

Abstract
Cerebral oedema is a devastating consequence of acute liver failure (ALF) and may be associated with the development of intracranial hypertension and death. In ALF, some patients may develop cerebral oedema and increased intracranial pressure but progression to life-threatening intracranial hypertension is less frequent than previously described, complicating less than one third of cases who have proceeded to coma since the advent of improved clinical care. The rapid onset of encephalopathy may be dramatic with the development of asterixis, delirium, seizures and coma. Cytotoxic and vasogenic oedema mechanisms have been implicated with a preponderance of experimental data favouring a cytotoxic mechanism. Astrocyte swelling is the most consistent neuropathological finding in humans with ALF and ammonia plays a definitive role in the development of cytotoxic brain oedema. The mechanism(s) by which ammonia induces astrocyte swelling remains unclear but glutamine accumulation within astrocytes has led to the osmolyte hypothesis. Current evidence also supports an alternate ‘Trojan horse’ hypothesis, with glutamine as a carrier of ammonia into mitochondria, where its accumulation results in oxidative stress, energy failure and ultimately astrocyte swelling. Although a complete breakdown of the blood-brain barrier is not evident in human ALF, increased permeation to water and other small molecules such as ammonia has been demonstrated resulting from subtle alterations in the protein composition of paracellular tight junctions. At present, there is no fully efficacious therapy for cerebral oedema other than liver transplantation and this reflects our incomplete knowledge of the precise mechanisms underlying this process which remain largely unknown.
Keywords: Cerebral oedema, Acute liver failure, Ammonia, Hepatic encephalopathy, Intracranial pressure, Intracranial hypertension, Cerebral blood flow
Core tip: Cytotoxic and vasogenic cerebral oedema have been implicated in acute liver failure (ALF) with a preponderance of experimental data favouring cytotoxic mechanisms. Astrocyte swelling is a consistent neuropathological finding in human ALF and ammonia plays a definitive role. The mechanism(s) by which ammonia induces astrocyte swelling remains unclear but glutamine plays a central role inducing oxidative stress, energy failure and ultimately astrocyte swelling. Although complete breakdown of the blood-brain barrier is not evident in human ALF, increased permeation to water and ammonia has been demonstrated. There is no efficacious therapy other than liver transplantation reflecting the incomplete knowledge base.
INTRODUCTION
Acute liver failure (ALF) is a complex clinical syndrome that results from a sudden and severe loss in hepatocyte function in a patient without pre-existing liver disease[1]. This rapid loss of function is the result of massive hepatocyte necrosis and is typically associated with hepatic encephalopathy (HE) and coagulopathy, the hallmark features of ALF. In many cases progressive multi-organ failure ensues. Although ALF is rare, with an incidence of one to six cases per million people every year in the United States and Western Europe, the mortality rate and the cost of treatment is high[2]. The majority of those affected are young adults.
ALF is sometimes referred to as fulminant hepatic failure (FHF), a term first used in 1970 by Trey and Davidson[3] who described a potentially reversible disorder resulting from severe hepatic injury, with an onset of encephalopathy within 8 wk of symptom appearance in the absence of chronic liver disease. Whilst the main features of this definition remain relevant today, O’Grady et al[4] proposed a new classification for adults with ALF, dividing them into three groups based on the time between the onset of jaundice to the development of encephalopathy: hyperacute (within 7 d), acute (8-28 d) and subacute (5-12 wk). This classification recognises that ALF complications and prognosis depend on the rate of evolution of the disorder. It has now gained wide acceptance in clinical and research studies. Those with a hyperacute presentation, such as following an acetaminophen overdose, are at highest risk of developing cerebral oedema.
The most reliable clinical signs of severe ALF include coagulopathy [international normalised ratio (INR) ≥ 1.5], which may become severe enough to cause spontaneous bleeding, and HE (any degree of altered mentation). HE presents with a rapid onset of initially subtle mental alterations such as minor confusion, disorientation and agitation, progressing to delirium, seizures and coma. When severe, HE is typically associated with the development of cerebral oedema[5].
Historically, cerebral oedema was thought to occur in up to 80% of patients with ALF and be the most common cause of death[6]. However, recent data following a review of 3300 patients presenting to a single tertiary liver centre has shown that the proportion of patients with intracranial hypertension (ICH) fell from 76% in 1984-1988 to 20% in 2004-2008 (P < 0.0001). In those who developed ICH, mortality fell from 95% to 55% (P < 0.0001). This mirrored a fall in the admission markers of disease severity and most likely reflects earlier illness recognition, improved intensive care, and use of salvage liver transplantation[7]. A further study from Bernal and colleagues from King’s College Hospital on 165 patients presenting with ALF and grade 3/4 HE found that only 29% showed evidence of ICH. However, only one third had intracranial bolts inserted which raises the possibility that some of this cohort may have developed ICH without showing clinical sequelae. Whether the development of cerebral oedema is similar or higher in patients with ALF in developing countries remains to be determined. Nevertheless, along with sepsis and multi-organ failure, it is one the leading causes of death in these patients[1,8].
Patients with ALF are acutely ill and are best managed in intensive care units within tertiary liver transplant centres. The armamentarium of treatments available to alleviate cerebral oedema include mannitol, hyperventilation, hypertonic sodium chloride, induced hypothermia and barbiturates which aim to decrease the total fluid volume within the brain either by reducing the interstitial fluid and/or by reducing cerebral blood flow[8]. However, at present, no fully efficacious medical therapy for ALF is available and the only effective treatment is an emergency liver transplantation[1]. Nevertheless, liver transplantation is not always an option, with co-morbidities, sepsis, multi-organ failure and graft availability posing a major obstacle to a patient qualifying for a life-saving liver transplant.
The pathophysiological mechanisms underpinning the development of cerebral oedema are complex and remain to be fully unravelled. The central role of ammonia in the pathogenesis of cerebral oedema in ALF however, remains undisputed. Indeed, arterial ammonia concentrations greater than 100 μmol/L have been shown to predict the onset of severe HE with 70% accuracy with ICH developing in 55% of patients with ALF with an arterial ammonia concentration > 200 μmol/L[9]. Furthermore, Clemmesen and colleagues have shown that blood ammonia levels in excess of 150 μmol/L predicted a greater likelihood of dying from brain herniation[10].
CEREBRAL OEDEMA AND ACUTE LIVER FAILURE: AN OVERVIEW
Cerebral oedema is a net increase in total brain water content. The rigid skull bone protecting the brain limits the compliance of the brain and as a consequence a small increase in fluid can cause a significant rise in intracranial pressure. ICH can lead to a decrease in cerebral perfusion pressure and capillary blood flow, culminating in ischaemia[11].
Cerebral oedema as a complication of massive hepatic necrosis was first described by Ware et al[12] in 1971 and was found to be present in 80% of comatose ALF patients on post-mortem examinations. Increased water content of brain tissue has been considered to be a cardinal feature of cerebral oedema in ALF. However, ICH caused by an increase in cerebral blood flow has also been demonstrated in experimental models of ALF[13] in addition to patients with ALF[14]. Impaired autoregulation of CBF is well documented in patients with ALF and can be explained by the presence of vasodilatation of cerebral arterioles resulting in increased intracranial blood volume (cerebral hyperemia or the so-called luxury perfusion)[15-17].
More recent studies have suggested that neuroinflammatory mediators, particularly pro-inflammatory cytokines such as the interleukins (IL)-1β and IL-6 and tumour necrosis factor-alpha (TNF-α), play an important role in the development of ICH[18] and progression of HE[19,20]. The presence of an infection or systemic inflammation (also known as ‘systemic inflammatory response syndrome’ or ‘SIRS’) is common in ALF and has been shown to be a major prognosticator of both the progression of HE and mortality in patients with ALF[21,22]. Moreover, evidence suggests that this inflammatory response may not only be peripheral but may arise within the brain itself[18,23,24]. Neuroinflammation is now widely considered to result from a direct interaction between microglia and ammonia[25,26]. The released pro-inflammatory cytokines from activated microglial cells and ammonia appear to act synergistically to induce cerebral oedema[27].
The neuropathological aspects of cerebral oedema were first described by Klatzo[28] in a presidential address classifying the underlying mechanisms of cerebral oedema into cytotoxic or vasogenic. This was further explored within the context of ALF by Ede et al[29]. In cytotoxic oedema the BBB is intact and there is intracellular swelling[30], whereas in vasogenic oedema there is breakdown of the BBB and water and plasma constituents accumulate in the extracellular space[31].
CYTOTOXIC OEDEMA AND ASTROCYTE SWELLING
The most prominent neuropathological finding from studies of brain autopsies of patients with ALF[32] and from animal models of cerebral oedema due to ALF is astrocyte swelling[30,33-35]. Astrocytes found within the gray matter are mainly affected and swelling of astrocytic foot processes rather than cell bodies is more commonly seen[32].
Magnetic resonance imaging (MRI) studies using diffusion tensor imaging (DTI) in humans support the view that astrocyte swelling, i.e., cytotoxic oedema, represents the major component of cerebral oedema in ALF[36]. A reduction in the apparent diffusion coefficient (ADC) has been demonstrated in patients with ALF, indicative of a reduction in the size of the extracellular space. This implies that the development of cerebral oedema in ALF results from the accumulation of intracellular fluid.
Approximately one third of the brain volume is made up of astrocytes. They have an important function supporting neurones and have many biochemical, neurochemical and regulatory roles. Swelling of astrocytes therefore impacts upon their function. Abnormal membrane depolarisation has been demonstrated which could affect the ability of astrocytes to maintain ionic gradients and regulate neurotransmitter uptake and processing[37,38]. Impairment of astrocytic function can have deleterious effects on the rest of the central nervous system (CNS) leading to impairment of neuronal excitability and function.
The precise mechanism by which astrocytes swell remains to be determined, although many factors have been implicated. The evidence is most compelling for a role for ammonia in the development of astrocyte swelling in ALF[35]. Whilst other factors, including cerebral blood flow, vaso paralysis, hyperthermia, hyponatremia, substances derived from the necrotic liver, infection, inflammatory cytokines, lactic acid and glutamate have all been implicated in astrocyte swelling, the data is insufficient and often conflicting[34,39]. These factors may all act synergistically to induce cytotoxic swelling with ammonia playing a central role[20].
AMMONIA-GLUTAMINE HYPOTHESIS
Ammonia is mainly produced in the small bowel by the enzyme glutaminase, which breaks down glutamine into ammonia and glutamate. Ammonia is metabolised to urea primarily by the liver and to a lesser extent by the kidneys. In ALF this detoxification pathway, known as the urea cycle, is impaired from the loss of hepatocytes and the concentration of ammonia in the blood rises. Arterial concentrations of ammonia have been shown to correlate with the development of intracranial hypertension[9] and cerebral herniation[10]. Numerous experimental models of ALF have unequivocally associated ammonia exposure with the induction of astrocyte swelling. Treatment of cultured astrocytes with ammonia has consistently caused astrocytes to swell[40]. In vivo animal models of hyperammonemia have also demonstrated the presence of astrocyte swelling[30,41]. Rose et al[42] treated rats in ALF with L-ornithine-L-aspartate, an ammonia-lowering agent which acts by stimulating the urea cycle, and found a reduction in plasma ammonia concentrations and, more importantly, a reduction in cerebral oedema. Lastly, in the absence of liver pathology, patients with genetic disorders of urea cycle enzymes culminating in hyperammonemia develop cerebral oedema, suggesting that elevated levels of ammonia alone are sufficient to cause brain swelling[43]. The precise mechanisms underlying ammonia-induced astrocyte swelling are still poorly understood. Ammonia is able to enter the brain by diffusion[44,45] and its increased uptake from the circulation[46,47] leads to disturbances in astrocyte function[48-50].
