

Abstract
AIM: To investigate the regulative effect of miRNA (miR)-221 on colorectal carcinoma (CRC) cell radiosensitivity and the underlying mechanisms.
METHODS: A human CRC-derived cell line was cultured conventionally and exposed to different doses of X-rays (0, 2, 4, 6 and 8 Gy). The total RNA and protein of the cells were extracted 24 h after irradiation, and the alteration of miR-221 and phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene mRNA expression was detected by real-time reverse transcriptase polymerase chain reaction (PCR). The protein alteration of PTEN in the cells was detected by Western blotting. Caco2 cells were pretreated with or without anti-PTEN-siRNA prior to the addition of pre-miR-221 or anti-miR-221 using Lipofectamine 2000. Colony formation assay and flow cytometry analysis were used to measure the surviving cell fraction and the sensitizing enhancement ratio after irradiation. Additionally, PTEN 3′-untranslated region fragment was PCR amplified and inserted into a luciferase reporter plasmid. The luciferase reporter plasmid construct was then transfected into CRC cells together with pre-miR-221 or anti-miR-221, and the luciferase activity in the transfected cells was detected.
RESULTS: The X-ray radiation dose had a significant effect on the expression of miR-221 and PTEN protein in human Caco2 cells in a dose-dependent manner. The miR-221 expression level improved gradually with the increase in irradiation dose, while the PTEN protein expression level reduced gradually. miR-221 expression was significantly reduced in the anti-miR-221 group compared with the pre-miR-221 and negative control groups (P < 0.01). Anti-miR-221 upregulated expression of PTEN protein and enhanced the radiosensitivity of Caco2 cells (P < 0.01). Moreover, the inhibitory effect was dramatically abolished by pretreatment with anti-PTEN-siRNA, suggesting that the enhancement of radiosensitivity was indeed mediated by PTEN. A significant increase of luciferase activity was detected in CRC cells that were cotransfected with the luciferase reporter plasmid construct and anti-miR-221 (P < 0.01).
CONCLUSION: Anti-miR-221 can enhance the radiosensitivity of CRC cells by upregulating PTEN.
Keywords: Colorectal carcinoma, miR-221, Phosphatase and tensin homolog deleted on chromosome 10, Radiosensitivity
Core tip: Previous studies have shown that miRNA (miR)-221 expression is elevated in radioresistant colorectal carcinoma (CRC) cells; however, it is unknown whether and how miR-221 controls cellular response to irradiation. We demonstrated that knockdown of miR-221 upregulated phosphatase and tensin homolog deleted on chromosome 10 (PTEN) expression, and PTEN was identified as a direct target of miR-221 in CRC. Upregulated PTEN expression suppressed AKT activity and increased radiation-induced cell death, enhancing radiosensitivity in CRC cells. This study provides evidence for antioncogenic activity of anti-miR-221 in the irradiation of CRC and may be a useful biomarker or therapeutic target in CRC.
INTRODUCTION
Colorectal carcinoma (CRC) is one of the most frequent cancers and a common cause of cancer-related death worldwide, with an increasing incidence expected in the next few decades[1]. The overall incidence of CRC is 5% in the general population and the 5-year survival rate ranges from 40% to 60%[2]. The national comprehensive cancer network guidelines on CRC treatment include radiotherapy as standard for patients with a high risk of recurrence (http://www.nccn.org/index.asp). However, the radiotherapeutic efficiency is often limited by the occurrence of radioresistance, reflected as a diminished susceptibility of the irradiated cells to undergo apoptosis. As a result, this therapeutic strategy cannot substantially improve the survival rate[3]. With the understanding of the molecular biology of CRC, it has been recognized that development of radioresistance is related to changes of tumor environment and the dysregulation of certain genes, including some genes involved in a variety of cell signaling pathways as well as oncogenes and tumor suppressor genes[4].
miRNAs are a new class of small noncoding RNAs that regulate the expression of target genes through translational repression or mRNA cleavage/decay[5]. Genome-wide studies have demonstrated that miRNA genes are frequently located at cancer-associated genomic regions, indicating the potential roles of miRNAs in tumorigenesis[6]. miRNAs play an important role in the multistep processes of carcinogenesis, either by oncogenic or tumor suppressor function in CRC[7]. However, only a few studies have determined the roles of miRNAs in radiation response in CRC[8]. miRNA (miR)-221, encoded in tandem from a gene cluster located on chromosome X, is a recently discovered miRNA and is involved in tumor development by regulating cell proliferation cycle[9]. In our previous study, we have demonstrated that miR-221 promotes CRC occurrence and progression, which makes it a potential antitumor candidate for treatment and prevention of CRC[10]. However, the miR-221 response for CRC to survive radiation-induced injury remains largely unknown.
The phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene, located at 10q23.3, encodes a central domain with homology to the catalytic region of protein tyrosine phosphatases[11]. This gene is an important regulator of protein phosphatases and 3′-phosphoinositol phosphatases. PTEN dephosphorylates phosphatidylinositol-3,4,5-triphosphate (PIP3), the second messenger produced by phosphoinositide 3-kinase, to regulate negatively the activity of the serine/threonine protein kinase, AKT[12]. PTEN is inactivated in some malignant tumors, resulting in AKT hyperactivation, thereby promoting cell proliferation, inhibition of apoptosis, and enhanced cell invasion and radioresistance[13]. miRNAs, specifically miR-221, have been established as regulators of PTEN expression[14]. However, how miR-221 affects PTEN in CRC radiation has not been elucidated. Therefore, in this study, we observed the effect of miR-221 on the radiosensitivity of CRC cells and its underlying mechanisms.
MATERIALS AND METHODS
Cell culture and transfection
Human CRC-derived cell lines, including HT-29, Lovo, SW-480, Caco2, and control human umbilical vein endothelial cells (HUVECs), provided by Shanghai Institutes For Biological Science, CAS, were resuscitated routinely, resuspended with RPMI-1640 supplemented with 10% (v/v) fetal bovine serum (FBS; Hyclone, Logan, UT, United States), 100 kU/mL penicillin G and 100 g/L streptomycin, and then planted in a 25-cm2 culture bottle and incubated in a 5% CO2 humidified atmosphere at 37 °C. The media were changed every 3 d and the cells were trypsinized using trypsin/edetic acid when they reached 80%-90% confluence. Cells aged at passages 4-8 were used for the experiments. The day before transfection, cells were seeded in antibiotic-free medium. Cells (1 × 104/well) were seeded in a 96-well plate, incubated for 24 h to allow them attach to the bottom of the well, and then transfected with 50 nmol/L negative control, pre-miR-221 or anti-miR-221 oligonucleotides, respectively (Shanghai GenePharma, Shanghai, China). Transfection of miRNAs was carried out using Lipofectamine 2000 in accordance with the manufacturer’s procedure (Invitrogen, Carlsbad, CA, United States). The above experiment was repeated at least three times.
Cell proliferation analysis of transfected cells by MTT assay
The status of cell proliferation was determined by 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl trozolium bromide (MTT; Amresco, Solon, OH, United States) assay. In short, exponentially growing CRC cells were adjusted to 1.5 × 104 cells/mL with RPMI-1640, planted in 96-well plates (Corning, Corning, NY, United States) at 200 μL/well, and incubated for 12 h. After transfection with 50 nmol/L pre-miR-221 or anti-miR-221 and incubation for 48 h (five duplicate wells for each sample), 20 μL/well MTT (5 g/L) was added to each well. The medium was removed after 4 h incubation and 100 μL/well dimethylsulfoxide was added to dissolve the reduced formazan product. Finally, the plate was read in an enzyme-linked immunity implement (Bio-Rad 2550, Hercules, CA, United States) at 490 nm. Cellular proliferation inhibition rate (CPIR) was calculated using the following formula: CPIR = (1 - average A value of experimental group/average A value of control group) × 100%. The above experiment was repeated at least three times.
miRNA target prediction
The analysis of miR-221-predicted targets was performed using the algorithms TargetScan (http://targetscan.org/), PicTar (http://pictar.mdc-berlin.de/) and MiRanda (http://www.microrna.org/microrna/home.do).
Luciferase activity assay
The human 3′-untranslated region (UTR) of the PTEN gene was amplified by PCR using the primers 5’-CGATTCTAGAAATCATGTTCTGGTGG-3’ for PTEN-3’-UTR-Forward and 5’-GCATTCTAGAATTCTGCACAGTAAGCATA-3’ for PTEN-3’-UTR-Reverse and cloned into the XbaI site of the pGL3-control vector (Promega, Madison, WI, United States), downstream of the luciferase gene, to generate the vector pGL3-PTEN. For luciferase assay, the CRC cells were cultured in 24-well plates and transfected with 500 ng of either pGL3-PTEN or pGL3-control vector and 50 pmol pre-miR-221, anti-miR-221 or negative control. Transfection was performed using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. At 24 h after transfection, firefly luciferase activity was measured using the Dual Luciferase Reporter Assay (Promega, Madison, WI, United States). The above experiment was repeated at least three times.
Radiation exposure
Irradiation was performed at room temperature in a linear accelerator (Varian 600; Palo Alto, CA, United States) at a dose rate of 3.2 Gy/min. Monolayer cells were plated into six-well plates, placed 100 cm from the source, and exposed to the specified dose (0, 2, 4, 6 and 8 Gy) of X-rays[14].
