyp1B1-deficient (cyp1b1−/−) vascular cells remain unknown.
Cytochrome P-450 enzymes, including Cyp2J2 and Cyp2B6, have been identified within the cardiovascular system (3, 43, 48). Cytochrome P-450 enzymes utilize endogenous substrates, such as retinoic acid and arachidonic acid, to generate intracellular messengers, such as cis-epoxyeicosatrienoic acids, mid-chain cis-trans-conjugated dienols, and hydroxyl epoxyeicosatrienoic acids, with important roles in the modulation of vascular tone, blood flow, and angiogenesis (11, 13, 14, 48). Cyp1B1 is mainly expressed in extrahepatic epithelia, steroidogenic tissues, and vascular EC and smooth muscle cells (12, 19, 20, 28, 32, 38, 39, 41). Cyp1B1 participates in the oxidative metabolism of xenobiotics and metabolizes substrates of endogenous origin, including retinol, melatonin, and dietary plant flavonoids (7, 37, 51).
Recent studies conducted in our laboratory established an important role for Cyp1B1 in the regulation of angiogenesis. In vivo, retinas from cyp1b1−/− mice exhibited reduced vascular density and failed to undergo neovascularization during oxygen-induced ischemic retinopathy. We also demonstrated, for the first time, that Cyp1B1 is constitutively expressed in EC and PC from the retina and vascular beds of other tissues (31, 39). The lack of Cyp1B1 resulted in attenuation of migration, endothelial nitric oxide (NO) synthase (eNOS) expression, and capillary morphogenesis of EC. More recently, we demonstrated that Cyp1B1 deficiency in retinal PC results in increased proliferation and migration, decreased vascular endothelial growth factor secretion, and increased Akt activation. These changes were concomitant with increased oxidative stress, sustained NF-κB activation, and TSP2 expression in EC and PC. Thus Cyp1B1 plays an essential role in maintaining the physiological function of vascular cells and angiogenesis. How Cyp1b1 deficiency leads to sustained activation of NF-κB and TSP2 expression needs further delineation.
Isothiocyanates are bioactive products produced from the breakdown of glucosinolates in cruciferous vegetables, such as broccoli, cabbage, and watercress (8). Cruciferous vegetable consumption is associated with reduction of cancer incidence through inhibition of phase I biotransformation enzymes, particularly the cytochrome P-450 family (29). Phenethyl isothiocyanate (PEITC), an important hydrolysis product of gluconasturtiin in watercress, has been shown to have antioxidant properties, inhibit NF-κB signaling, and induce apoptosis of various human cancer cells (15, 44, 47). Another compound demonstrated to inhibit NF-κB and to have antioxidant and anti-inflammatory effects is pyrrolidine dithiocarbamate (PDTC), a member of the dithiocarbamate family (6, 34). Dithiocarbamates have a widespread range of use, from pesticide applications to antiviral and antitumor drugs and removal of metals from organisms (6, 25, 34, 36). PDTC exerts protective effects against acute lung injury in rabbits by inhibiting NF-κB, resulting in partial inhibition of TNF-α production and upregulation of ICAM-1 (42).
In the present study we examine the effect of PEITC and PDTC, with antioxidant and NF-κB inhibitory activities, in cyp1b1−/− vascular cells. We demonstrate that administration of PEITC and PDTC inhibits NF-κB activation, abrogates oxidative stress, and restores migration and capillary morphogenesis of cyp1b1−/− vascular cells without reducing TSP2 expression. Knockdown of TSP2 in cyp1b1−/− EC and PC and overexpression of TSP2 in wild-type (cyp1b1+/+) EC and PC demonstrate an important role for TSP2 in NF-κB activation in these cells. Together, our studies indicate that, in the absence of Cyp1b1, increased oxidative stress and TSP2 expression attenuate angiogenesis through sustained activation of NF-κB.
MATERIALS AND METHODS
Experimental animals.
All experiments were carried out in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health. Immortomice expressing a temperature-sensitive simian virus-40 large-T antigen (Charles River Laboratories, Wilmington, MA) were backcrossed into C57BL/6 mice in our laboratory, further crossed with cyp1b1−/− mice, and generated in a C57BL/6 background, as previously described (39). A line of TSP2 reporter transgenic mice was generated by insertion of an enhanced green fluorescent protein (GFP) cDNA downstream of the TSP2 promoter in a bacterial artificial chromosome clone. This transgene allows expression of GFP from the endogenous TSP2 promoter.
Tissue preparation and culture of retinal vascular cells.
Retinal EC and PC were isolated from cyp1b1+/+ and cyp1b1−/− immortomice, the identity of the cells was confirmed, and the cells were cultured as previously described (31, 39).
Visualization of the retinal vasculature.
The retinal vascular pattern was analyzed using retinal whole mounts stained with rabbit anti-GFP antibody (Ab 10145, EMD Millipore, Billerica, MA) and rat anti-platelet endothelial cell adhesion molecule 1 (PECAM-1; catalog no. 553370, clone MEC13.3, BD Biosciences, San Jose, CA). At designated postnatal time points, the mouse eyes were enucleated, fixed for 4 min in 4% paraformaldehyde, and then placed in 70% ethanol for ≥24 h at −20°C. Retinas were dissected in PBS (Sigma) and then washed with PBS three times for 10 min each. After incubation with blocking buffer (50% FCS and 20% normal goat serum in PBS) for 2 h, the retinas were incubated with rabbit anti-GFP and rat anti-PECAM-1 at 4°C overnight. Retinas were then washed three times with PBS for 10 min each and incubated with the secondary antibodies anti-rabbit Cy3 and anti-rat Cy2 (Jackson ImmunoResearch, West Grove, PA; 1:500 dilution prepared in PBS containing 20% FCS and 20% normal goat serum) for 2 h at room temperature. After incubation, retinas were washed four times with PBS for 30 min each and mounted on a slide using PBS-glycerol. Retinas were viewed by fluorescence microscopy, and images were captured in digital format using a Zeiss microscope (Carl Zeiss, Chester, VA).
Cell viability and dose range determinations.
Cell viability with various concentrations of PEITC and PDTC was determined using the CellTiter 96 AQueous Non-Radioactive Cell Viability Assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega, Madison, WI]. The cyp1b1+/+ and cyp1b1−/− retinal EC and PC were plated at 8 × 103 cells per well of a 96-well plate and incubated for 24 h with PEITC or PDTC (Sigma Aldrich, St. Louis, MO) in growth medium. Viability was determined by measurement of absorbance at 490 nm using a microplate reader (Thermomax, Molecular Devices, Sunnyvale, CA) and determined as a percentage of control untreated cells. All samples were prepared in triplicate and repeated at least three times with similar results.
Real time-PCR analysis.
The cyp1b1+/+ and cyp1b1−/− retinal cells were allowed to reach 80% confluence and incubated for 24 h with PEITC or PDTC in growth medium. Cells were rinsed twice with PBS, scraped from 60-mm tissue culture plates, and transferred to microcentrifuge tubes. Cells were centrifuged, immediately frozen in liquid nitrogen, and stored at −80°C until analysis. Total RNA was extracted using the mirVana PARIS kit (Ambion) according to the manufacturer's instructions. cDNA was synthesized from 1 μg of total RNA using the Sprint RT Complete-Double PrePrimed kit (Clontech, Mountain View, CA). For quantitative PCR (qPCR) assays, 1 μl of each cDNA (dilution 1:10) was used as template. qPCR assays were performed in triplicate using three biological replicates on Mastercycler Realplex (Eppendorf, Hauppauge, NY) using the SYBR qPCR Premix (Clontech). Amplification parameters were as follows: 95°C for 2 min, 40 cycles of amplification (95°C for 15 s, 60°C for 40 s), and dissociation curve step (95°C for 15 s, 60°C for 15 s, 95°C for 15 s). Standard curves were generated from known quantities for each target gene of linearized plasmid DNA. Ten-times-dilution series were used for each known target, which was amplified using SYBR-Green qPCR. The linear regression line for nanograms of DNA was determined from relative fluorescence units at a threshold fluorescence value (Ct) to quantify gene targets from cell extracts by comparison of the relative fluorescence units at the Ct with the standard curve and normalized by the simultaneous amplification of Rpl13A, which was used as a housekeeping gene to normalize all samples. TSP2 forward (5′-CCCCAAACTGCCAAATTCC-3′) and reverse (5′-TCGTCACAAGCATCTCCGATT-3′) primers were used.
Apoptosis assays.