The exclusive localisation within astrocytes of glutamine synthetase[51], a cytosolic enzyme which converts ammonia to glutamine, has led to the ‘osmolyte’ or ‘ammonia-glutamine’ hypothesis. Ammonia is detoxified to glutamine within the astrocyte, a precursor for the neurotransmitter glutamate. In addition to causing astrocyte swelling, ammonia has been shown to increase cerebral glutamine levels in the ALF setting[41]. Elevated glutamine levels have been found in brain tissue from animal models of HE[52] and in cerebrospinal fluid (CSF) and brain from patients with HE due to ALF[53,54]. These findings collectively suggest a potential role for glutamine in the development of astrocyte swelling, with hyperammonemia causing increased synthesis and accumulation of glutamine in astrocytes, resulting in astrocyte swelling[55,56].
Originally, it was thought that glutamine acted as an organic osmolyte increasing the intracellular osmolarity, resulting in an influx of water into the cell and culminating in astrocyte swelling and dysfunction. In order to verify whether glutamine accumulation induces astrocyte swelling in hyperammonemic states, studies utilising L-methionine S-sulfoximine (MSO), an irreversible inhibitor of glutamine synthetase, have been performed[57]. Firstly, MSO lowers glutamine in normal brains[58] and prevents cerebral oedema in ammonia-infused healthy rats[59]. Subsequently, it was found to significantly diminish astrocyte swelling both in vivo[41] and in cell culture[60]. Therefore, inhibition of glutamine synthesis may have a protective effect, preventing glutamine accumulation, astrocyte swelling and thus cerebral oedema.
Although these findings suggest that glutamine accumulation within astrocytes plays an important role in cerebral oedema, more recent studies have questioned the glutamine-osmolyte hypothesis. In rats with ALF, glutamine concentrations do not correlate well with the degree of encephalopathy and associated cerebral oedema[61]. In two experimental models of ALF, rats were cooled to reduce brain swelling. Although cerebral oedema was ameliorated by mild hypothermia, it was not accompanied by a similar decrease in glutamine level[62,63]. Jayakumar et al[64] further tested the hypothesis using cultured astrocytes exposed to ammonia. They found no direct correlation between astrocyte swelling and glutamine levels. More importantly, astrocyte swelling was absent when glutamine levels peaked and cell swelling was maximal when glutamine levels were low. Furthermore, the duration and persistence of hyperammonemia, rather than its absolute level is most likely to determine brain glutamine levels and correlate with the development of cerebral oedema and raised intracranial pressure[65]. This delay in astrocyte swelling in relation to an increase in cellular glutamine content is not consistent with the concept of glutamine acting as an osmolyte in ALF and suggests that astrocyte swelling may not be the result of a direct osmotic effect of glutamine.
The ‘Trojan horse’ hypothesis has recently been proposed as an alternative theory by Albrecht et al[66] to explain the development of astrocyte swelling and brain oedema and suggests an important role for both ammonia and glutamine. The excess glutamine synthesised within astrocytes is transported into mitochondria where it is metabolised by phosphate-activated glutaminase (PAG) to ammonia and glutamate[67]. Glutamine, the “Trojan horse”, thereby acts as a carrier of ammonia into mitochondria, where its accumulation can lead to oxidative stress and ultimately astrocyte swelling (Figure 1).
Figure 1.
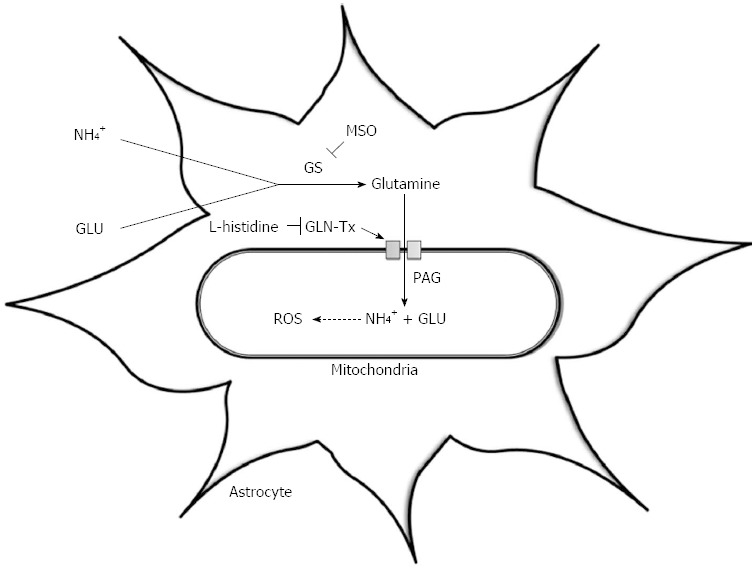
The ‘Trojan Horse’ hypothesis. This illustrates the synthesis of glutamine via the enzyme glutamine synthetase; its transport into mitochondria via the glutamine transporter (GLN-Tx); its hydrolysis by phosphate-activated glutaminase (PAG) resulting in glutamate (GLU) and ammonia (NH4+) production and the subsequent generation of reactive oxygen species (ROS). MSO: L-methionine S-sulfoximinel; GS: Glutamine synthetase.
OXIDATIVE STRESS, MITOCHONDRIAL PERMEABILITY TRANSITION AND ENERGY FAILURE
Oxidative stress has been implicated as an important factor in the pathophysiology of ammonia-induced neurotoxicity[68]. O’Connor et al[69] first suggested oxidative stress might play a role in the pathogenesis of HE when they found evidence of lipid peroxidation in hyperammonemic mice. Norenberg et al[70] subsequently described the concept that protein peroxidation as well as lipid peroxidation may occur in astrocytes treated with ammonia. Further studies revealed that ammonia was able to generate free radicals such as superoxide in cultured astrocytes[71] and in vivo[72]. Ammonia also increases mRNA levels of heme-oxygenase-1 (HO-1), which is considered to be one of the best markers of oxidative stress, in a portacaval shunt rat model of HE[73]. Finally, decreased activity of the antioxidant enzymes glutathione peroxidase, superoxide dismutase and catalase were described in rats exposed to ammonia toxicity adding to the burden of oxidative stress[72].
Oxidative stress has been shown to be a key component of the cerebral oedema which develops in rats following hepatic devascularization[74]. Moreover, administration of antioxidants such as superoxide dismutase, catalase and vitamin E have been shown to inhibit the ammonia-induced astrocyte swelling[64]. Although most evidence supporting the development of oxidative stress in ALF comes from animal and cell culture studies, clinically, the antioxidant and anti-inflammatory agent N-acetylcysteine has proven to be beneficial in the management of patients with ALF[75-77], and agents such as mannitol and sodium benzoate, which are occasionally used in the treatment of ALF, have also been shown to have antioxidant effects[78].
Nitrosative stress is also considered to play an important role in ammonia-induced neurotoxicity. Data from experimental models of HE revealed increased nitric oxide synthase (NOS) gene expression and activity in the brain[79,80]. Inhibition of NOS by nitroarginine significantly reduced deaths in mice exposed to ammonia neurotoxicity[81]. In line with these findings, nitric oxide (NO), was shown to have increased in brains of portacaval-shunted rats given continuous ammonia infusions[82]. This animal model is a well-standardised paradigm of cerebral oedema which occurs in the absence of ALF.
Free radicals such as NO and superoxide can be categorised into reactive nitrogen and oxygen species (RNOS), respectively. In cultured astrocytes and in rat brain in vivo, ammonia triggers their formation through N-methyl-D-aspartate (NMDA)-receptor and calcium (Ca2+)-dependent mechanisms[71,83-86]. Activation of the NMDA receptor is thought to result from the depolarisation-induced removal of the magnesium blockade, which can be induced by ammonia and swelling of the cell itself. Ammonia induces glutamate release from cultured astrocytes[87] and NMDA receptor activity can be further amplified by subsequent Ca2+-dependent astroglial glutamate release and autocrine NMDA receptor stimulation[88]. There is a close relationship between oxidative stress and astrocyte swelling which makes it difficult to separate them temporally as both events are causally interlinked[85,89,90]. This suggests a self-amplifying cycle[91] whereby on the one hand, astrocyte swelling induces oxidative/nitrosative stress through NMDA receptor and Ca2+-dependent mechanisms, and on the other, NMDA receptor activation and oxidative stress trigger astrocyte swelling.
Exactly how ammonia-induced free radicals lead to cell swelling and cerebral oedema is not known. One possibility is that they cause direct damage to proteins and lipids in the membranes of cells and organelles such as mitochondria, thereby altering membrane permeability by affecting ion transport systems. In mitochondria, oxidative injury could lead to altered bioenergetics. Controlled ion transport systems and energy production are essential in maintaining normal cell volume, and alterations in their activity could lead to disturbed volume regulation.
One critical consequence of oxidative and nitrosative stress is induction of the mitochondrial permeability transition[92]. The MPT usually develops in response to an increase in mitochondrial Ca2+ levels and results in a sudden opening of the permeability transition pore (PTP), a large non-selective permeability pore in the inner mitochondrial membrane. This leads to increased permeability of the inner mitochondrial membrane to protons, ions and other small solutes. As a result, the inner mitochondrial membrane potential dissipates causing mitochondrial dysfunction. The MPT is therefore associated with movement of metabolites across the inner mitochondrial membrane, swelling of the mitochondrial matrix, defective oxidative phosphorylation and adenosine triphosphate (ATP) production, and generation of free radicals[93]. Production of free radicals through MPT induction further aggravates the MPT, resulting in a vicious cycle. Induction of the MPT was described in cultured astrocytes exposed to ammonia[60]. The mechanism underlying MPT induction most likely involves oxidative stress, as antioxidants including superoxide dismutase, catalase and vitamin E were able to inhibit the development of the MPT by ammonia[94].
Cyclosporine A (CsA) blocks ammonia-induced astrocyte swelling in culture during the evolution of swelling[95]. Nevertheless, the mechanism(s) by which the MPT mediates astrocyte swelling in hyperammonemia remains unclear. Interestingly, glutamine is capable of inducing the MPT in cultured astrocytes[55] as well as causing mitochondrial swelling in isolated rat cerebral mitochondria[96]. It is notable that, like ammonia, glutamine has been shown to induce oxidative stress by forming free radicals[97]. How glutamine acts to induce oxidative stress, the MPT and consequent astrocyte swelling, is less clear however although it has been suggested that glutamine mediates its deleterious effects through ammonia. Glutamine is hydrolysed in the mitochondria by PAG to yield high levels of ammonia which leads to oxidative stress and the MPT. In support of this concept is the finding that inhibition of PAG by 6-diazo-5-oxo-L-norleucine (DON) blocks free radical production[97], MPT formation[98] as well as ammonia-induced astrocyte swelling[64]. In a further study, L-histidine, an inhibitor of mitochondrial glutamine transport, was further used to study the role of mitochondrial glutamine in a rat model of ALF[99]. L-histidine was found to inhibit HO-1 overexpression, the MPT and brain oedema, supporting the involvement of glutamine in the development of oxidative stress. Taken together, the above data supports the key role of glutamine transport into mitochondria and subsequent metabolism to ammonia in the pathogenesis of cerebral oedema in ALF. Furthermore, these findings support the “Trojan horse” theory, which suggests that glutamine acts as a “stealth” carrier of ammonia in ammonia-induced neurotoxicity.