Detection of miR-221 and PTEN mRNA expression by real-time reverse transcriptase polymerase chain reaction
Total RNA was extracted with routine Trizol reagent (Invitrogen). The precipitation was dissolved in diethylpyrocarbonate-treated water. Nucleic acid protein analyzer (Beckman Coulter, Fullerton, CA, United States) was used to determine RNA concentration. The purity and integrity of RNA were identified by two aspects: A260nm/A280nm ≥ 1.8, and a band ratio of 28S to 18S RNA ≥ 1.5 in formaldehyde denaturing gel electrophoresis. miR-221 and PTEN mRNA was quantified as described previously[10]. The comparative 2-ΔΔCT method was used for relative quantification and statistical analysis. The above experiment was repeated at least three times.
Detection of target protein expression by Western blotting
The cells were rinsed twice with cold phosphate buffered solution (PBS) buffer, and were then lysed in an ice-cold lysis buffer containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.6), 0.1% SDS, 1% Nonidet P-40, and protease inhibitor cocktail (Boehringer Mannheim, Lewes, Sussex, United Kingdom). The samples were cleared by centrifugation at 13000 g for 10 min. Fifty micrograms of protein from the tissue was subjected to SDS-PAGE and electrotransferred to polyvinylidine fluoride membranes (Immobilon, Bedford, MA). After blocking in 20 mmol/L Tris-HCl, pH 7.6 (containing 150 mmol/L NaCl, 0.1% Tween-20, and 5% nonfat dry milk), membranes were incubated with primary antibodies against target protein or β-actin (used as a sample loading control) overnight at 4 °C and then incubated with horseradish-peroxidase-conjugated secondary antibody. The blot was developed using the ECL detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, United States) according to the manufacturer’s instructions and the protein imprinting band was obtained. The above experiment was repeated at least three times.
Colony formation assay
At 24 h after irradiation, all cells were trypsinized and counted. Corresponding numbers of cells were seeded into 10-cm dishes containing RPMI-1640 supplemented with 10% FBS, in triplicate, incubated for 14 d to allow colony growth, and colonies were stained with crystal violet. Colonies containing ≥ 50 cells were counted. The plating efficiency was calculated by dividing the average number of colonies per dish by the number of cells plated. Survival fractions were calculated by normalization to the plating efficiency of appropriate control groups. The above experiment was repeated at least three times.
Flow cytometry assay
The effects of miR-221 and irradiation on CRC cell death were examined by flow cytometry. Pretreated CRC cells were harvested and washed twice with PBS, fixed with 70% ethanol at -20 °C for 30 min, and stored at 4 °C overnight, then washed with PBS again, treated with 100 mL 100 mg/L RNase at 37 °C for 30 min, and stained with 100 mL 50 mg/L propidium iodide at 4 °C for 30 min in the dark. The multiplication cycle and apoptotic rate were assayed using flow cytometry, and the data were analyzed using CellQuest software. The percentages of necrotic and apoptotic cells were measured by calculating the ratio of the number of corresponding cells to the number of total cells. For each sample, 10000 cells were measured.
Statistical analysis
All data in the experiment are presented as mean ± SD. Comparisons between groups were analyzed with one-way ANOVA and Student-Newman-Keuls Q test using SPSS version 15.0 (SPSS, Chicago, IL, United States). P < 0.05 was considered to be statistically significant.
RESULTS
Modulation of miR-221 expression in CRC cell lines
To study the expression pattern of miR-221 in CRC cells, we performed real-time RT-PCR to detect miR-221 expression in four CRC-derived cell lines. The expression values of miR-221 in HT-29, Lovo, SW-480 and Caco2 were 4.094 ± 0.208, 1.122 ± 0.138, 3.927 ± 0.232 and 1.831 ± 0.149, respectively. A significant overexpression of miR-221 was observed in all four CRC cell lines relative to HUVEC (0.223 ± 0.047, P < 0.01). The Caco2 cell line was chosen for both pre-miR-221 and anti-miR-221 transfection in the successive experiment because it exhibits, among the four cell lines tested, an intermediate miR-221 expression level.
To determine the biological impact of miR-221 in the CRC-derived cell line, Caco2 cells were transfected with pre-miR-221 or anti-miR-221 to increase or reduce miR-221 level, respectively. Real-time RT-PCR analysis revealed that introduction of pre-miR-221 caused a significant increase of miR-221 value; conversely, anti-miR-221 caused a significant decrease of miR-221 value (Figure 1A, P < 0.01). These strategies were then used as the basis of the remaining experiments.
Figure 1.