Apoptosis was determined by measurement of caspase activation using the Caspase-Glo 3/7 assay kit, as recommended by the supplier (Promega). The assay provides caspase-3/7 DEVD-aminoluciferin substrate, and caspase-3/7 activity is detected by luminescent signal. For the assay, cyp1b1+/+ and cyp1b1−/− retinal EC and PC were plated at 8 × 103 cells per well of a 96-well plate and incubated for 24 h with inhibitors in growth medium. Caspase activity was detected using a luminescent microplate reader (Victa2 1420 Multilabel Counter, PerkinElmer, Waltham, MA). All samples were prepared in triplicate and repeated at least twice with similar results.
Determination of reactive oxygen species production.
Reactive oxygen species (ROS) levels were determined by staining cells with dihydroethidium (DHE; Life Technologies, Grand Island, NY). DHE is oxidized to red-fluorescent ethidium by O2·− in the cytosol and intercalates in the DNA. Cells were plated at 3 × 104 cells per well of a four-well chamber slide (Lab-TEK, NUNC, Rochester, NY) coated with 2 μg/ml fibronectin (BD Biosciences) and incubated with inhibitors for 24 h in growth medium. Cells were loaded with 10 μM DHE for 20 min, washed with growth medium, and returned to growth medium twice for two 30-min recovery periods. Fluorescence intensity was analyzed with a fluorescence microscope (Carl Zeiss Optics, Germany), and images were captured in digital format. Three independent experiments were performed. For quantitative assessment, the mean cellular (nuclear) fluorescence intensities were determined, and representative images are shown.
NF-κB luciferase reporter assay.
Retinal EC (5 × 105) were plated in 60-mm tissue culture plates and, on the next day, cotransfected with 10 μg of pNF-κBLuc reporter plasmid (16) (NF-κB-dependent luciferase reporter, 3× κB-Luc, kindly provided by Dr. Shigeki Miyamoto, University of Wisconsin-Madison), 10 μg of β-galactosidase plasmid, and 20 μl of Lipofectin (Life Technologies) according to the manufacturer's instructions. A corresponding empty vector was included in all transfections as control. Growth medium was refreshed after the first 24 h. Cells were treated with PEITC or PDTC for 24 h in EC growth medium at 48 h posttransfection. Lysates were prepared using cell culture lysis buffer (Promega), and luciferase and β-galactosidase activities were measured according to the manufacturer's instructions (Promega).
Retinal PC (5 × 105) were plated in 60-mm tissue culture plates and, on the next day, cotransfected with 10 μg of pNF-κBLuc reporter plasmid, 10 μg of β-galactosidase plasmid, and 15 μl of FuGENE 6 (Promega) for 48 h. Cells were treated and lysates were prepared as stated above. Luminometric reactions were monitored using the Monolight 310 luminomether (BD Biosciences). Luciferase activity was corrected for transfection efficiency using β-galactosidase activity measured by a colorimetric assay according to the manufacturer's instructions (Promega).
TSP2 expression studies.
To overexpress TSP2 in cyp1b1+/+ retinal EC, cells (5 × 105) were plated on 60-mm tissue culture dishes. On the next day, adenoviruses encoding TSP2 or GFP (40) (100 plaque-forming units/cell) and Lipofectin (15 μl; Life Technologies) were diluted with 0.75 ml of Opti-MEM (Life Technologies) separately and incubated for 30 min at room temperature. After incubation, the diluted adenoviruses and Lipofectin were gently mixed and allowed to incubate at room temperature for 10 min. After incubation, the tissue culture plates were removed from the incubator, washed twice in serum-free DMEM, and incubated with 1.5 ml of the adenovirus-Lipofectin mixture overnight. On the next day, the adenovirus-Lipofectin-containing medium was removed, and the cells were gently washed twice with DMEM containing 10% FBS and incubated with EC growth medium for 2 days before they were used for experiments.
To express TSP2 in cyp1b1+/+ PC, cells (5 × 105) were plated on 60-mm tissue culture dishes. On the next day, adenoviruses encoding TSP2 or GFP (100 plaque-forming units/cell) and FuGENE 6 (15 μl; Promega) were diluted in 0.5 ml of Opti-MEM and incubated for 15 min at room temperature. After incubation, the tissue culture plates were removed from the incubator and rinsed with DMEM containing 10% FBS and 2.5 ml of growth medium was added. The diluted adenovirus mixture was added drop-wise to the plate, and the plates were incubated for 2 days before they were used for experiments.
Expression of IκBα.
The hemagglutinin-tagged S32A/S36A mutant IκBα [dominant-negative (DN)] and empty expression constructs were provided by Dr. Shigeki Miyamoto and are described elsewhere (17, 27). The cyp1b1−/− retinal EC and PC were transfected with DN-mIκBα or empty vector using transfection reagents as described above for 48 h and used for further analysis.
TSP2 knockdown.
Lentiviruses expressing a gene-specific small interfering RNA (siRNA) for mouse TSP2 (Sigma) were evaluated for knockdown expression. We tested six clones for knockdown of TSP2: TRCN0000065399, TRCN0000065400, TRCN0000065402, TRCN00000351965, TRCN00000351966, and TRCN00000351967. Cells (5 × 104) were plated in 12-well tissue culture plates. On the next day, lentiviral particles were thawed on ice and briefly centrifuged. Hexadimethrine bromide (8 μg/ml; Sigma) was added to fresh growth medium containing 10% FBS and used for infection. Lentiviral particles were added to cells at multiplicity of infection (MOI) of 5 or 10 in duplicate in 1 ml of the above-described medium. On the following day, the medium was replaced with fresh growth medium. On day 4, the medium was removed and replaced with fresh growth medium containing puromycin (5 μg/ml; Sigma). Cells were fed fresh growth medium containing puromycin every 3 days until wells were nearly confluent. Cells were subsequently passaged to larger plates and maintained in growth medium. Knockdown efficiency was analyzed by Western blot analysis. Clones were selected on the basis of expression of TSP2. We observed >75% knockdown in a selected group of clones, and some clones that showed little or no decreased expression were used as control. Clones TRCN0000065400 and TRCN0000065402 at MOI of 10 were selected for cyp1b1−/− retinal EC, and clones TRCN0000065399 and TRCN0000065402 at MOI of 10 were selected for cyp1b1−/− retinal PC. Stable cell populations expressing a specific TSP2 siRNA were used for further analysis.
Indirect immunofluorescence.
Retinal cells (1 × 105) were plated in four-well chamber slides (Lab-TEK, NUNC) coated with 2 μg/ml fibronectin (BD Biosciences) and incubated with inhibitors for 24 h in growth medium. On the next day, chambers were washed with PBS, fixed with methanol for 15 min on ice, and blocked with 1% BSA in Tris-buffered saline (TBS) at 37°C for 20 min. Slides were washed with TBS and incubated with anti-p65 NF-κB (1:200 dilution; Santa Cruz Biotechnology) in TBS containing 1% BSA at 37°C for 1 h. After they were washed with TBS, the cells were incubated with appropriate Cy3-conjugated secondary antibody (1:500 dilution in TBS containing 1% BSA) at 37°C for 40 min. Cells were washed with TBS three times, mounted with a 1:1 TBS-glycerol solution with 4′,6-diamidino-2-phenylindole, and analyzed with a fluorescence microscope (Carl Zeiss Optics). Images were captured in digital format. Three independent experiments were performed, and representative images are shown.
Transwell migration assay.
Transwell filters (Corning, Acton, MA) were coated with 2 μg/ml fibronectin in PBS and incubated overnight at 4°C. The bottom of the Transwell filter was rinsed with PBS and blocked with 2% BSA in PBS for 1 h at room temperature. The Transwell filter was rinsed with PBS, and 500 μl of serum-free DMEM were added to the bottom of each well. Cells were incubated with PEITC or PDTC in growth medium for 24 h prior to the experiment. Cells, at a concentration of 1 × 105 in 100 μl of serum-free medium, were added to the top of the Transwell membrane. Inhibitors were added to the top and bottom of the Transwell filter. After 4 h in a 33°C tissue culture incubator, the cells and medium were aspirated, and the upper side of the membrane was wiped with a cotton swab. The cells that had migrated through the membrane were fixed with 4% paraformaldehyde, stained with hematoxylin-eosin, and mounted on a slide. Ten high-power (×200) fields of cells were counted for each condition, and the average and standard error of the means were determined. All samples were prepared in duplicate, and the experiment was repeated at least twice with similar results.
Western blot analysis.