In terms of a timeline, it was shown that exposure of cultured rat astrocytes and mice brain slices to ammonia results in rapid ROS formation and astrocyte swelling[89,90], whereas MPT-induction and glutamine accumulation occurs later[60,64] implying astrocyte swelling occurs primarily through oxidative/nitrosative stress and is then further aggravated by glutamine accumulation in astrocytes[100].
Cell volume regulation is an energy-dependent process and involves ion homeostasis through ionic transporters and exchangers and extrusion of osmotically active amino acids[101]. In particular, the Na/K/Cl cotransporter-1 (NKCC1) was found to be implicated in astrocyte swelling. NKCC1 expression and activity was increased in cultured astrocytes exposed to ammonia and its activation appears to be mediated by oxidative/nitrosative stress[102]. Energy failure following MPT induction is another possible mechanism underlying cell swelling. Ammonia is thought to interfere with mitochondrial energy metabolism and several studies have reported depletion of ATP in vitro and in vivo models of ammonia neurotoxicity[103]. The implications of energy failure in ALF have largely been ignored despite the presence of higher lactate levels in patients with ALF, which is a consequence of energy failure[104,105]. Indeed, Zwingmann et al[104] in an experimental ALF rodent model showed that in the early (pre-coma) stages of encephalopathy there was a significant 2 to 4.5-fold increase in total brain glutamine and lactate but in the severe (coma) stages of encephalopathy and brain oedema there was a further significant increase in brain lactate but no such increase in glutamine suggesting that impaired glucose oxidative pathways rather than intracellular glutamine accumulation per se may play a more dominant role[101]. This is supported by data by Bernal et al[105] that unequivocally shows lactate to be an important prognostic marker in ALF[102] and data from Rose et al[106] in a pig model of ALF which demonstrated using cerebral microdialysis that ALF animals had increased levels of lactate dehydrogenase activity and mitochondrial complex IV activity.
Mitogen-activated protein kinases (MAPKs) are activated by oxidative/nitrosative stress in cultured astrocytes exposed to ammonia and inhibition of MAPK phosphorylation abrogates astrocyte swelling[107]. Activation of MAPKs may therefore play an important protective role in cell volume regulation through phosphorylation of key proteins.
Water flow across cell membranes in astrocytes is largely dependent on aquaporin 4 (AQP4)[108]. Upregulation of AQP4 has been found to precede cell swelling in cultured astrocytes treated with ammonia and CsA can inhibit this upregulation, indicating that MPT induction is a key step in AQP4 upregulation in ammonia-induced astrocyte swelling[109]. Although it has been suggested that AQP4 is important in initiating signalling events associated with cerebral oedema[110], Wright et al[111] in a rat model of ALF could not find any association of the expression of AQP4 with the development of brain oedema, hyperammonemia or sepsis. The exact role of AQP4 in ALF therefore remains hotly debated.
In recent years, ammonia-induced and swelling-induced oxidative/nitrosative stress has been shown to result in multiple functional consequences. In addition to protein phosphorylation, oxidative/nitrosative stress can trigger protein tyrosine nitration, RNA oxidation and altered zinc metabolism, which can lead to changes in gene expression, intracellular signalling and synaptic plasticity[112]. Furthermore, nitration of glutamine synthetase inactivates the enzyme[113], which suggests this regulatory mechanism leads to reduced glutamine production and therefore astrocyte swelling (Figure 2).
Figure 2.
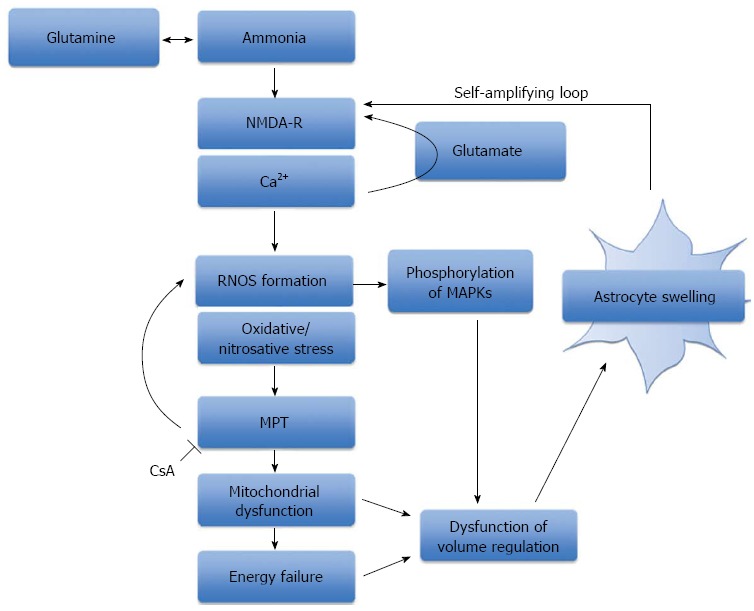
The role of oxidative stress, mitochondrial permeability transition and energy failure in ammonia-induced neurotoxicity. A schematic representation of the central role that ammonia plays in the production of oxidative/nitrosative stress and astrocyte swelling. Ammonia-induced astrocyte swelling is mediated by oxidative and nitrosative stress resulting in the induction of the MPT, activation of intracellular signaling kinases and alterations in gene expression. Mitochondrial dysfunction and energy failure culminates in astrocytes failing to regulate their cell volume, thereby resulting in astrocyte swelling. NMDA-R: N-methyl-D-aspartate-receptor; RNOS: Reactive nitrogen and oxygen species; MPT: Mitochondrial permeability transition; MAPKs: Mitogen-activated protein kinases; CsA: Cyclosporine A.
VASOGENIC OEDEMA AND BLOOD-BRAIN BARRIER DYSFUNCTION
The BBB plays a critical role in establishing and maintaining homeostasis of the brain. It exerts tight control over any exchange of metabolites between the circulating blood and the central nervous system. The BBB consists of brain capillary endothelial cells, pericytes and the enveloping end foot processes of astrocytes. Together they form a neurovascular unit capable of regulating the special composition of the CNS fluid[114]. The main structural constituent of the BBB, and the first to come into direct contact with potentially toxic substances, is the endothelial cell. By spreading itself to cover the entire luminal surface of the capillary and sealing its two surface edges with junctional complexes known as tight junctions (TJ), the endothelial cell forms a physical barrier. These tight junctions consist of transmembrane proteins, including junctional adhesion molecules (JAM), occludin, claudins and intracellular proteins [zona occludin (ZO)-1, -2, and -3] linked to the cytoskeleton which control the stability and functioning of the TJ. Together with adherens junctions located in the basal region below the TJ, they prevent circulating compounds from freely entering the brain parenchyma and limit paracellular diffusion of small molecules. Transport of larger molecules into the brain occurs in a transcellular fashion utilising specific transport systems within endothelial cells. Water is able to diffuse through the bilayer of endothelial cell plasma membranes but can also enter the brain through water channels known as aquaporins, the predominant one in the brain being AQP4[108].
Recent MRI studies of patients with ALF demonstrate evidence of interstitial brain oedema as well as cytotoxic oedema, implying there may be a vasogenic component to the cerebral oedema in ALF[115,116]. In an animal model of ALF, astrocyte swelling, extravascular and interstitial oedema have been described. However, brain capillary endothelial cells and their tight junctions appeared intact[30,117]. Similar findings were also reported in patients who died of ALF[32]. Apart from an increase in cytoplasmic vesicles, suggesting altered transcellular transport across the BBB, no gross structural damage was found in capillary endothelial cells. Similarly, Nguyen[118] has described physically intact tight junctions in ALF, but these were lengthened and tortuous in shape. Thus, electron microscopic examination of the BBB reveals only minimal ultrastructural changes in the brain capillaries of animals and humans with ALF.
Nevertheless, subtle increases in BBB transport of amino acids and energy metabolites have been widely described in the context of hyperammonemia[119]. Changes in BBB penetration of ammonia itself have also been reported in hyperammonemic states. However, the results of these reports, which used PET with 13N-labeled ammonia to study BBB passage of ammonia, are inconsistent[47,120,121]. Nevertheless, in animal models of ALF, ammonia uptake into the brain is thought to increase[122]. Investigating possible changes in BBB permeability to ammonia has been hampered by the recent discovery that ammonia may be able to cross the BBB via two possible routes, and it is not known which of the two may be affected in hyperammoneamic states. Circulating ammonia is largely present as a cation (NH4+) and transport across the BBB was originally considered to occur via diffusion in its gaseous form (NH3), the amount of which is rather small at physiological pH levels[45]. In ALF, due to the acidosis caused by lactic acid, the amount of NH3 and hence its diffusion across the BBB would be expected to be reduced still further. The electric charge of the ionic form was thought to prevent ammonia transport across the BBB, but now an alternative, transcellular route, through potassium channels and transporters, has been suggested[123]. This transcellular transport of ammonia may be affected in ALF, resulting in increased ammonia concentrations within the CNS. Pathological increases in BBB permeability could also result in gaseous ammonia entering the brain via a paracellular route.
Although there has been little evidence for a complete BBB breakdown, findings from more recent studies suggest vasogenic oedema may still contribute to the development of cerebral oedema in ALF. Nguyen and colleagues used Evans blue dye, which binds to albumin and is normally unable to penetrate the BBB, and injected it into the circulation of mice with azoxymethane-induced ALF to assess brain extravasation. They found that BBB permeability to Evans blue dye and water was significantly increased in mice with experimentally-induced ALF. Under electron microscopy, they noted that the leakage of Evans blue dye, i.e., extravasation, occurred mostly in the surrounding region of the brain capillaries. Consistent with previous findings, the BBB and tight junctions were found to be structurally intact[124]. Furthermore, they were able to demonstrate that BBB permeability and brain water was reduced in ALF mice given monoclonal antibodies specific for active matrix metalloproteinase-9 (MMP-9), a member of the matrix metalloproteinase (MMP) family of endopeptidase enzymes that degrade the extracellular matrix in normal and disease states. MMP-9 in particular, causes protein degradation of tight junctions and is upregulated in the liver of ALF mice. Increased blood concentrations of MMP-9 can also be found. These findings collectively show increased BBB permeation to water and plasma constituents in experimental ALF mice and suggest that BBB dysfunction is associated with protein deregulation in tight junctions but not necessarily with a structural breakdown. Circulating MMP-9 derived from the necrotic liver contributes to fine perturbation in BBB integrity and increased brain extravasation in mice with azoxymethane-induced ALF and inhibition of MMP-9 may be useful in preventing the development of brain oedema. Chen et al[125] further demonstrated that MMP-9 induces significant degradation of the TJ proteins occludin and claudin-5 in brain endothelial cells in vitro and in mice with azoxymethane-induced ALF; these alterations in TJ proteins correlated with increased BBB permeability and were reversed by inhibiting MMP-9. Chen et al[126]went on to demonstrate that MMP-9 induces activation of the epidermal growth factor receptor (EGFR) and p38 mitogen activated protein kinase/nuclear factor-κB (MAPK/NFκB) in brain endothelial cells. Activation of this pathway in turn leads to degradation of the TJ protein occludin and deregulation of the TJ. Taken together, these findings suggest that substances derived from the injured liver, such as MMP-9, reach the BBB and induce increased permeability through subtle changes in TJ composition (Figure 3). An important role for a vasogenic mechanism in the development of cerebral oedema in ALF is thus supported by these studies.