Retinal EC and PC were plated at 7 × 105 in 60-mm culture dishes and, on the next day, incubated with PEITC or PDTC for 24 h in growth medium. For conditioned medium collection, the cells were plated and, on the next day, rinsed twice with serum-free DMEM and incubated with serum-free growth medium with PEITC or PDTC for 2 days. Conditioned medium was collected and clarified by centrifugation. Cells were rinsed once in 0.04% EDTA in PBS and lysed in 100 μl of lysis buffer [50 mM HEPES (pH 7.5), 100 mM NaCl, 0.1 M EDTA, 1 mM CaCl2, 1 mM MgCl2, 1% Triton X-100, 1% NP-40, 0.5% deoxycholate, and protease inhibitor cocktail; Roche Biochemicals, Mannheim, Germany], briefly sonicated, and centrifuged at 14,000 g for 10 min at 4°C.
In some cases, total protein lysates were prepared from these samples in a modified lysis buffer (2 mM orthovanadate and 2 mM sodium fluoride) to determine phosphorylation status. Retinal EC and PC (7 × 105) were plated in 60-mm culture dishes and, on the next day, incubated with PEITC or PDTC for 24 h in growth medium. Protein concentrations were determined using the bicinchoninic acid method (Pierce, Rockford, IL). Samples were adjusted for protein content (50 μg), mixed with an appropriate volume of 6× SDS-sample buffer, and analyzed by SDS-PAGE (4–20% Tris-glycine gels; Invitrogen). Proteins were transferred to a nitrocellulose membrane and incubated in blocking buffer (0.05% Tween 20 and 5% skim milk in TBS) for 1 h at room temperature. Membranes were then incubated with mouse anti-TSP2 (BD Biosciences), mouse anti-β-actin (Thermo Fisher Scientific), rabbit anti-phosphorylated (Ser1177) eNOS, rabbit anti-Akt, rabbit anti-phosphorylated (Ser473) Akt, rabbit anti-IκBα (Cell Signaling), rabbit anti-p65, rabbit anti-phosphorylated (Ser311) p65, rabbit anti-RelB, rabbit anti-phosphorylated (Ser552) RelB, mouse anti-phosphorylated (Ser32) IκBα, and rabbit anti-eNOS (Santa Cruz Biotechnology). Membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibody (1:5,000 dilution; Jackson ImmunoResearch Laboratories) for 1 h at room temperature, and the protein was visualized according to the chemiluminescence procedure (chemiluminescence reagent, GE Biosciences). The mean band intensities were measured densitometrically using ImageJ 1.46a (National Institutes of Health, Bethesda, MD).
Capillary morphogenesis assays.
Tissue culture plates (35 mm) were coated with 0.5 ml of Matrigel (10 mg/ml; BD Biosciences) and allowed to harden by incubation at 37°C for ≥30 min. Cells were incubated with PEITC or PDTC for 8 h prior to assay in growth medium. Cells were removed by trypsin-EDTA, washed with DMEM containing 10% FBS, and resuspended at 1 × 105 cells/ml in EC growth medium without FBS. Cells in a 2-ml volume were applied to the Matrigel-coated plates, incubated with PEITC or PDTC, and photographed after 18 h using a Nikon microscope in a digital format. For quantitative assessment of the data, the mean number of branch points was determined by counting the number of branch points in five high-power (×100) fields.
Statistical analysis.
Statistical differences between samples were evaluated with Student's unpaired t-test (2-tailed) or two-way ANOVA with Bonferroni's correction for multiple comparisons when appropriate. Values are means ± SE. Each result is representative of at least three independent experiments. All statistical assessments were evaluated at the 0.05 level of significance. Statistical analyses were performed with GraphPad Prism statistical software (GraphPad Software, La Jolla, CA).
RESULTS
TSP2 expression in retinal vasculature is mainly attributed to the perivascular supporting cells.
In 1993, Iruela-Arispe et al. (18) demonstrated that TSP2 is highly expressed in blood vessels of the developing embryo. TSP2 is also highly expressed by mesenchymal progenitor cells and mesenchymally derived tissues such as bone and adipose tissue (35). We previously demonstrated that PC isolated from the retinal vasculature have mesenchymal stem cell characteristics (31, 33). However, the major source of TSP2 in the retinal vasculature is unclear. Using retinal whole mounts from TSP2-GFP transgenic mice, we stained the retinal vasculature for PECAM-1 and GFP. In Fig. 1A, left, PECAM-1 staining was used to identify EC. We previously showed that PC do not express PECAM-1 (31, 33). Staining using an antibody against GFP to identify sites of TSP2 expression and localization is depicted in Fig. 1A, middle. Expression of GFP localizing to cells on the abluminal surface of the blood vessels is shown in Fig. 1A, right. Modest overlap between GFP and PECAM-1 staining was observed. The GFP-positive cells are PC or vascular smooth muscle cells, in the case of larger vessels, based on their abluminal localization on retinal vasculature.
Fig. 1.
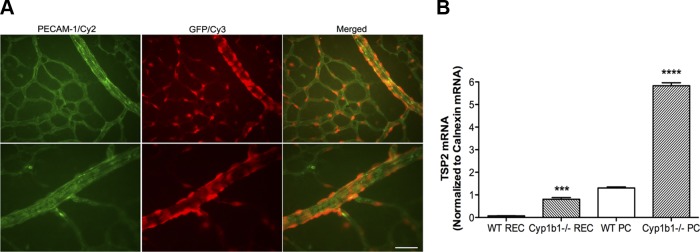
Thrombospondin 2 (TSP2) expression is localized to retinal pericytes (PC). A: retinal whole mounts were prepared from P7 TSP2-green fluorescent protein (GFP) transgenic mice and stained with anti-platelet endothelial cell adhesion molecule 1 (PECAM-1) and anti-GFP antibodies. Top and bottom show different fields of view. Scale bar, 20 μm. B: real-time quantitative PCR (qPCR) analysis of relative mRNA expression of TSP2 in wild-type (WT, cyp1b1+/+) and cytochrome P-450 1B1-deficient (cyp1b1−/−) retinal endothelial cells (REC) and PC (n = 9). ***P < 0.001, ****P < 0.0001.
We next examined TSP2 expression in cyp1b1+/+ EC and PC (Fig. 1B). Expression of TSP2 was very low in cyp1b1+/+ retinal EC compared with PC. We next examined TSP2 expression in cyp1b1−/− retinal EC and PC. Lack of Cyp1B1 in both cell types increased TSP2 expression. However, lack of Cyp1B1 in PC resulted in a fourfold increase in TSP2 expression. These results are consistent with our previous report of increased TSP2 expression in cell lysates and conditioned medium of cyp1b1−/− EC and PC (31, 39). Thus TSP2 is predominantly expressed by retinal perivascular supporting cells, including PC and vascular smooth muscle cells.
PEITC and PDTC inhibit NF-κB activation.
We previously reported that cyp1b1−/− retinal EC and PC have sustained NF-κB activation and that the p65 subunit is the predominant NF-κB protein family member in the retina (31, 39). We used PEITC and PDTC to study the role of NF-κB activation in cyp1b1−/− vascular cells. Phosphorylation and total p65 expression were studied by Western blot analysis. PEITC (1 μM) and PDTC (10 nM) decreased the level of phosphorylated (Ser311) p65 in EC and PC (Fig. 2, A and C). Quantitative assessment of the data is shown in Fig. 2, B and D.
Fig. 2.

Phenethyl isothiocyanate (PEITC) and pyrrolidine dithiocarbamate (PDTC) suppress NF-κB activation in cyp1b1−/− retinal vascular cells. A: cyp1b1−/− EC were incubated with PEITC (500 nM and 1 μM) or PDTC (5, 10, and 50 nM), and lysates were analyzed by Western blot analysis for phosphorylated p65 (p-p65), total p65, and β-actin. B: quantitative assessment of data in A (n = 3). **P < 0.01, ***P < 0.001. C: cyp1b1−/− PC were incubated with PEITC (500 nM and 1 μM) or PDTC (5, 10, and 50 nM), and lysates were analyzed by Western blot analysis for phosphorylated p65, total p65, and β-actin. D: quantitative assessment of data in C (n = 3). *P < 0.05. E and F: NF-κB activity was determined in cyp1b1+/+ and cyp1b1−/− EC and PC using an NF-κB luciferase reporter assay in the presence or absence of PEITC or PDTC (n = 3). **P < 0.01, ***P < 0.001, ****P < 0.0001. G: indirect immunofluorescent staining using p65 was performed to demonstrate p65 NF-κB localization; 4′,6-diaminido-2-phenylindole was used to stain cell nuclei. Scale bar, 50 μm.
To examine the effect of PEITC and PDTC on NF-κB activity, we transfected cyp1b1−/− cells with NF-κB luciferase reporter plasmids and then incubated the cells with PEITC and PDTC for 24 h, as described in materials and methods. PEITC and PDTC inhibited NF-κB activity in cyp1b1−/− retinal PC and EC (Fig. 2, E and F). We observed a 1.5-fold decrease with 1 μM PEITC and a 2-fold decrease with 10 nM PDTC in retinal EC. In retinal PC, 1 μM PEITC and 10 nM PDTC decreased NF-κB activity by 25% and 33%, respectively. Using immunofluorescence staining, we next investigated the localization and expression of total p65. We observed increased nuclear and cytoplasmic staining of p65 in cyp1b1−/− EC and PC compared with wild-type vascular cells, consistent with our Western blot analysis. Total p65 protein levels decreased after incubation with PEITC and PDTC in cyp1b1−/− vascular cells (Fig. 2G). Hence, PEITC and PDTC inhibited the sustained p65 NF-κB activity and total expression in cyp1b1−/− EC and PC.
Inhibition of NF-κB alters apoptosis in cyp1b1−/− retinal EC.
Many studies using various cancer cell lines have demonstrated the potential of PEITC and PDTC to inhibit proliferation through cell cycle arrest and induction of apoptosis (21, 23, 49). We previously demonstrated that loss of Cyp1B1 in retinal vascular cells confers resistance to apoptosis by decreasing caspase-3/7 activation. We determined the concentration range of PEITC (100–500 nM) and PDTC (50 nM-1 μM) cytotoxicity in cyp1b1−/− retinal vascular cells (not shown). Final concentrations were chosen on the basis of cell viability results after 24 h of exposure and the ability to inhibit NF-κB p65 levels. In retinal EC, 50 nM PDTC exhibited an ∼20% decrease in cell viability, while PEITC did not have an impact. In retinal PC, increasing concentrations of PEITC and PDTC decreased cell viability, with the highest concentration decreasing viability by ∼20%. Unless otherwise stated, 1 μM PEITC and 10 nM PDTC were used for all experiments. Apoptotic cell death was determined by evaluation of the activation status of caspase-3/7. In Fig. 3A, cyp1b1−/− retinal EC exhibited a decrease in basal caspase-3/7 activity compared with cyp1b1+/+ cells that was restored by PEITC and PDTC; cyp1b1−/− PC also exhibited less caspase-3/7 activity than cyp1b1+/+ cells, but these inhibitors did not restore caspase-3/7 levels in these cells (Fig. 3B).
Fig. 3.

Inhibition of NF-κB alters apoptosis in retinal EC. A and B: rate of apoptosis was determined by measuring caspase activity with luminescent signal from caspase-3/7 DEVD-aminoluciferin substrate. In A, cyp1b1−/− EC demonstrated an ∼2-fold decrease in basal levels of caspase-3/7 and a 1.5-fold increase when incubated with PEITC or PDTC. In B, cyp1b1−/− PC demonstrated a 3.5-fold decrease in basal levels of caspase-3/7 and no change when incubated with PEITC or PDTC. **P < 0.01, ***P < 0.001; #P < 0.05 vs. cyp1b1−/− (control). C and E: cyp1b1−/− EC (C) and PC (E) were incubated with PEITC (500 nM and 1 μM) or PDTC (5, 10, and 50 nM), and lysates were analyzed by Western blot analysis for phosphorylated Akt (p-Akt), total Akt, and β-actin. D and F: quantitative assessment of phosphorylated Akt relative to total Akt (n = 2). *P < 0.05.
To further confirm our observations, we examined the phosphorylation status of Akt by Western blot analysis. Akt, a serine/threonine-specific kinase, is a regulator of multiple downstream processes, including apoptosis and cell proliferation. We previously reported that cyp1b1−/− EC and PC display increased proliferation, which may in part be due to significant increases in phosphorylated Akt signaling. The cyp1b1−/− retinal EC and PC displayed increased Akt phosphorylation on Ser473. Quantification of Akt phosphorylation showed an initial, not statistically significant, decrease after PEITC and PDTC incubation in cyp1b1−/− EC (Fig. 3D). We did not observe this decrease in PC, thereby confirming the caspase-3/7 results (Fig. 3E). Thus our results suggest a cell-specific role for PEITC and PDTC in apoptosis.
Restoration of capillary morphogenesis and migration in cyp1b1−/− vascular cells.
Angiogenesis is a fundamental process in vascular development and remodeling during tissue injury. Formation of capillary-like structures is an important feature of EC that distinguishes them from other cell types. Most EC are able to differentiate and organize into a capillary-like network in Matrigel. We previously reported that loss of Cyp1B1 in retinal EC results in attenuation of capillary morphogenesis in Matrigel. To investigate whether sustained activation of NF-κB contributes to attenuation of capillary morphogenesis, cyp1b1−/− retinal EC were incubated with PEITC or PDTC. Both inhibitors were able to restore capillary morphogenesis in cyp1b1−/− retinal EC (Fig. 4A). Quantitative assessment of the mean number of branch points is shown in Fig. 4B. Further confirmation of the role of NF-κB in capillary morphogenesis was observed in coculture models using retinal EC and PC (Fig. 4C). The cyp1b1−/− EC and PC, similar to cyp1b1−/− EC alone, showed attenuation of capillary morphogenesis (Fig. 4C, image c). Incubation with PEITC and PDTC restored capillary morphogenesis (Fig. 4C, images d and e). Addition of wild-type PC to cyp1b1−/− EC also restored capillary morphogenesis (Fig. 4C, image b), indicating a cell autonomous role for PC.
Fig. 4.

NF-κB suppression restores capillary morphogenesis and migration. A: cyp1b1+/+ and cyp1b1−/− retinal EC were incubated with PEITC and PDTC, plated on Matrigel, and photographed after 18 h. Scale bar, 100 μm. B: mean number of branch points from 5 high-power (×40) fields (n = 3). ****P < 0.0001. C: capillary morphogenesis of retinal EC and PC was assessed by coculturing cells in Matrigel for 18 h. Representative images show cyp1b1+/+ EC + PC (a), cyp1b1−/− EC + cyp1b1+/+ PC (b), cyp1b1−/− EC + cyp1b1−/− PC (c), cyp1b1−/− EC + cyp1b1−/− PC + 1 μM PEITC (d), and cyp1b1−/− EC + cyp1b1−/− PC + 10 nM PDTC (e). Scale bar, 500 μm. D: mean number of branch points (n = 3). **P < 0.01, ****P < 0.0001. E and F: Transwell migration assays after 24 h of incubation of cyp1b1+/+ and cyp1b1−/− EC (E) and PC (F) with PEITC and PDTC (n = 3). Note 30% decrease in migration of cyp1b1−/− compared with cyp1b1+/+ EC, 20% increase in migration of cyp1b1−/− EC incubated with PEITC and PDTC, 75% increase in migration of cyp1b1−/− compared with cyp1b1+/+ PC, 40% decrease in migration of cyp1b1−/− PC incubated with PEITC, and 33% decrease in migration of cyp1b1−/− PC incubated with PDTC. **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.05 vs. cyp1b1−/− (control).
Proliferation and migration of EC is crucial to formation of patent vessels. The ability of PC to migrate and embed into the vascular basement membrane of the endothelium is also essential for the maturation and stabilization of newly forming vessels. We previously showed that loss of Cyp1B1 has differential effects in both EC and PC phenotypes. To provide evidence for NF-κB involvement in migration, cyp1b1−/− vascular cells were exposed to PEITC or PDTC for 24 h, and their migration was assessed using a Transwell migration assay. The cyp1b1−/− EC were less migratory, and their migration was restored by incubation with PEITC and PDTC (Fig. 4E). Differentially, cyp1b1−/− PC displayed increased migration compared with cyp1b1+/+ cells. PEITC and PDTC decreased the rate of migration in cyp1b1−/− PC, comparable with cyp1b1+/+ cells (Fig. 4F). These results are consistent with restoration of capillary morphogenesis of cyp1b1−/− EC and PC incubated with these compounds.
PEITC and PDTC alleviate increased ROS in cyp1b1−/− vascular cells.