Figure 3.
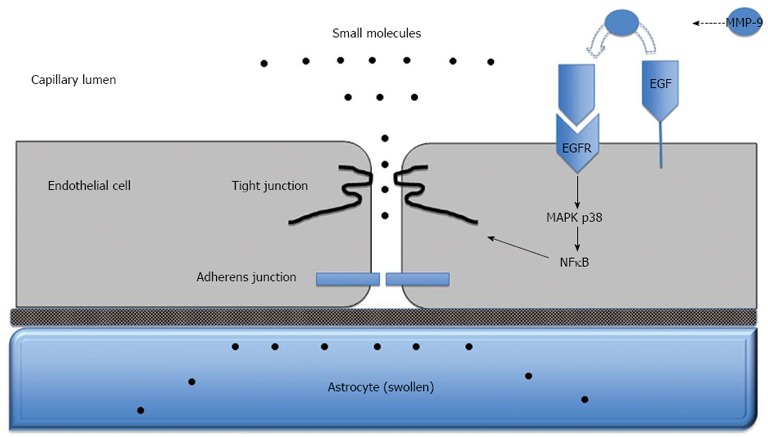
Blood-brain barrier dysfunction in acute liver failure. Anatomy of the blood-brain barrier (BBB) created by the brain capillary endothelial cell and its paracellular tight junction and adherens junction. In acute liver failure, activation of epidermal growth factor receptor (EGFR) and other signaling pathways results in a loss of BBB tight junction integrity. Tight junctional proteins are altered, resulting in increased permeability to small molecules, leading to astrocyte swelling. MMP-9: Matrix metalloproteinase-9; MAPK p38: Mitogen activated protein kinase p38; NFκB: Nuclear factor-κB.
Interestingly, activation of the p38 MAPK pathway as a result of oxidative/nitrosative stress is also thought to mediate ammonia-induced astrocyte swelling[107]. The p38 MAPK pathway and subsequent phosphorylation of key proteins appears to play an important role in the pathophysiology of cell swelling[127,128] and thus cerebral oedema, and therefore, this pathway may be a potential therapeutic target.
In recent years, there has been some controversy as to whether ALF per se causes the changes seen within the BBB integrity or whether these changes are due to secondary complications associated with ALF such as infection and sepsis[129]. Consistent with this viewpoint is the evidence that neurosteroid biosynthesis is increased in the brains of rats with ALF[130] and that these neurosteroids protect against BBB breakdown induced by ammonia[131]. Jayakumar et al[132] have also reported neuroprotective effects of neurosteroids in some models of ALF but not in all suggesting that there may be differences in outcomes depending on which hepatotoxin-induced ALF model is used and that this may explain the inconsistent reports on BBB breakdown in ALF.
TREATMENT OF CEREBRAL OEDEMA IN ACUTE LIVER FAILURE
Management principles
In the absence of overt HE patients in the early stages of ALF may be observed and managed conservatively. However such patients are susceptible to extrahepatic manifestations including the development of multiorgan dysfunction, acute kidney injury and infections[21] both of which can accelerate the development of advanced HE and brain oedema[8]. Frequent clinical and neurological examinations, concentrating on pupil size, coma grade, evidence of delirium and reflexes, are imperative to detect features which may herald the development of brain oedema. The development of grade 3/4 coma, indicative of impending raised intracranial pressure (ICP), typically necessitates intubation and ventilation[133]. ICH should be suspected in patients with sudden onset systemic hypertension, changes in pupillary reactivity, abnormal oculovestibular reflexes or decerebrate posturing. ICH becomes problematic when the ICP is above 20 mmHg due to the risk of compromising cerebral perfusion pressure. Ultimately severe ICH can result in brain stem compression causing ischaemia, haemorrhage and death[134].
Transcranial doppler ultrasonography is a non-invasive device which can continuously measure middle cerebral artery blood flow velocity, producing a velocity-time waveform that indirectly monitors changes in cerebral hemodynamics, including ICP avoiding the complications associated with more invasive monitoring devices which include haemorrhage and infection. In a small retrospective study of 16 patients with ALF four features in the waveform were found to capture the cerebral hemodynamic state and potentially can be used to predict dynamic changes in ICP or CPP. This included the slope of the Windkessel upstroke, the slope of the Windkessel downstroke, the slope of the diastolic downstroke, and the angle between the end systolic downstroke and start diastolic upstroke[135]. ICP monitoring, involving intracranial bolt insertion, is used in patients who are at high risk for the development of ICH. ICP monitoring is indicated in a subset of patients with grade 3/4 coma[136] (Glasgow Coma Scale < 8) who also display a combination of the following features; fever and tachycardia, arterial ammonia > 150 μmol/L, hyponatraemia, seizures or pupillary abnormalities, acute/hyperacute liver failure, vasopressor requirement, are less than age 40 or have jugular venous oxygen saturations or have middle cerebral artery doppler monitoring indicative of a very high or very low cerebral blood flow[8]. Additionally reverse jugular vein oxygen saturation should also be monitored, which gives an indication of cerebral oxygenation and metabolism which is often reduced as a result of the loss of CBF autoregulation in patients with ALF[15]. In terms of imaging the brain for evidence of cerebral oedema, computed tomography is only of benefit if cerebral herniation or intracranial bleeding is suspected and has no role in the routine surveillance. Electroencephalography is very useful for the detection of subclinical seizures and to measure brain activity in comatose patients, but due to its lack of specificity it is not employed routinely to diagnose encephalopathy or cerebral oedema[8].
Metabolic changes contributing to the development of raised ICP in ALF can be monitored utilizing in vivo cerebral microdialysis and have been documented in research settings in human ALF but this technique is currently only reserved for experimental studies and is not used in routine clinical settings[137].
Specific therapies
The treatment of cerebral oedema is aimed at preventing infection, reducing or controlling inflammation, ensuring sufficient sedation and correcting hypo-osmolality. The objective of ICH management is to maintain the ICP at less than 20 mmHg and to keep the cerebral perfusion pressure over 70 mmHg although this can be very difficult to practically achieve and the evidence base in human ALF to support such strategies is very limited. Patients are nursed in the 20°-30° head-up position favouring venous drainage to reduce ICP whilst maintaining cerebral perfusion pressure. Hypoxaemia should be avoided with target arterial oxygenation of above 95%. Patients with grade 3 encephalopathy and above should be intubated and ventilated. Propofol and other short acting sedatives are commonly utilised to ease mechanical ventilation and reduce seizure risk. Opiates, such as fentanyl, are often used for analgesia[133]. Most patients are normoventilated but hyperventilation is employed in those displaying signs indicative of imminent cerebral herniation, such as pupillary dilatation and extensor posturing. Hyperventilation results in reduced ICP by inducing hypocapnia which causes precapillary vasoconstriction decreasing CBF[17].
Patients should be adequately fluid resuscitated. Plasma volume expansion results in a significant reduction in plasma ammonia concentration by increasing urinary ammonia excretion[138]. Hypertension can reduce cerebral perfusion pressure by increasing intracranial blood volume and is best avoided; sedation can help to combat this. Arterial hypotension, especially in the presence of reduced cerebral blood flow autoregulation, will also compromise cerebral perfusion pressure. Theoretically, diastolic blood pressure should be kept > 40 mmHg higher than the ICP in patients with severe cerebral oedema and ICH who have ICP bolt monitoring in situ to guarantee adequate CBF but again this is often hard to achieve in practice[139]. Vasopressors, commonly noradrenaline, may be necessary to maintain this.
Hyponatraemia should be corrected. Background hypertonic saline (30%) infusions are used to induce and maintain serum sodium levels between 145 and 150 mmol/L thus maintaining the BBB osmotic pressure gradient[140]. Hypertonic saline acts as a dehydrating agent reducing brain water content and subsequently lowers ICP. Mannitol may also be used for the same purpose but it may be more rational to use hypertonic saline instead of mannitol as the BBB has a reflection coefficient of 1 for sodium chloride vs 0.9 for mannitol making it more efficient to exclude saline from the brain. It is also recommended that serum osmolarity be maintained at < 320 mOsm/L. Boluses of hypertonic saline or mannitol are used for sustained increases in ICP (> 25 mmHg) but resistant rises in ICP may be treated with indomethacin[141] or hypothermia[142]. Indomethacin (a non-selective cyclo-oxygenase inhibitor) induces cerebral vasoconstriction by inhibiting the endothelial cyclooxygenase pathway, reducing cerebral temperature and modifying extracellular pH. However, it has a number of adverse effects, including nephrotoxicity, platelet dysfunction and gastrointestinal bleeding, and therefore its use in ALF patients is limited to when all other management options to reduce ICP have been exhausted[8]. Moderate hypothermia (32-34 °C) may be useful in patients with resistant ICH awaiting liver transplantation by decreasing brain ammonia uptake and also through its role in reducing brain cytokine production, OS and CBF[143]. Barbiturates are postulated to reduce brain metabolism and consequently lead to a decrease in cerebral blood volume. Thiopental infusion has been shown to be efficacious in 14 patients with ALF as measured by extradural transducers with minimal side effects although additional data in the context of human ALF is scarce[144]. However, due to their hepatic metabolism and negative inotropic effects they are only used to reduce ICP surges as a last resort[8].
ALF has many similarities to septic shock[145] and there is evidence that patients exhibiting a systemic inflammatory response progress more rapidly to severe encephalopathy[21]. Broad spectrum intravenous antibiotics and antifungals are therefore used empirically to reduce the risk of sepsis and development of severe encephalopathy.
Intravenous N-acetylcysteine (NAC) is now considered as standard of care in the treatment of acetaminophen-induced and non-acetaminophen induced ALF as it acts as both as an antioxidant and anti-inflammatory agent. Early administration of intravenous NAC after an overdose of acetaminophen replenishes glutathione stores and helps to alleviate hepatic necrosis[146]. NAC also has beneficial hemodynamic effects and has been shown to improve cerebral perfusion pressure[147] mediated by enhanced activity of the nitric oxide soluble cyclic GMP system[76].
Ultimately emergency liver transplantation reverses cerebral oedema, although a variety of neurological manifestations including intracerebral haemorrhage and seizures, precipitated by cerebral hypoperfusion, coagulopathy and transfusion may still occur post-operatively. If the graft is functioning well ICH is expected to resolve 48 h post-transplant[148,149].