Many vascular pathologies and subsequent diseases present with alterations in the cellular oxidative state. Studies suggest a link between increased ROS and NF-κB activity (5). Cyp1B1 catalyzes oxidation and reduction reactions that may play a significant role in modulation of the cellular reductive state. Because many studies have demonstrated the antioxidant functionality of PEITC and PDTC, we next investigated whether PEITC and PDTC decreased intracellular accumulation of ROS in cyp1b1−/− cells. Using DHE staining, we analyzed production of ROS in the basal state and during incubation with PEITC and PDTC. Retinal EC and PC exhibited increased fluorescence, indicating increased ROS production, as previously reported (31, 39). Incubation with PEITC and PDTC decreased staining in cyp1b1−/− EC and PC by approximately twofold, indicating reduced levels of ROS (Fig. 5, A and C). Quantitative assessment of the data is shown in Fig. 5, B and D.
Fig. 5.

Abrogation of oxidative stress in cyp1b1−/− vascular cells incubated with PEITC and PDTC. A and C: oxidative stress was measured by dihydroethidium staining of cyp1b1+/+ and cyp1b1−/− EC (A) and PC (C) after 24 h of incubation with PEITC and PDTC. Representative images are shown. Scale bar, 50 μm. B and D: quantitative assessment of mean fluorescence intensity in A and C, respectively (n = 3). ****P < 0.0001; #P < 0.05 vs. cyp1b1−/− (control).
Inhibition of NF-κB does not alter TSP2 levels.
TSP2 is a large homotrimeric multidomain glycoprotein that modulates cell adhesion, proliferation, migration, and apoptosis with antiangiogenic activity (4). TSP2-null mice display obvious abnormalities in the extracellular matrix of skin, tendons, and ligaments and also exhibit an altered foreign body response, increased inflammation and extracellular matrix deposition, and leakage of the blood-brain barrier (22). Despite the plethora of growing knowledge regarding the function of TSP2, very little is known about its expression during vascular development and pathological angiogenesis. We hypothesized that sustained NF-κB activation is responsible for the increase in TSP2 levels in the cyp1b1−/− retinal vascular cells. To test whether inhibition of NF-κB altered TSP2 levels, Western blot analysis and RT-qPCR were performed after 24 h of incubation of cyp1b1−/− vascular cells with PEITC and PDTC. PEITC and PDTC did not alter TSP2 levels in cell lysates or conditioned medium from cyp1b1−/− EC or PC, respectively (Fig. 6, A and B). We also did not observe significant changes in TSP2 mRNA transcripts (Fig. 6C).
Fig. 6.

Suppression of NF-κB activity does not alter TSP2 levels. A and B: cyp1b1−/− EC and PC were incubated with PEITC or PDTC in serum-free medium for 48 h, and lysates and conditioned medium were collected and evaluated for TSP2 and β-actin expression by Western blot analysis. C: relative TSP2 mRNA level was analyzed using real-time qPCR in cyp1b1+/+ and cyp1b1−/− EC and PC after incubation with PEITC and PDTC (n = 9). ***P < 0.001. D–G: cyp1b1−/− PC were transfected with empty vector or dominant-negative (DN)-mIκBα plasmid for 48 h, and lysates were evaluated for phosphorylated p65, total p65, phosphorylated IκBα (p-IκBα), total IκBα, phosphorylated RelB (p-RelB), total RelB, p105/p50, p100/p52, and β-actin. WT, cyp1b1+/+; Vector, cyp1b1−/− empty vector; IκBα, cyp1b1−/− DN-mIκBα plasmid. H: cyp1b1−/− PC were transfected with empty vector or DN-mIκBα plasmid for 48 h and incubated in serum-free medium for 48 h. Lysates and conditioned medium were collected and evaluated for TSP2 and β-actin expression by Western blot analysis. Note lack of effect on TSP2 level in cyp1b1−/− PC expressing DN-mIκBα compared with vector control. I: cyp1b1−/− EC were transfected with empty vector or DN-mIκBα plasmid for 48 h. Capillary morphogenesis was determined by plating transfected cells on Matrigel and capturing images in digital format after 18 h. Representative images are shown. J: mean number of branch points (n = 3). ****P < 0.0001; #P < 0.05 vs. cyp1b1−/− (control). K: oxidative stress was measured by dihydroethidium (DHE) staining in cyp1b1+/+ and cyp1b1−/− EC and PC 48 h posttransfection using empty vector (cyp1b1−/−) or DN-mIκBα plasmid (IκBα). Representative images are shown. Scale bar, 50 μm. L: quantitative assessment of mean fluorescence intensity (n = 3). Note lack of effect on oxidative stress levels in cells expressing DN-mIκBα compared with control cells. ****P < 0.0001.
To further analyze the contribution of NF-κB to enhanced TSP2 levels, we used an IκBα mutant (DN-mIκBα) in which Ser32/36 is mutated to prevent phosphorylation at these residues, suppressing NF-κB activation in cyp1b1−/− EC and PC. This allowed us to solely consider the contribution of sustained NF-κB activation to TSP2 expression without considering other modes of PEITC and PDTC action. Retinal EC and PC were transfected with DN-mIκBα or empty vector (control) for 48 h, and Western blot analysis was used to assess NF-κB suppression in the lysates. We observed significant downregulation of phosphorylated p65 NF-κB in EC and PC (only PC data are shown for simplicity; Fig. 6D). We also analyzed other NF-κB family members, including RelB, p105/p50 and p100/p52, and IκBα. We observed decreased phosphorylated (Ser32) IκBα, decreased phosphorylated (Ser552) RelB, and decreased p105, as expected, after inhibition of IκBα (Fig. 6, E–G). Levels of p100 were very low in these cells. Western blot analysis of cell lysates and conditioned medium from cells transfected with DN-mIκBα did not reveal alterations in TSP2 levels (Fig. 6H). Thus NF-κB activity does not impact TSP2 expression.
To investigate if suppression of NF-κB by expression of DN-mIκBα also restores capillary morphogenesis, cyp1b1−/− retinal EC were transfected for 48 h and subsequently plated in Matrigel for a tube formation assay (Fig. 6I). We observed a twofold increase in capillary morphogenesis in retinal EC following transfection with the plasmid vs. control (Fig. 6J). Thus suppression of NF-κB activation in the cyp1b1−/− retinal EC restored capillary morphogenesis, similar to that observed with the NF-κB inhibitors PEITC and PDTC.
To determine whether sustained activation of NF-κB in the cyp1b1−/− retinal vascular cells contributes to the increased ROS, retinal EC and PC were transfected with DN-mIκBα for 48 h and stained using DHE. Suppression of NF-κB activation did not decrease oxidative stress in cyp1b1−/− retinal EC or PC (Fig. 6, K and L). Thus NF-κB activation is downstream of ROS and TSP2 expression.
TSP2 knockdown in cyp1b1−/− retinal EC and PC restores capillary morphogenesis but does not decrease oxidative stress.
To demonstrate that changes in cell functions described above are due specifically to the increase in TSP2 expression, we generated stable cyp1b1−/− retinal EC and PC in which TSP2 was effectively knocked down using lentiviral siRNA (>75%). Figure 7, A and B, shows that these viruses effectively knocked down TSP2 expression in cyp1b1−/− EC and PC. Clones 400 and 402 were chosen for negative and positive controls for retinal EC, respectively, while clones 966 and 402 were chosen as negative and positive controls for retinal PC, respectively. We next determined whether downregulation of TSP2 in cyp1b1−/− retinal EC restored capillary morphogenesis. Figure 7C shows that decreased TSP2 expression in cyp1b1−/− EC significantly improved their ability to undergo capillary morphogenesis (Fig. 7C, image c). Cells in which TSP2 knockdown was minimal were used as control, and these cells did not undergo capillary morphogenesis, as expected (Fig. 7C, image b). A similar result was previously observed in cyp1b1−/− EC with use of transient knockdown of TSP2 (39).
Fig. 7.

TSP2 knockdown in cyp1b1−/− EC and PC restores capillary morphogenesis but does not alleviate oxidative stress. A and B: stable lines of cyp1b1−/− EC (A) and PC (B) were generated by lentiviral small interfering RNA (siRNA) knockdown of TSP2. For EC, clone 400 was chosen as a negative control and clone 402 was chosen as a positive control for TSP2 knockdown. For PC, clone 966 was chosen as a negative control and clone 402 was chosen as a positive for TSP2 knockdown. Note decrease in TSP2 levels in clones 402 for EC and PC (>75% knockdown) compared with parental cells and those expressing a siRNA inefficient in TSP2 knockdown. C: capillary morphogenesis determined by plating TSP2 siRNA knockdown EC on Matrigel. Images were captured in digital format after 18 h. Representative images show cyp1b1+/+ EC (a), cyp1b1−/− EC clone 400 (b), and cyp1b1−/− EC clone 402 (c). Scale bar, 100 μm. For quantification, mean number of branch points was counted (n = 3). ****P < 0.0001; #P < 0.05 vs. cyp1b1−/− (control). D: oxidative stress was measured by DHE staining in cyp1b1+/+, cyp1b1−/− control, and cyp1b1−/− TSP2 siRNA knockdown EC and PC. Representative images are shown. Note no effect on DHE staining of cyp1b1−/− infected with TSP2 specific siRNAs compared with cyp1b1−/− control.