Bernal et al[7] reviewed 3305 patients with acute liver dysfunction from 1973-2008 and found a significant reduction in the proportion of patients with ICH (from 76% in 1984-1988 to 20% in 2004-2008 (P < 0.0001)). Furthermore, mortality of patients with ICH decreased from 95% to 55% (P < 0.0001). The cause for this improvement is likely to be multifactorial. Patients now present and are diagnosed earlier and the prompt use of N-acetylcysteine, fluid resuscitation, empirical antibiotics and renal replacement therapy may have reduced the incidence of cerebral oedema and ICH by modulating principal contributory factors. Such approaches may also limit hepatotoxicity, reduce plasma ammonia levels and prevent sepsis. The more timely use of emergency liver transplantation for those at greater risk may also have contributed to the reduction in ICH.
NOVEL THERAPIES IN DEVELOPMENT
Minocycline
Jiang et al[74] studied the use of minocycline, a broad-spectrum tetracycline antibiotic which has been shown to attenuate lipopolysaccharide-induced neuroinflammation[150], in an experimental model of ALF[24]. They were able to demonstrate that it delayed the progression of HE and brain oedema by exerting a potent inhibitory action on microglial activation independently of its antimicrobial properties.
NMDA receptor antagonists
NMDA receptor antagonists have been shown to prevent the oxidative stress induced by acute ammonia intoxication[83]. This is likely to be the case as the production of ROS is mediated by NMDA-receptor activation in hyperammonemic states[151]. Memantine is a non-competitive NMDA-receptor antagonist and has been shown to improve EEG activity, clinical grading, ICP and brain water content in portocaval shunted rats infused with ammonia, and in rats with ALF induced by ischaemia which was independent of ammonia concentration[152].
Endotoxin removal
An albumin replacement system with a novel endotoxin ligation (ARSeNEL) function has been developed at University College London and tested in an ALF pig model. Early data have reported an improvement in survival, endotoxemia and ICP index which warrant further studies in clinical settings[153].
Novel anti-inflammatory agents
It make sense that if agents were used to reduce the systemic inflammation that is frequently seen in ALF and which sensitises the brain to the effects of ammonia, then we might be able to prevent the development of cerebral oedema. Unfortunately however, the proinflammatory response which develops in the wake of acute liver injury is also key in initiating liver repair and regeneration. One would postulate therefore that it may be detrimental to use agents and antibodies which target prominent pro-inflammatory mediators. Neutrophil malfunction, akin to that seen in septic shock is a consistent finding in patients with ALF and recent data support an intimate relationship with hyperammonemia[127,145]. Strategies that target innate and adaptive immune dysfunction in ALF including TLR expression and production of ROS would certainly be of therapeutic interest and warrant further study. Granulocyte colony stimulating factor (GCSF) has been shown in 2 small studies to improve neutrophil phagocytic capability in patients with ALF[154,155] and as such may have utility in the prevention of advanced HE.
Plasmapheresis
Larsen et al[156] have previously shown that high volume plasmapheresis can alleviate brain oedema in some patients with ALF with favourable changes in systemic hemodynamics despite increasing cerebral blood flow. Plasmapheresis may also have a positive impact on alleviating the systemic immune dysfunction and endothelial dysfunction that commonly develops. This was the stimulus for performing a randomised clinical trial of high volume plasmapheresis of which the preliminary analysed data suggests that it may improve survival in patients unsuitable for liver transplantation (verbal communication-Dr Finn Stolze Larsen).
CONCLUSION
This detailed review has unequivocally presented the evidence to support the critical role of the neurotoxin ammonia in the development of astrocyte swelling and cytotoxic oedema in ALF. Although a generalised breakdown of the BBB cannot be demonstrated in patients with ALF, more recent studies have described a “leaky” BBB resulting from subtle changes in the integrity of the tight junctions, supporting a role of a vasogenic component in the pathophysiology of cerebral oedema in ALF.
Exactly how both cytotoxic and vasogenic mechanisms interact to bring about cerebral oedema in ALF, and the extent of their involvement, remains unknown. Moreover, the sequence of events is unclear. Is BBB dysfunction the result of a cytotoxic insult or is cytotoxic oedema a consequence of increased BBB permeability? It has been postulated that increased BBB permeability to small molecules such as water and ammonia arises as a result of BBB dysfunction as an initial event in the pathophysiology of brain oedema in ALF. Increased BBB permeability may then invoke vasogenic oedema. The subsequent development of ammonia neurotoxicity and cytotoxic oedema may then occur as a downstream manifestation. This sequence of events is supported by the observation that in rats with ALF an early increase in BBB permeability correlates with increased ICP and results in vasogenic oedema followed by a progressive increase in brain ammonia and glutamine levels[157]. However, it is difficult to determine to what extent these data definitively support a vasogenic mechanism in ALF. For example, the potency of mannitol in treating ICH in the context of cerebral oedema in patients with ALF supports the BBB being intact and the predominant mechanism being a cytotoxic one. Another possibility however, is that certain brain areas may behave respond differently to others. For example, Cauli et al[157] were able to show in a rodent experimental ALF model that the mechanism and time course of the appearance of brain oedema differed between 12 different brain regions with the cerebellum showing predominantly vasogenic oedema whilst the frontal cortex exhibited cytotoxic oedema.
The syndrome of ALF arises in the context of various aetiological toxic insults to the liver and is frequently associated with the development of multiple organ dysfunction and sepsis. It is not known how these manifestations independently impact on BBB integrity and function. Furthermore, the liver toxins utilised in the various animal ALF models could directly affect the BBB independently of ALF. Changes to the BBB in ALF are very different in nature to that seen in brain ischemia or traumatic brain injury, where complete BBB breakdown is commonly observed. The mechanisms underpinning cerebral oedema in ALF are therefore also different and therapeutic interventions that are beneficial in other types of brain injury may not be useful in the treatment of ALF.
It is clear that the development of more effective therapies in ALF will require further knowledge of the pathophysiology of cerebral oedema, which is a devastating and frequently fatal feature of ALF. Greater knowledge of the sequence of events and key mediators involved in the development of brain oedema will allow for specific targets to be identified.
Footnotes
Supported by Medical Research Council (MRC) Centre for Transplantation, King’s College London, United Kingdom-MRC grant No. MR/J006742/1; The National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London
P- Reviewers: Felipo V, Llompart-Pou J, Shimizu Y, Sun XJ S- Editor: Cui XM L- Editor: A E- Editor: Zhang DN
References
- 1.Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 2.Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102:2459–2463. doi: 10.1111/j.1572-0241.2007.01388.x. [DOI] [PubMed] [Google Scholar]
- 3.Trey C, Davidson CS. The management of fulminant hepatic failure. Prog Liver Dis. 1970;3:282–298. [PubMed] [Google Scholar]
- 4.O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342:273–275. doi: 10.1016/0140-6736(93)91818-7. [DOI] [PubMed] [Google Scholar]
- 5.Blei AT, Larsen FS. Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol. 1999;31:771–776. doi: 10.1016/s0168-8278(99)80361-4. [DOI] [PubMed] [Google Scholar]
- 6.Blei AT. Medical therapy of brain edema in fulminant hepatic failure. Hepatology. 2000;32:666–669. doi: 10.1053/jhep.2000.17923. [DOI] [PubMed] [Google Scholar]
- 7.Bernal W, Hyyrylainen A, Gera A, Audimoolam VK, McPhail MJ, Auzinger G, Rela M, Heaton N, O’Grady JG, Wendon J, et al. Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J Hepatol. 2013;59:74–80. doi: 10.1016/j.jhep.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Shawcross DL, Wendon JA. The neurological manifestations of acute liver failure. Neurochem Int. 2012;60:662–671. doi: 10.1016/j.neuint.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Bernal W, Hall C, Karvellas CJ, Auzinger G, Sizer E, Wendon J. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844–1852. doi: 10.1002/hep.21838. [DOI] [PubMed] [Google Scholar]
- 10.Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648–653. doi: 10.1002/hep.510290309. [DOI] [PubMed] [Google Scholar]
- 11.Larsen FS. Cerebral circulation in liver failure: Ohm’s law in force. Semin Liver Dis. 1996;16:281–292. doi: 10.1055/s-2007-1007241. [DOI] [PubMed] [Google Scholar]
- 12.Ware AJ, D’Agostino AN, Combes B. Cerebral edema: a major complication of massive hepatic necrosis. Gastroenterology. 1971;61:877–884. [PubMed] [Google Scholar]
- 13.Cordoba J, Gottstein J, Blei AT. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: implications for ammonia-induced brain edema. Hepatology. 1996;24:919–923. doi: 10.1002/hep.510240427. [DOI] [PubMed] [Google Scholar]
- 14.Jalan R, Olde Damink SW, Deutz NE, Hayes PC, Lee A. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001;34:50–54. doi: 10.1053/jhep.2001.25386. [DOI] [PubMed] [Google Scholar]
- 15.Larsen FS, Adel Hansen B, Pott F, Ejlersen E, Secher NH, Paulson OB, Knudsen GM. Dissociated cerebral vasoparalysis in acute liver failure. A hypothesis of gradual cerebral hyperaemia. J Hepatol. 1996;25:145–151. doi: 10.1016/s0168-8278(96)80066-3. [DOI] [PubMed] [Google Scholar]
- 16.Larsen FS, Strauss G, Møller K, Hansen BA. Regional cerebral blood flow autoregulation in patients with fulminant hepatic failure. Liver Transpl. 2000;6:795–800. doi: 10.1053/jlts.2000.18705. [DOI] [PubMed] [Google Scholar]