Given the nature of reactions catalyzed by Cyp1B1, we previously demonstrated that loss of Cyp1B1 increased ROS. However, it is unknown whether TSP2 plays a role during oxidative stress in these cells. Knockdown of TSP2 in cyp1b1−/− EC and PC did not decrease ROS production determined by DHE staining. Representative images are shown in Fig. 7D. It is plausible that ROS are produced from products of cellular respiration that are not removed due to lack of Cyp1B1 and, further, that TSP2 status does not impact ROS generation. Thus increased TSP2 expression may be independent of ROS production as a result of Cyp1B1 deficiency.
Enhanced TSP2 expression activates the NF-κB pathway.
Whether a relationship exists between TSP2 expression and sustained NF-κB activation is unknown. In the absence of Cyp1B1, we hypothesized that TSP2 upregulation may constitutively activate the NF-κB pathway, thereby promoting the antiangiogenic state. We observed decreased p65 NF-κB phosphorylation in the stable TSP2 knockdown EC and PC by Western blot analysis (Fig. 8, A and B). Using indirect immunofluorescence, we showed that p65 expression was decreased in the cyp1b1−/− TSP2 knockdown cells compared with the cyp1b1−/− vascular cells (not shown). To demonstrate TSP2 activation of the NF-κB pathway, we used adenoviruses encoding TSP2 cDNA or GFP to overexpress mouse TSP2 in cyp1b1+/+ retinal EC and PC. Figure 8C shows that this virus effectively overexpressed TSP2 in retinal EC and PC. We first demonstrated that overexpression in retinal EC changes eNOS expression in wild-type retinal EC. We previously showed decreased eNOS expression in cyp1b1−/− retinal EC (38). Figure 8D shows that overexpression of TSP2 decreased eNOS and phosphorylated (Ser1177) eNOS levels. Using Western blot analysis, we next determined whether overexpression of TSP2 affects phosphorylation of p65NF-κB and increases p105/p50. Figure 8, E and F, shows increased p65 phosphorylation (Ser311) following expression of TSP2 in wild-type retinal EC. Similarly, we showed increased p65 phosphorylation as well as p105/p50 after expressing TSP2 in cyp1b1+/+ PC (Fig. 8, G and H). Thus TSP2 levels may directly affect activation of the NF-κB pathway.
Fig. 8.

TSP2 levels and NF-κB activation are linked. A and B: cyp1b1−/− EC (A) and PC (B) infected with TSP2-specific siRNA were lysed and analyzed for phosphorylated and total p65 by Western blot analysis; β-actin was used as a loading control. Note decreased levels of phosphorylated p65 in cyp1b1−/− [knockout (KO)] EC and PC expressing TSP2-specific siRNA clone 402 with reduced TSP2 level compared with parental cells or those expressing siRNA clone 400 or 966. Also note decreased levels of phosphorylated p65 NF-κB in cyp1b1−/− TSP2 knockdown cells expressing siRNA clone 402 compared with parental control or siRNA clone 400 and 966. C: cyp1b1+/+ EC and PC were transfected using TSP2 expressing adenoviruses. After transfection, conditioned medium and lysates were prepared from EC and PC and analyzed for TSP2 by Western blot analysis. WT, cyp1b1+/+ cells infected with GFP-adenovirus; TSP2, cyp1b1+/+ cells infected with TSP2 adenovirus; KO, cyp1b1−/− cells. D–F: Western blot analysis of phosphorylated endothelial nitric oxide synthase (eNOS), total eNOS, phosphorylated and total p65, and p105/p50 in cyp1b1+/+ EC. Note reduced level of eNOS and phosphorylated eNOS in cyp1b1+/+ cells expressing TSP2 compared with control cells. G and H: Western blot analysis of phosphorylated and total p65 and p105/p50 in cyp1b1+/+ PC expressing TSP2. Note increased phosphorylated p65 NF-κB in cyp1b1+/+ PC expressing TSP2 compared with control.
DISCUSSION
The studies presented here establish a novel relationship between TSP2 expression and NF-κB activation as regulators of angiogenesis. The major focus of the present study was to determine the role of NF-κB and TSP2 in vascular defects associated with Cyp1B1 deficiency. We previously demonstrated that cyp1b1−/− retinal vascular cells display increased oxidative stress, sustained NF-κB activation, and increased TSP2 levels, which result in attenuation of angiogenesis in vivo and altered vascular cell function in vitro. Here we show that Cyp1b1 deficiency results in increased ROS and TSP2 levels in retinal vascular cells upstream of NF-κB. Our studies demonstrate a novel role for TSP2 in the activation of NF-κB, independent of increased oxidative stress, and attenuation of angiogenesis and altered vascular cell function.
NF-κB is a ubiquitous, heterodimeric transcription factor that plays pivotal roles in survival pathways, chemotaxis, and inflammation (1, 2). In quiescent cells, NF-κB is sequestered in the cytosol by the IκB proteins. Phosphorylation of IκB by the IκB kinase family of proteins leads to proteasome-mediated degradation, thereby allowing NF-κB nuclear translocation and subsequent activation of transcription. Angiogenesis is a complex process requiring the coordinated regulation of many pro- and antiangiogenic factors, as well as the interplay between EC and PC. The activation of NF-κB has long been implicated in the dysregulation of angiogenesis, and current therapeutic approaches include use of pharmacological inhibition of NF-κB, including isothiocyanates and dithiocarbamates.
In this study we used the pharmacological inhibitors PEITC and PDTC to inhibit NF-κB, and we addressed the effects of PEITC and PDTC on capillary morphogenesis, migration, oxidative stress, and TSP2 expression in the cyp1b1−/− vascular cell. We demonstrated that PEITC and PDTC inhibited p65 NF-κB activity and reduced total p65 expression in cyp1b1−/− retinal EC and PC and improved capillary morphogenesis and migration, while abrogating oxidative stress in these cells. We showed differential effects of these inhibitors on cyp1b1−/− retinal EC and PC, demonstrating that sustained NF-κB activation affects the proangiogenic properties in a cell-specific manner.
One of the unclear phenotypic evaluations that remains to be resolved is the increase in cellular apoptosis through activation of caspase-3/7 in the cyp1b1−/− EC, but not PC, incubated with PEITC or PDTC. It is unclear which pathways contribute to the enhanced cell survival and how these pathways, which are potentially responsible for activation of caspase-3/7 and subsequent apoptosis, are interdependent. We previously demonstrated increased phosphorylated Akt in cyp1b1−/− vascular cells. Further evaluation of intracellular signaling pathways demonstrated that PEITC or PDTC did not alter the Akt signaling pathway in either cell type. Thus PEITC and PDTC incubation may affect other signaling pathways that influence the survival of retinal EC. In a model of Alzheimer's disease, PDTC was observed to activate Akt. However, this was not associated with changes in NF-κB activity or oxidative stress. Thus Akt activation may differ significantly among these models (25). We previously showed that proapoptotic protein Bax mRNA and protein expression is decreased in the cyp1b1−/− PC. However, incubation of these cells with PEITC or PDTC did not alter Bax mRNA levels (not shown). These results suggest that complex cell type-specific mechanisms are involved, and their identities are the subject of future investigation.
Restoration of capillary morphogenesis in cyp1b1−/− retinal EC and PC may be due to abrogation of oxidative stress, as we previously showed that the antioxidant N-acetylcysteine restores the ability of cyp1b1−/− retinal EC to undergo capillary morphogenesis (39). Another potential mechanism for restoration of capillary morphogenesis may reside with eNOS and NO production. We previously showed that loss of Cyp1B1 is associated with decreased eNOS expression and NO production (38). We were unable to demonstrate a clear increase in eNOS signaling or NO production after incubation with PEITC or PDTC. We also found that inhibition of NF-κB expression and activity did not alter TSP2 expression. MacLauchlan et al. (24) showed that TSP2 is modulated by NO. The eNOS knockout mice exhibit increased TSP2 levels in cutaneous wounds and hindlimb ischemia. The in vitro studies of MacLauchlan et al. confirmed that NO represses TSP2 promoter activity. We recently demonstrated that loss of Cyp1B1 in EC decreases eNOS expression and increases TSP2 (38). However, reexpression of eNOS or use of a NO donor in cyp1b1−/− EC was not sufficient to decrease the TSP2 level. Retinal PC do not express eNOS and/or produce NO under normal conditions. How TSP2 becomes upregulated in PC and the role of TSP2 in the attenuation of their proangiogenic properties are unclear.