- 17.Strauss G, Hansen BA, Knudsen GM, Larsen FS. Hyperventilation restores cerebral blood flow autoregulation in patients with acute liver failure. J Hepatol. 1998;28:199–203. doi: 10.1016/0168-8278(88)80006-0. [DOI] [PubMed] [Google Scholar]
- 18.Wright G, Shawcross D, Olde Damink SW, Jalan R. Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab Brain Dis. 2007;22:375–388. doi: 10.1007/s11011-007-9071-4. [DOI] [PubMed] [Google Scholar]
- 19.Jalan R, Pollok A, Shah SH, Madhavan K, Simpson KJ. Liver derived pro-inflammatory cytokines may be important in producing intracranial hypertension in acute liver failure. J Hepatol. 2002;37:536–538. doi: 10.1016/s0168-8278(02)00240-4. [DOI] [PubMed] [Google Scholar]
- 20.Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol. 2004;41:613–620. doi: 10.1016/j.jhep.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 22.Vaquero J, Polson J, Chung C, Helenowski I, Schiodt FV, Reisch J, Lee WM, Blei AT. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755–764. doi: 10.1016/s0016-5085(03)01051-5. [DOI] [PubMed] [Google Scholar]
- 23.Butterworth RF. Hepatic encephalopathy: a central neuroinflammatory disorder? Hepatology. 2011;53:1372–1376. doi: 10.1002/hep.24228. [DOI] [PubMed] [Google Scholar]
- 24.Jiang W, Desjardins P, Butterworth RF. Cerebral inflammation contributes to encephalopathy and brain edema in acute liver failure: protective effect of minocycline. J Neurochem. 2009;109:485–493. doi: 10.1111/j.1471-4159.2009.05981.x. [DOI] [PubMed] [Google Scholar]
- 25.Rodrigo R, Cauli O, Gomez-Pinedo U, Agusti A, Hernandez-Rabaza V, Garcia-Verdugo JM, Felipo V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology. 2010;139:675–684. doi: 10.1053/j.gastro.2010.03.040. [DOI] [PubMed] [Google Scholar]
- 26.Zemtsova I, Görg B, Keitel V, Bidmon HJ, Schrör K, Häussinger D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology. 2011;54:204–215. doi: 10.1002/hep.24326. [DOI] [PubMed] [Google Scholar]
- 27.Rama Rao KV, Jayakumar AR, Tong X, Alvarez VM, Norenberg MD. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J Neuroinflammation. 2010;7:66. doi: 10.1186/1742-2094-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klatzo I. Presidental address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967;26:1–14. doi: 10.1097/00005072-196701000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Ede RJ, Williams RW. Hepatic encephalopathy and cerebral edema. Semin Liver Dis. 1986;6:107–118. doi: 10.1055/s-2008-1040594. [DOI] [PubMed] [Google Scholar]
- 30.Traber PG, Dal Canto M, Ganger DR, Blei AT. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: ultrastructure and integrity of the blood-brain barrier. Hepatology. 1987;7:1272–1277. doi: 10.1002/hep.1840070616. [DOI] [PubMed] [Google Scholar]
- 31.Cui W, Sun CM, Liu P. Alterations of blood-brain barrier and associated factors in acute liver failure. Gastroenterol Res Pract. 2013;2013:841707. doi: 10.1155/2013/841707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato M, Hughes RD, Keays RT, Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992;15:1060–1066. doi: 10.1002/hep.1840150615. [DOI] [PubMed] [Google Scholar]
- 33.Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am J Physiol. 1999;277:G455–G462. doi: 10.1152/ajpgi.1999.277.2.G455. [DOI] [PubMed] [Google Scholar]
- 34.Blei AT, Olafsson S, Therrien G, Butterworth RF. Ammonia-induced brain edema and intracranial hypertension in rats after portacaval anastomosis. Hepatology. 1994;19:1437–1444. [PubMed] [Google Scholar]
- 35.Norenberg MD. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest. 1977;36:618–627. [PubMed] [Google Scholar]
- 36.Saksena S, Rai V, Saraswat VA, Rathore RS, Purwar A, Kumar M, Thomas MA, Gupta RK. Cerebral diffusion tensor imaging and in vivo proton magnetic resonance spectroscopy in patients with fulminant hepatic failure. J Gastroenterol Hepatol. 2008;23:e111–e119. doi: 10.1111/j.1440-1746.2007.05158.x. [DOI] [PubMed] [Google Scholar]
- 37.Kimelberg HK, Kettenmann H. Swelling-induced changes in electrophysiological properties of cultured astrocytes and oligodendrocytes. I. Effects on membrane potentials, input impedance and cell-cell coupling. Brain Res. 1990;529:255–261. doi: 10.1016/0006-8993(90)90835-y. [DOI] [PubMed] [Google Scholar]
- 38.Kimelberg HK, Anderson E, Kettenmann H. Swelling-induced changes in electrophysiological properties of cultured astrocytes and oligodendrocytes. II. Whole-cell currents. Brain Res. 1990;529:262–268. doi: 10.1016/0006-8993(90)90836-z. [DOI] [PubMed] [Google Scholar]
- 39.Norenberg MD. Astrocytic-ammonia interactions in hepatic encephalopathy. Semin Liver Dis. 1996;16:245–253. doi: 10.1055/s-2007-1007237. [DOI] [PubMed] [Google Scholar]
- 40.Norenberg M. Hepatic Encephalopathy: Studies with astrocyte cultures. In: Norenberg M, Hertz L, Schousboe A, editors. The Biochemical Pathology of Astrocytes. New York: Alan R Liss; 1988. pp. 451–464. [Google Scholar]
- 41.Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71:589–599. doi: 10.1016/0306-4522(95)00462-9. [DOI] [PubMed] [Google Scholar]
- 42.Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology. 1999;30:636–640. doi: 10.1002/hep.510300311. [DOI] [PubMed] [Google Scholar]
- 43.Batshaw ML. Inborn errors of urea synthesis. Ann Neurol. 1994;35:133–141. doi: 10.1002/ana.410350204. [DOI] [PubMed] [Google Scholar]
- 44.Cooper AJ, Mora SN, Cruz NF, Gelbard AS. Cerebral ammonia metabolism in hyperammonemic rats. J Neurochem. 1985;44:1716–1723. doi: 10.1111/j.1471-4159.1985.tb07159.x. [DOI] [PubMed] [Google Scholar]
- 45.Cooper AJ, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev. 1987;67:440–519. doi: 10.1152/physrev.1987.67.2.440. [DOI] [PubMed] [Google Scholar]
- 46.Dejong CH, Kampman MT, Deutz NE, Soeters PB. Cerebral cortex ammonia and glutamine metabolism during liver insufficiency-induced hyperammonemia in the rat. J Neurochem. 1992;59:1071–1079. doi: 10.1111/j.1471-4159.1992.tb08349.x. [DOI] [PubMed] [Google Scholar]
- 47.Lockwood AH, Yap EW, Wong WH. Cerebral ammonia metabolism in patients with severe liver disease and minimal hepatic encephalopathy. J Cereb Blood Flow Metab. 1991;11:337–341. doi: 10.1038/jcbfm.1991.67. [DOI] [PubMed] [Google Scholar]
- 48.Hindfelt B, Plum F, Duffy TE. Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J Clin Invest. 1977;59:386–396. doi: 10.1172/JCI108651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lockwood AH, McDonald JM, Reiman RE, Gelbard AS, Laughlin JS, Duffy TE, Plum F. The dynamics of ammonia metabolism in man. Effects of liver disease and hyperammonemia. J Clin Invest. 1979;63:449–460. doi: 10.1172/JCI109322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehrlich M, Plum F, Duffy TE. Blood and brain ammonia concentrations after portacaval anastomosis. Effects of acute ammonia loading. J Neurochem. 1980;34:1538–1542. doi: 10.1111/j.1471-4159.1980.tb11238.x. [DOI] [PubMed] [Google Scholar]
- 51.Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- 52.Hilgier W, Olson JE. Brain ion and amino acid contents during edema development in hepatic encephalopathy. J Neurochem. 1994;62:197–204. doi: 10.1046/j.1471-4159.1994.62010197.x. [DOI] [PubMed] [Google Scholar]
- 53.Larsen FS, Gottstein J, Blei AT. Cerebral hyperemia and nitric oxide synthase in rats with ammonia-induced brain edema. J Hepatol. 2001;34:548–554. doi: 10.1016/s0168-8278(00)00069-6. [DOI] [PubMed] [Google Scholar]
- 54.Albrecht J, Sonnewald U, Waagepetersen HS, Schousboe A. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332–343. doi: 10.2741/2067. [DOI] [PubMed] [Google Scholar]
- 55.Rama Rao KV, Jayakumar AR, Norenberg MD. Induction of the mitochondrial permeability transition in cultured astrocytes by glutamine. Neurochem Int. 2003;43:517–523. doi: 10.1016/s0197-0186(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 56.Yudkoff M, Nissim I, Pleasure D. Astrocyte metabolism of [15N]glutamine: implications for the glutamine-glutamate cycle. J Neurochem. 1988;51:843–850. doi: 10.1111/j.1471-4159.1988.tb01820.x. [DOI] [PubMed] [Google Scholar]
- 57.Tanigami H, Rebel A, Martin LJ, Chen TY, Brusilow SW, Traystman RJ, Koehler RC. Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience. 2005;131:437–449. doi: 10.1016/j.neuroscience.2004.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Folbergrova J. Free glutamine level in the rat brain in vivo after methionine sulphoximine administration. Physiol Bohemoslov. 1964;13:21–27. [PubMed] [Google Scholar]
- 59.Takahashi H, Koehler RC, Brusilow SW, Traystman RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol. 1991;261:H825–H829. doi: 10.1152/ajpheart.1991.261.3.H825. [DOI] [PubMed] [Google Scholar]
- 60.Bai G, Rama Rao KV, Murthy CR, Panickar KS, Jayakumar AR, Norenberg MD. Ammonia induces the mitochondrial permeability transition in primary cultures of rat astrocytes. J Neurosci Res. 2001;66:981–991. doi: 10.1002/jnr.10056. [DOI] [PubMed] [Google Scholar]
- 61.Zwingmann C, Chatauret N, Rose C, Leibfritz D, Butterworth RF. Selective alterations of brain osmolytes in acute liver failure: protective effect of mild hypothermia. Brain Res. 2004;999:118–123. doi: 10.1016/j.brainres.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 62.Chatauret N, Zwingmann C, Rose C, Leibfritz D, Butterworth RF. Effects of hypothermia on brain glucose metabolism in acute liver failure: a H/C-nuclear magnetic resonance study. Gastroenterology. 2003;125:815–824. doi: 10.1016/s0016-5085(03)01054-0. [DOI] [PubMed] [Google Scholar]
- 63.Córdoba J, Crespin J, Gottstein J, Blei AT. Mild hypothermia modifies ammonia-induced brain edema in rats after portacaval anastomosis. Gastroenterology. 1999;116:686–693. doi: 10.1016/s0016-5085(99)70191-5. [DOI] [PubMed] [Google Scholar]
- 64.Jayakumar AR, Rao KV, Murthy ChR, Norenberg MD. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem Int. 2006;48:623–628. doi: 10.1016/j.neuint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 65.Tofteng F, Hauerberg J, Hansen BA, Pedersen CB, Jørgensen L, Larsen FS. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2006;26:21–27. doi: 10.1038/sj.jcbfm.9600168. [DOI] [PubMed] [Google Scholar]
- 66.Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology. 2006;44:788–794. doi: 10.1002/hep.21357. [DOI] [PubMed] [Google Scholar]
- 67.Sonnewald U, Westergaard N, Jones P, Taylor A, Bachelard HS, Schousboe A. Metabolism of [U-13C5] glutamine in cultured astrocytes studied by NMR spectroscopy: first evidence of astrocytic pyruvate recycling. J Neurochem. 1996;67:2566–2572. doi: 10.1046/j.1471-4159.1996.67062566.x. [DOI] [PubMed] [Google Scholar]