Appropriate migration of vascular cells is critical to the creation and stabilization of patent vessels. Loss of Cyp1B1 differentially impacted the migration of retinal EC and PC, resulting in decreased and increased migration, respectively. Interestingly, PEITC and PDTC restored appropriate migration in each of these cell types. PEITC was shown to have antimetastatic effects in many studies using in vitro cancer models, including prostate and gastric cell lines, by inhibiting focal adhesion kinase, RhoA, matrix metalloproteinase (MMP-9) expression, and a plethora of signaling molecules and pathways (21, 23, 49). This evidence substantiates the effects of PEITC in our cyp1b1−/− PC, in which we saw reduced migration; however, the mechanism through which this occurs and how PEITC increases migration of cyp1b1−/− EC are unclear.
Since PEITC and PDTC are potent inhibitors of NF-κB activation in vitro, as demonstrated by many studies, the changes we demonstrate suggest that NF-κB activation in part is responsible. However, other mechanisms of action for these pharmacological inhibitors have also been described (6, 34, 36, 44, 46, 50). Other investigators have demonstrated functions for PEITC and PDTC that include metal chelation and antioxidant inhibition of NF-κB (6, 34, 36, 44, 46, 50). Several studies investigating the mechanisms of PDTC revealed that it is the prooxidant properties, rather than any antioxidant effects, that are responsible for the inhibition of NF-κB (6). Other reports also describe a prooxidant effect that is responsible for generating increased oxidative stress (30). Inhibition of NF-κB decreased ROS levels in the cyp1b1−/− retinal EC and PC; however, whether this is due to inhibition of NF-κB or solely to the antioxidant properties of these compounds warrants further investigation using other methods to inhibit NF-κB.
Using a DN repressor of NF-κB activation to exclude the contribution of multiple intracellular signaling pathways, we performed additional studies that examined the contribution of NF-κB (26, 45). Transfection of this repressor resulted in decreased NF-κB activation in cyp1b1−/− retinal PC and EC, as well as decreased levels of phosphorylated RelB, p105/p50, p100/p52, and phosphorylated IκBα, as expected (Fig. 6, D–G; retinal EC data not shown for simplicity). We were also unable to demonstrate that activation of NF-κB contributes to the increased ROS in these cells. These observations provide further evidence that the decrease in ROS is not due to the inhibition of NF-κB by PEITC and PDTC, but by the antioxidant properties of these two compounds. Although we observed restoration of capillary morphogenesis, similar to the NF-κB inhibitors, we did not observe a change in TSP2 protein levels (Fig. 6H). Thus modulation of TSP2 expression is independent of NF-κB activation.
To further elucidate the role of TSP2, we knocked down TSP2 expression in cyp1b1−/− retinal EC and PC. The knockdown studies revealed restoration of capillary morphogenesis but no change in the ROS levels in the cells (Fig. 7, C and D). These results suggest that TSP2 does not influence the oxidative state of the cell but, rather, our initial hypothesis that loss of Cyp1B1 enzymatic activity results in ROS accumulation. We further demonstrated that TSP2 knockdown results in a decrease in NF-κB activation in the absence of Cyp1B1. Furthermore, expression of TSP2 in cyp1b1+/+ retinal EC and PC confirmed our hypothesis that TSP2 expression is sufficient to, directly or indirectly, activate the NF-κB pathway. A direct κB binding site has not been identified in the TSP2 promoter region. There is very little evidence to suggest a link between NF-κB and TSP2. De Stefano et al. (9) investigated the role of NF-κB in the angiogenic and fibrogenic response induced by λ-carrageenin in a rat model of chronic inflammation. They showed that inhibition of NF-κB/DNA binding activity downregulated cyclooxygenase-2, MMP-9, and TNF-α. Blockage of NF-κB activity prevented the development of granulation tissue induced by a λ-carrageenin-soaked sponge implant by upregulating Bax, TSP1, and TSP2 expression. This study suggested that TSP2 is a gene target of NF-κB; however, we were unable to demonstrate that TSP2 is a direct transcriptional target of NF-κB but, rather, that TSP2 influences NF-κB activity in our retinal vascular cells.
In summary, we show that sustained activation of NF-κB results in altered proangiogenic properties of retinal EC and PC. Furthermore, enhanced TSP2 expression contributes to the activation of NF-κB in the cyp1b1−/− vascular cells, and impeding the expression of endogenous angiogenesis inhibitor TSP2 restores angiogenesis. Thus targeting of TSP2 may provide a method to control NF-κB activation and its associated vascular dysfunctions.
GRANTS
This work was supported by National Institutes of Health Grants R01 EY-016995, R01 EY-018179, RC4 EY-021357, T32 ES-007015, and P30 EY-016665, National Cancer Institute University of Wisconsin Paul P. Carbone Cancer Center Support Grant P30 CA-014520, and an unrestricted departmental award from Research to Prevent Blindness. N. Sheibani is a recipient of American Diabetes Association and Retina Research Foundation Research Award 1-10-BS-160. C. M. Sorenson is supported by American Heart Association Grant 0950057G. T. L. Palenski is the recipient of National Eye Institute Kirschstein-National Research Service Award Fellowship F31 EY-021091 and a Science and Medicine Graduate Research Scholar at the University of Wisconsin-Madison.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.L.P., C.M.S., and N.S. are responsible for conception and design of the research; T.L.P., Z.G., C.M.S., and K.D.H. performed the experiments; T.L.P., Z.G., C.M.S., and N.S. analyzed the data; T.L.P., Z.G., C.M.S., and N.S. interpreted the results of the experiments; T.L.P. prepared the figures; T.L.P. drafted the manuscript; T.L.P., C.M.S., K.D.H., and N.S. edited and revised the manuscript; T.L.P., Z.G., C.M.S., K.D.H., and N.S. approved the final version of the manuscript.