- 68.Kosenko E, Venediktova N, Kaminsky Y, Montoliu C, Felipo V. Sources of oxygen radicals in brain in acute ammonia intoxication in vivo. Brain Res. 2003;981:193–200. doi: 10.1016/s0006-8993(03)03035-x. [DOI] [PubMed] [Google Scholar]
- 69.O’Connor JE, Costell M. New roles of carnitine metabolism in ammonia cytotoxicity. Adv Exp Med Biol. 1990;272:183–195. doi: 10.1007/978-1-4684-5826-8_12. [DOI] [PubMed] [Google Scholar]
- 70.Norenberg MD, Jayakumar AR, Rama Rao KV, Panickar KS. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219–234. doi: 10.1007/s11011-007-9062-5. [DOI] [PubMed] [Google Scholar]
- 71.Murthy CR, Rama Rao KV, Bai G, Norenberg MD. Ammonia-induced production of free radicals in primary cultures of rat astrocytes. J Neurosci Res. 2001;66:282–288. doi: 10.1002/jnr.1222. [DOI] [PubMed] [Google Scholar]
- 72.Kosenko E, Kaminsky Y, Kaminsky A, Valencia M, Lee L, Hermenegildo C, Felipo V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic Res. 1997;27:637–644. doi: 10.3109/10715769709097867. [DOI] [PubMed] [Google Scholar]
- 73.Warskulat U, Görg B, Bidmon HJ, Müller HW, Schliess F, Häussinger D. Ammonia-induced heme oxygenase-1 expression in cultured rat astrocytes and rat brain in vivo. Glia. 2002;40:324–336. doi: 10.1002/glia.10128. [DOI] [PubMed] [Google Scholar]
- 74.Jiang W, Desjardins P, Butterworth RF. Minocycline attenuates oxidative/nitrosative stress and cerebral complications of acute liver failure in rats. Neurochem Int. 2009;55:601–605. doi: 10.1016/j.neuint.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 75.Harrison PM, Wendon JA, Gimson AE, Alexander GJ, Williams R. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med. 1991;324:1852–1857. doi: 10.1056/NEJM199106273242604. [DOI] [PubMed] [Google Scholar]
- 76.Harrison P, Wendon J, Williams R. Evidence of increased guanylate cyclase activation by acetylcysteine in fulminant hepatic failure. Hepatology. 1996;23:1067–1072. doi: 10.1053/jhep.1996.v23.pm0008621135. [DOI] [PubMed] [Google Scholar]
- 77.Stravitz RT, Sanyal AJ, Reisch J, Bajaj JS, Mirshahi F, Cheng J, Lee WM. Effects of N-acetylcysteine on cytokines in non-acetaminophen acute liver failure: potential mechanism of improvement in transplant-free survival. Liver Int. 2013;33:1324–1331. doi: 10.1111/liv.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Norenberg MD. Oxidative and nitrosative stress in ammonia neurotoxicity. Hepatology. 2003;37:245–248. doi: 10.1053/jhep.2003.50087. [DOI] [PubMed] [Google Scholar]
- 79.Rao VL, Audet RM, Butterworth RF. Increased nitric oxide synthase activities and L-[3H]arginine uptake in brain following portacaval anastomosis. J Neurochem. 1995;65:677–678. doi: 10.1046/j.1471-4159.1995.65020677.x. [DOI] [PubMed] [Google Scholar]
- 80.Rao VL, Audet RM, Butterworth RF. Increased neuronal nitric oxide synthase expression in brain following portacaval anastomosis. Brain Res. 1997;765:169–172. doi: 10.1016/s0006-8993(97)00652-5. [DOI] [PubMed] [Google Scholar]
- 81.Kosenko E, Kaminsky Y, Grau E, Miñana MD, Grisolía S, Felipo V. Nitroarginine, an inhibitor of nitric oxide synthetase, attenuates ammonia toxicity and ammonia-induced alterations in brain metabolism. Neurochem Res. 1995;20:451–456. doi: 10.1007/BF00973101. [DOI] [PubMed] [Google Scholar]
- 82.Master S, Gottstein J, Blei AT. Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology. 1999;30:876–880. doi: 10.1002/hep.510300428. [DOI] [PubMed] [Google Scholar]
- 83.Kosenko E, Kaminski Y, Lopata O, Muravyov N, Felipo V. Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Radic Biol Med. 1999;26:1369–1374. doi: 10.1016/s0891-5849(98)00339-6. [DOI] [PubMed] [Google Scholar]
- 84.Schliess F, Görg B, Fischer R, Desjardins P, Bidmon HJ, Herrmann A, Butterworth RF, Zilles K, Häussinger D. Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J. 2002;16:739–741. doi: 10.1096/fj.01-0862fje. [DOI] [PubMed] [Google Scholar]
- 85.Schliess F, Foster N, Görg B, Reinehr R, Häussinger D. Hypoosmotic swelling increases protein tyrosine nitration in cultured rat astrocytes. Glia. 2004;47:21–29. doi: 10.1002/glia.20019. [DOI] [PubMed] [Google Scholar]
- 86.Görg B, Bidmon HJ, Keitel V, Foster N, Goerlich R, Schliess F, Häussinger D. Inflammatory cytokines induce protein tyrosine nitration in rat astrocytes. Arch Biochem Biophys. 2006;449:104–114. doi: 10.1016/j.abb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 87.Rose C, Kresse W, Kettenmann H. Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes, an effect of pH. J Biol Chem. 2005;280:20937–20944. doi: 10.1074/jbc.M412448200. [DOI] [PubMed] [Google Scholar]
- 88.Ohara K, Aoyama M, Fujita M, Sobue K, Asai K. Prolonged exposure to ammonia increases extracellular glutamate in cultured rat astrocytes. Neurosci Lett. 2009;462:109–112. doi: 10.1016/j.neulet.2009.06.090. [DOI] [PubMed] [Google Scholar]
- 89.Alvarez-Leefmans FJ, Herrera-Pérez JJ, Márquez MS, Blanco VM. Simultaneous measurement of water volume and pH in single cells using BCECF and fluorescence imaging microscopy. Biophys J. 2006;90:608–618. doi: 10.1529/biophysj.105.069450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reinehr R, Görg B, Becker S, Qvartskhava N, Bidmon HJ, Selbach O, Haas HL, Schliess F, Häussinger D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia. 2007;55:758–771. doi: 10.1002/glia.20504. [DOI] [PubMed] [Google Scholar]
- 91.Schliess F, Görg B, Häussinger D. Pathogenetic interplay between osmotic and oxidative stress: the hepatic encephalopathy paradigm. Biol Chem. 2006;387:1363–1370. doi: 10.1515/BC.2006.171. [DOI] [PubMed] [Google Scholar]
- 92.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 93.Zoratti M, Szabò I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 94.Rama Rao KV, Jayakumar AR, Norenberg MD. Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem Int. 2005;47:31–38. doi: 10.1016/j.neuint.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 95.Rama Rao KV, Chen M, Simard JM, Norenberg MD. Suppression of ammonia-induced astrocyte swelling by cyclosporin A. J Neurosci Res. 2003;74:891–897. doi: 10.1002/jnr.10755. [DOI] [PubMed] [Google Scholar]
- 96.Ziemińska E, Dolińska M, Lazarewicz JW, Albrecht J. Induction of permeability transition and swelling of rat brain mitochondria by glutamine. Neurotoxicology. 2000;21:295–300. [PubMed] [Google Scholar]
- 97.Jayakumar AR, Rama Rao KV, Schousboe A, Norenberg MD. Glutamine-induced free radical production in cultured astrocytes. Glia. 2004;46:296–301. doi: 10.1002/glia.20003. [DOI] [PubMed] [Google Scholar]
- 98.Rama Rao KV, Jayakumar AR, Norenberg MD. Differential response of glutamine in cultured neurons and astrocytes. J Neurosci Res. 2005;79:193–199. doi: 10.1002/jnr.20295. [DOI] [PubMed] [Google Scholar]
- 99.Rama Rao KV, Reddy PV, Tong X, Norenberg MD. Brain edema in acute liver failure: inhibition by L-histidine. Am J Pathol. 2010;176:1400–1408. doi: 10.2353/ajpath.2010.090756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lachmann V, Görg B, Bidmon HJ, Keitel V, Häussinger D. Precipitants of hepatic encephalopathy induce rapid astrocyte swelling in an oxidative stress dependent manner. Arch Biochem Biophys. 2013;536:143–151. doi: 10.1016/j.abb.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 101.Kimelberg HK, Mongin AA. Swelling-activated release of excitatory amino acids in the brain: relevance for pathophysiology. Contrib Nephrol. 1998;123:240–257. doi: 10.1159/000059916. [DOI] [PubMed] [Google Scholar]
- 102.Jayakumar AR, Liu M, Moriyama M, Ramakrishnan R, Forbush B, Reddy PV, Norenberg MD. Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem. 2008;283:33874–33882. doi: 10.1074/jbc.M804016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rao KV, Norenberg MD. Cerebral energy metabolism in hepatic encephalopathy and hyperammonemia. Metab Brain Dis. 2001;16:67–78. doi: 10.1023/a:1011666612822. [DOI] [PubMed] [Google Scholar]
- 104.Zwingmann C, Chatauret N, Leibfritz D, Butterworth RF. Selective increase of brain lactate synthesis in experimental acute liver failure: results of a [H-C] nuclear magnetic resonance study. Hepatology. 2003;37:420–428. doi: 10.1053/jhep.2003.50052. [DOI] [PubMed] [Google Scholar]
- 105.Bernal W, Donaldson N, Wyncoll D, Wendon J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: a cohort study. Lancet. 2002;359:558–563. doi: 10.1016/S0140-6736(02)07743-7. [DOI] [PubMed] [Google Scholar]
- 106.Rose C, Ytrebø LM, Davies NA, Sen S, Nedredal GI, Belanger M, Revhaug A, Jalan R. Association of reduced extracellular brain ammonia, lactate, and intracranial pressure in pigs with acute liver failure. Hepatology. 2007;46:1883–1892. doi: 10.1002/hep.21877. [DOI] [PubMed] [Google Scholar]
- 107.Jayakumar AR, Panickar KS, Murthy ChR, Norenberg MD. Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci. 2006;26:4774–4784. doi: 10.1523/JNEUROSCI.0120-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Agre P, Nielsen S, Ottersen OP. Towards a molecular understanding of water homeostasis in the brain. Neuroscience. 2004;129:849–850. doi: 10.1016/j.neuroscience.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 109.Rama Rao KV, Chen M, Simard JM, Norenberg MD. Increased aquaporin-4 expression in ammonia-treated cultured astrocytes. Neuroreport. 2003;14:2379–2382. doi: 10.1097/00001756-200312190-00018. [DOI] [PubMed] [Google Scholar]
- 110.Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Rangroo Thrane V, Enger R, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci USA. 2011;108:846–851. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wright G, Soper R, Brooks HF, Stadlbauer V, Vairappan B, Davies NA, Andreola F, Hodges S, Moss RF, Davies DC, et al. Role of aquaporin-4 in the development of brain oedema in liver failure. J Hepatol. 2010;53:91–97. doi: 10.1016/j.jhep.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 112.Häussinger D, Görg B. Interaction of oxidative stress, astrocyte swelling and cerebral ammonia toxicity. Curr Opin Clin Nutr Metab Care. 2010;13:87–92. doi: 10.1097/MCO.0b013e328333b829. [DOI] [PubMed] [Google Scholar]