REFERENCES
- 1. Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell 87: 13–20, 1996 [DOI] [PubMed] [Google Scholar]
- 2. Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol 12: 141–179, 1994 [DOI] [PubMed] [Google Scholar]
- 3. Bernauer U, Heinrich-Hirsch B, Tönnies M, Peter-Matthias W, Gundert-Remy U. Characterisation of the xenobiotic-metabolizing cytochrome P450 expression pattern in human lung tissue by immunochemical and activity determination. Toxicol Lett 164: 278–288, 2006 [DOI] [PubMed] [Google Scholar]
- 4. Bornstein P, Armstrong LC, Hankenson KD, Kyriakides TR, Yang Z. Thrombospondin 2, a matricellular protein with diverse functions. Matrix Biol 19: 557–568, 2000 [DOI] [PubMed] [Google Scholar]
- 5. Bowie A, O'Neill LA. Oxidative stress and nuclear factor-κB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol 59: 13–23, 2000 [DOI] [PubMed] [Google Scholar]
- 6. Brennan P, O'Neill LA. 2-Mercaptoethanol restores the ability of nuclear factor κB (NFκB) to bind DNA in nuclear extracts from interleukin 1-treated cells incubated with pyrrolidine dithiocarbamate (PDTC). Evidence for oxidation of glutathione in the mechanism. Biochem J 320: 975–981, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chambers D, Wilson L, Maden M, Lumsden A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 134: 1369–1383, 2007 [DOI] [PubMed] [Google Scholar]
- 8. Chung FL, Juchatz A, Vitarius J, Hecht SS. Effects of dietary compounds on α-hydroxylation of N-nitrosopyrrolidine and N′-nitrosonornicotine in rat target tissues. Cancer Res 44: 2924–2928, 1984 [PubMed] [Google Scholar]
- 9. De Stefano D, Nicolaus G, Maiuri MC, Cipolletta D, Galluzzi L, Cinelli MP, Tajana G, Iuvone T, Carnuccio R. NF-κB blockade upregulates Bax, TSP-1, and TSP-2 expression in rat granulation tissue. J Mol Med 87: 481–492, 2009 [DOI] [PubMed] [Google Scholar]
- 10. DeBusk LM, Massion PP, Lin PC. IκB kinase-α regulates endothelial cell motility and tumor angiogenesis. Cancer Res 68: 10223–10228, 2008 [DOI] [PubMed] [Google Scholar]
- 11. Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol 48: 331–341, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doshi M, Marcus C, Bejjani BA, Edward DP. Immunolocalization of CYP1B1 in normal, human, fetal and adult eyes. Exp Eye Res 82: 24–32, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Fleming I. Cytochrome P450 and vascular homeostasis. Circ Res 89: 753–762, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Gauthier KM, Yang W, Gross GJ, Campbell WB. Roles of epoxyeicosatrienoic acids in vascular regulation and cardiac preconditioning. J Cardiovasc Pharmacol 50: 601–608, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Gong A, He M, Krishna Vanaja D, Yin P, Karnes RJ, Young CY. Phenethyl isothiocyanate inhibits STAT3 activation in prostate cancer cells. Mol Nutr Food Res 53: 878–886, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IκBα controls cytoplasmic localization of inactive NF-κB/IκBα complexes. Proc Natl Acad Sci USA 97: 1014–1019, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang TT, Wuerzberger-Davis SM, Seufzer BJ, Shumway SD, Kurama T, Boothman DA, Miyamoto S. NF-κB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J Biol Chem 275: 9501–9509, 2000 [DOI] [PubMed] [Google Scholar]
- 18. Iruela-Arispe ML, Liska DJ, Sage EH, Bornstein P. Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev Dyn 197: 40–56, 1993 [DOI] [PubMed] [Google Scholar]
- 19. Jennings BL, Anderson LJ, Estes AM, Yaghini FA, Fang XR, Porter J, Gonzalez FJ, Campbell WB, Malik KU. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension 59: 348–354, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kerzee JK, Ramos KS. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: role of the Ahr bHLH/PAS transcription factor. Circ Res 89: 573–582, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Kim SH, Sehrawat A, Sakao K, Hahm ER, Singh SV. Notch activation by phenethyl isothiocyanate attenuates its inhibitory effect on prostate cancer cell migration. PLos One 6: e26615, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CE, Ginns EI, Bornstein P. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol 140: 419–430, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lai KC, Hsu SC, Kuo CL, Ip SW, Yang JS, Hsu YM, Huang HY, Wu SH, Chung JG. Phenethyl isothiocyanate inhibited tumor migration and invasion via suppressing multiple signal transduction pathways in human colon cancer HT29 cells. J Agric Food Chem 58: 11148–11155, 2010 [DOI] [PubMed] [Google Scholar]
- 24. MacLauchlan S, Yu J, Parrish M, Asoulin TA, Schleicher M, Krady MM, Zeng J, Huang PL, Sessa WC, Kyriakides TR. Endothelial nitric oxide synthase controls the expression of the angiogenesis inhibitor thrombospondin 2. Proc Natl Acad Sci USA 108: E1137–E1145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, Salminen A, Auriola S, Van Groen T, Tanila H, Koistinaho J. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting β-amyloid burden. J Neurosci 27: 3712–3721, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miyamoto S, Chiao PJ, Verma IM. Enhanced IκBα degradation is responsible for constitutive NF-κB activity in mature murine B-cell lines. Mol Cell Biol 14: 3276–3282, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyamoto S, Seufzer BJ, Shumway SD. Novel IκBα proteolytic pathway in WEHI231 immature B cells. Mol Cell Biol 18: 19–29, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murray GI, Melvin WT, Greenlee WF, Burke MD. Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu Rev Pharmacol Toxicol 41: 297–316, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos 29: 1110–1113, 2001 [PubMed] [Google Scholar]
- 30. Nobel CS, Burgess DH, Zhivotovsky B, Burkitt MJ, Orrenius S, Slater AF. Mechanism of dithiocarbamate inhibition of apoptosis: thiol oxidation by dithiocarbamate disulfides directly inhibits processing of the caspase-3 proenzyme. Chem Res Toxicol 10: 636–643, 1997 [DOI] [PubMed] [Google Scholar]
- 31. Palenski TL, Sorenson CM, Jefcoate CR, Sheibani N. Lack of Cyp1b1 promotes the proliferative and migratory phenotype of perivascular supporting cells. Lab Invest 93: 646–662, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ragavan N, Hewitt R, Cooper LJ, Ashton KM, Hindley AC, Nicholson CM, Fullwood NJ, Matanhelia SS, Martin FL. CYP1B1 expression in prostate is higher in the peripheral than in the transition zone. Cancer Lett 215: 69–78, 2004 [DOI] [PubMed] [Google Scholar]
- 33. Scheef EA, Sorenson CM, Sheibani N. Attenuation of proliferation and migration of retinal pericytes in the absence of thrombospondin-1. Am J Physiol Cell Physiol 296: C724–C734, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J Exp Med 175: 1181–1194, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shitaye HS, Terkhorn SP, Combs JA, Hankenson KD. Thrombospondin-2 is an endogenous adipocyte inhibitor. Matrix Biol 29: 549–556, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Snyder JG, Prewitt R, Campsen J, Britt LD. PDTC and Mg132, inhibitors of NF-κB, block endotoxin induced vasodilation of isolated rat skeletal muscle arterioles. Shock 17: 304–307, 2002 [DOI] [PubMed] [Google Scholar]
- 37. Takemura H, Itoh T, Yamamoto K, Sakakibara H, Shimoi K. Selective inhibition of methoxyflavonoids on human CYP1B1 activity. Bioorg Med Chem 18: 6310–6315, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Tang Y, Scheef EA, Gurel Z, Sorenson CM, Jefcoate CR, Sheibani N. CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am J Physiol Cell Physiol 298: C665–C678, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, Sheibani N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 113: 744–754, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taylor DK, Meganck JA, Terkhorn S, Rajani R, Naik A, O'Keefe RJ, Goldstein SA, Hankenson KD. Thrombospondin-2 influences the proportion of cartilage and bone during fracture healing. J Bone Miner Res 24: 1043–1054, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsuchiya Y, Nakajima M, Kyo S, Kanaya T, Inoue M, Yokoi T. Human CYP1B1 is regulated by estradiol via estrogen receptor. Cancer Res 64: 3119–3125, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Wang M, Liu T, Wang D, Zheng Y, Wang X, He J. Therapeutic effects of pyrrolidine dithiocarbamate on acute lung injury in rabbits. J Transl Med 9: 61, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther 314: 522–532, 2005 [DOI] [PubMed] [Google Scholar]
- 44. Wu CL, Huang AC, Yang JS, Liao CL, Lu HF, Chou ST, Ma CY, Hsia TC, Ko YC, Chung JG. Benzyl isothiocyanate (BITC)- and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J Orthop Res 29: 1199–1209, 2011 [DOI] [PubMed] [Google Scholar]
- 45. Wuerzberger-Davis SM, Chen Y, Yang DT, Kearns JD, Bates PW, Lynch C, Ladell NC, Yu M, Podd A, Zeng H, Huang TT, Wen R, Hoffmann A, Wang D, Miyamoto S. Nuclear export of the NF-κB inhibitor IκBα is required for proper B cell and secondary lymphoid tissue formation. Immunity 34: 188–200, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiao D, Powolny AA, Moura MB, Kelley EE, Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B, Singh SV. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem 285: 26558–26569, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiao D, Zeng Y, Choi S, Lew KL, Nelson JB, Singh SV. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res 11: 2670–2679, 2005 [DOI] [PubMed] [Google Scholar]
- 48. Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev 63: 597–609, 2011 [DOI] [PubMed] [Google Scholar]
- 49. Yang MD, Lai KC, Lai TY, Hsu SC, Kuo CL, Yu CS, Lin ML, Yang JS, Kuo HM, Wu SH, Chung JG. Phenethyl isothiocyanate inhibits migration and invasion of human gastric cancer AGS cells through suppressing MAPK and NF-κB signal pathways. Anticancer Res 30: 2135–2143, 2010 [PubMed] [Google Scholar]
- 50. Zhang JJ, Xu ZM, Zhang CM, Dai HY, Ji XQ, Wang XF, Li C. Pyrrolidine dithiocarbamate inhibits nuclear factor-κB pathway activation, and regulates adhesion, migration, invasion and apoptosis of endometriotic stromal cells. Mol Hum Reprod 17: 175–181, 2011 [DOI] [PubMed] [Google Scholar]
- 51. Zhang L, Savas U, Alexander DL, Jefcoate CR. Characterization of the mouse Cyp1B1 gene. Identification of an enhancer region that directs aryl hydrocarbon receptor-mediated constitutive and induced expression. J Biol Chem 273: 5174–5183, 1998 [DOI] [PubMed] [Google Scholar]