- 113.Görg B, Qvartskhava N, Voss P, Grune T, Häussinger D, Schliess F. Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett. 2007;581:84–90. doi: 10.1016/j.febslet.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 114.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 115.Rai V, Nath K, Saraswat VA, Purwar A, Rathore RK, Gupta RK. Measurement of cytotoxic and interstitial components of cerebral edema in acute hepatic failure by diffusion tensor imaging. J Magn Reson Imaging. 2008;28:334–341. doi: 10.1002/jmri.21438. [DOI] [PubMed] [Google Scholar]
- 116.Kale RA, Gupta RK, Saraswat VA, Hasan KM, Trivedi R, Mishra AM, Ranjan P, Pandey CM, Narayana PA. Demonstration of interstitial cerebral edema with diffusion tensor MR imaging in type C hepatic encephalopathy. Hepatology. 2006;43:698–706. doi: 10.1002/hep.21114. [DOI] [PubMed] [Google Scholar]
- 117.Gove CD, Hughes RD, Ede RJ, Williams R. Regional cerebral edema and chloride space in galactosamine-induced liver failure in rats. Hepatology. 1997;25:295–301. doi: 10.1002/hep.510250207. [DOI] [PubMed] [Google Scholar]
- 118.Nguyen JH. Blood-brain barrier in acute liver failure. Neurochem Int. 2012;60:676–683. doi: 10.1016/j.neuint.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Skowrońska M, Albrecht J. Alterations of blood brain barrier function in hyperammonemia: an overview. Neurotox Res. 2012;21:236–244. doi: 10.1007/s12640-011-9269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dam G, Keiding S, Munk OL, Ott P, Vilstrup H, Bak LK, Waagepetersen HS, Schousboe A, Sørensen M. Hepatic encephalopathy is associated with decreased cerebral oxygen metabolism and blood flow, not increased ammonia uptake. Hepatology. 2013;57:258–265. doi: 10.1002/hep.25995. [DOI] [PubMed] [Google Scholar]
- 121.Lockwood AH. Molecules in mammalian brain that interact with the colchicine site on tubulin. Proc Natl Acad Sci USA. 1979;76:1184–1188. doi: 10.1073/pnas.76.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis. 2002;17:221–227. doi: 10.1023/a:1021989230535. [DOI] [PubMed] [Google Scholar]
- 123.Ott P, Larsen FS. Blood-brain barrier permeability to ammonia in liver failure: a critical reappraisal. Neurochem Int. 2004;44:185–198. doi: 10.1016/s0197-0186(03)00153-0. [DOI] [PubMed] [Google Scholar]
- 124.Nguyen JH, Yamamoto S, Steers J, Sevlever D, Lin W, Shimojima N, Castanedes-Casey M, Genco P, Golde T, Richelson E, et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol. 2006;44:1105–1114. doi: 10.1016/j.jhep.2005.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen F, Ohashi N, Li W, Eckman C, Nguyen JH. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology. 2009;50:1914–1923. doi: 10.1002/hep.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen F, Hori T, Ohashi N, Baine AM, Eckman CB, Nguyen JH. Occludin is regulated by epidermal growth factor receptor activation in brain endothelial cells and brains of mice with acute liver failure. Hepatology. 2011;53:1294–1305. doi: 10.1002/hep.24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shawcross DL, Wright GA, Stadlbauer V, Hodges SJ, Davies NA, Wheeler-Jones C, Pitsillides AA, Jalan R. Ammonia impairs neutrophil phagocytic function in liver disease. Hepatology. 2008;48:1202–1212. doi: 10.1002/hep.22474. [DOI] [PubMed] [Google Scholar]
- 128.Häussinger D, Schliess F, Dombrowski F, Vom Dahl S. Involvement of p38MAPK in the regulation of proteolysis by liver cell hydration. Gastroenterology. 1999;116:921–935. doi: 10.1016/s0016-5085(99)70076-4. [DOI] [PubMed] [Google Scholar]
- 129.Bémeur C, Butterworth RF. Liver-brain proinflammatory signalling in acute liver failure: role in the pathogenesis of hepatic encephalopathy and brain edema. Metab Brain Dis. 2013;28:145–150. doi: 10.1007/s11011-012-9361-3. [DOI] [PubMed] [Google Scholar]
- 130.Ahboucha S, Gamrani H, Baker G. GABAergic neurosteroids: the “endogenous benzodiazepines” of acute liver failure. Neurochem Int. 2012;60:707–714. doi: 10.1016/j.neuint.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 131.Ishrat T, Sayeed I, Atif F, Hua F, Stein DG. Progesterone and allopregnanolone attenuate blood-brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp Neurol. 2010;226:183–190. doi: 10.1016/j.expneurol.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jayakumar AR, Ruiz-Cordero R, Tong XY, Norenberg MD. Brain edema in acute liver failure: role of neurosteroids. Arch Biochem Biophys. 2013;536:171–175. doi: 10.1016/j.abb.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bernal W, Auzinger G, Sizer E, Wendon J. Intensive care management of acute liver failure. Semin Liver Dis. 2008;28:188–200. doi: 10.1055/s-2008-1073118. [DOI] [PubMed] [Google Scholar]
- 134.Mohsenin V. Assessment and management of cerebral edema and intracranial hypertension in acute liver failure. J Crit Care. 2013;28:783–791. doi: 10.1016/j.jcrc.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 135.Aggarwal S, Brooks DM, Kang Y, Linden PK, Patzer JF. Noninvasive monitoring of cerebral perfusion pressure in patients with acute liver failure using transcranial doppler ultrasonography. Liver Transpl. 2008;14:1048–1057. doi: 10.1002/lt.21499. [DOI] [PubMed] [Google Scholar]
- 136.Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 137.Tofteng F, Larsen FS. Monitoring extracellular concentrations of lactate, glutamate, and glycerol by in vivo microdialysis in the brain during liver transplantation in acute liver failure. Liver Transpl. 2002;8:302–305. doi: 10.1053/jlts.2002.32283. [DOI] [PubMed] [Google Scholar]
- 138.Jalan R, Kapoor D. Enhanced renal ammonia excretion following volume expansion in patients with well compensated cirrhosis of the liver. Gut. 2003;52:1041–1045. doi: 10.1136/gut.52.7.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Larsen FS, Strauss G, Knudsen GM, Herzog TM, Hansen BA, Secher NH. Cerebral perfusion, cardiac output, and arterial pressure in patients with fulminant hepatic failure. Crit Care Med. 2000;28:996–1000. doi: 10.1097/00003246-200004000-00014. [DOI] [PubMed] [Google Scholar]
- 140.Murphy N, Auzinger G, Bernel W, Wendon J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39:464–470. doi: 10.1002/hep.20056. [DOI] [PubMed] [Google Scholar]
- 141.Tofteng F, Larsen FS. The effect of indomethacin on intracranial pressure, cerebral perfusion and extracellular lactate and glutamate concentrations in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2004;24:798–804. doi: 10.1097/01.WCB.0000125648.03213.1D. [DOI] [PubMed] [Google Scholar]
- 142.Jalan R, O Damink SW, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet. 1999;354:1164–1168. doi: 10.1016/s0140-6736(98)12440-6. [DOI] [PubMed] [Google Scholar]
- 143.Jalan R, Olde Damink SW, Deutz NE, Davies NA, Garden OJ, Madhavan KK, Hayes PC, Lee A. Moderate hypothermia prevents cerebral hyperemia and increase in intracranial pressure in patients undergoing liver transplantation for acute liver failure. Transplantation. 2003;75:2034–2039. doi: 10.1097/01.TP.0000066240.42113.FF. [DOI] [PubMed] [Google Scholar]
- 144.Forbes A, Alexander GJ, O’Grady JG, Keays R, Gullan R, Dawling S, Williams R. Thiopental infusion in the treatment of intracranial hypertension complicating fulminant hepatic failure. Hepatology. 1989;10:306–310. doi: 10.1002/hep.1840100309. [DOI] [PubMed] [Google Scholar]
- 145.Taylor NJ, Nishtala A, Manakkat Vijay GK, Abeles RD, Auzinger G, Bernal W, Ma Y, Wendon JA, Shawcross DL. Circulating neutrophil dysfunction in acute liver failure. Hepatology. 2013;57:1142–1152. doi: 10.1002/hep.26102. [DOI] [PubMed] [Google Scholar]
- 146.Harrison PM, Keays R, Bray GP, Alexander GJ, Williams R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335:1572–1573. doi: 10.1016/0140-6736(90)91388-q. [DOI] [PubMed] [Google Scholar]
- 147.Ytrebø LM, Korvald C, Nedredal GI, Elvenes OP, Nielsen Grymyr OJ, Revhaug A. N-acetylcysteine increases cerebral perfusion pressure in pigs with fulminant hepatic failure. Crit Care Med. 2001;29:1989–1995. doi: 10.1097/00003246-200110000-00023. [DOI] [PubMed] [Google Scholar]
- 148.Stracciari A, Guarino M. Neuropsychiatric complications of liver transplantation. Metab Brain Dis. 2001;16:3–11. doi: 10.1023/a:1011698526025. [DOI] [PubMed] [Google Scholar]
- 149.Guarino M, Benito-Leon J, Decruyenaere J, Schmutzhard E, Weissenborn K, Stracciari A. EFNS guidelines on management of neurological problems in liver transplantation. Eur J Neurol. 2006;13:2–9. doi: 10.1111/j.1468-1331.2006.01353.x. [DOI] [PubMed] [Google Scholar]
- 150.Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marcaida G, Felipo V, Hermenegildo C, Miñana MD, Grisolía S. Acute ammonia toxicity is mediated by the NMDA type of glutamate receptors. FEBS Lett. 1992;296:67–68. doi: 10.1016/0014-5793(92)80404-5. [DOI] [PubMed] [Google Scholar]
- 152.Vogels BA, Maas MA, Daalhuisen J, Quack G, Chamuleau RA. Memantine, a noncompetitive NMDA receptor antagonist improves hyperammonemia-induced encephalopathy and acute hepatic encephalopathy in rats. Hepatology. 1997;25:820–827. doi: 10.1002/hep.510250406. [DOI] [PubMed] [Google Scholar]
- 153.Thiel K, Proven A, Davies N, Leckie P, Frieg R, Storr M, Koenigsrainer A, Schenk M. The development and testing of the University College London liver support device (UCL-ARSeNEL): improvement of survival in paracetamol-induced acute liver failure pigs. Gut. 2010;59:A4. [Google Scholar]
- 154.Rolando N, Clapperton M, Wade J, Panetsos G, Mufti G, Williams R. Granulocyte colony-stimulating factor improves function of neutrophils from patients with acute liver failure. Eur J Gastroenterol Hepatol. 2000;12:1135–1140. doi: 10.1097/00042737-200012100-00011. [DOI] [PubMed] [Google Scholar]
- 155.Rolando N, Clapperton M, Wade J, Wendon J. Administering granulocyte colony-stimulating factor to acute liver failure patients corrects neutrophil defects. Eur J Gastroenterol Hepatol. 2000;12:1323–1328. doi: 10.1097/00042737-200012120-00010. [DOI] [PubMed] [Google Scholar]
- 156.Larsen FS, Hansen BA, Ejlersen E, Secher NH, Clemmesen JO, Tygstrup N, Knudsen GM. Cerebral blood flow, oxygen metabolism and transcranial Doppler sonography during high-volume plasmapheresis in fulminant hepatic failure. Eur J Gastroenterol Hepatol. 1996;8:261–265. doi: 10.1097/00042737-199603000-00014. [DOI] [PubMed] [Google Scholar]
- 157.Cauli O, López-Larrubia P, Rodrigo R, Agusti A, Boix J, Nieto-Charques L, Cerdán S, Felipo V. Brain region-selective mechanisms contribute to the progression of cerebral alterations in acute liver failure in rats. Gastroenterology. 2011;140:638–645. doi: 10.1053/j.gastro.2010.10.043. [DOI] [PubMed] [Google Scholar]