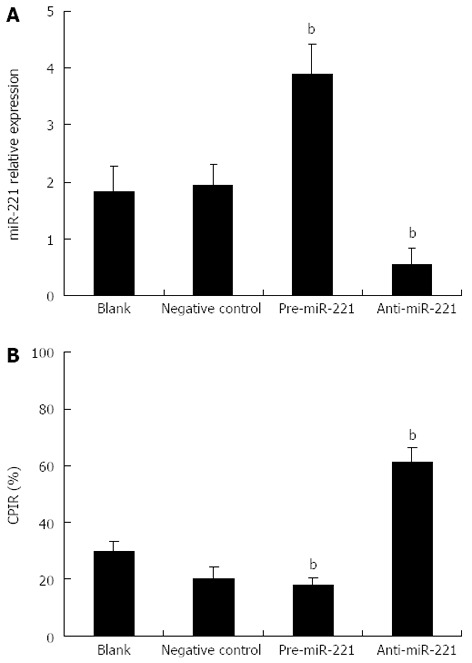
Modulation of miR-221 expression and cell proliferation in Caco2 cells. A: Expression of miR-221 was detected by real-time RT-PCR. Expression of U6 snRNA was used as internal control; B: The status of cell proliferation was determined by MTT assay. CPIR in the presence of pre-miR-221 or anti-miR-221 was compared with those of controls. n = 6, mean ± SD; bP < 0.01 vs control group.
We further tested whether the cell proliferation potential of the transfected CRC cells was modified and the status of cell proliferation was determined by MTT assay. We observed a significant increase in proliferation after transfection of pre-miR-221. In contrast, anti-miR-221 significantly decreased cell proliferation (Figure 1B, P < 0.01). These data indicate that cell proliferation can be significantly enhanced by increase of miR-221 expression, a result which strongly supports the potential oncogenic activity of miR-221 in CRC.
PTEN is a target of miR-221 in CRC
Most miRs are thought to control gene expression by base-pairing with the miR-recognizing elements found in their messenger target. We utilized all three currently available major prediction programs, TargetScan, Miranda and PicTar, to analyze the potential interactions between miR-221 and PTEN. All these algorithms reveal a potential miR-221 target site in the PTEN mRNA 3’-UTR region (Figure 2A). To demonstrate the direct interaction between miR-221 and PTEN mRNA, we cloned PTEN-3’-UTR segment, which includes a potential target site for miR-221, downstream of the pGL3 luciferase reporter gene, to generate the pGL3-PTEN vector. This vector was cotransfected into Caco2 cells together with pre-miR-221 or anti-miR-221. Luciferase activity in Caco2 cells cotransfected with pGL3-PTEN vector and miR-221 was decreased markedly compared with negative controls. On the contrary, luciferase activity in Caco2 cells transfected with anti-miR-221 was increased significantly compared with negative controls (Figure 2B). These results support the bioinformatic prediction indicating the 3’-UTR of PTEN mRNA as a target for miR-221.
Figure 2.
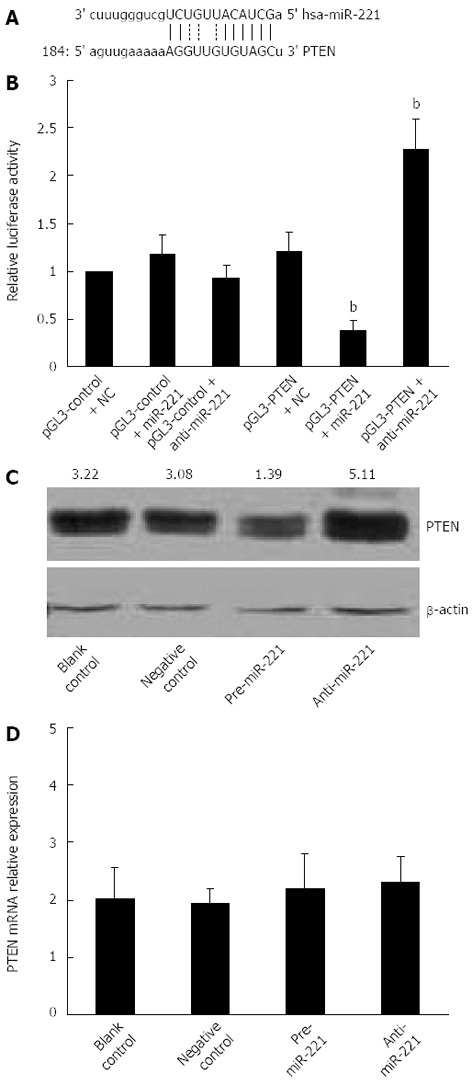
miR-221 regulated expression of phosphatase and tensin homolog deleted on chromosome 10 in colorectal carcinoma-derived cells. A: Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) 3’-UTR site potentially targeted by miR-221 as predicted by Miranda; B: Luciferase activity assay showing direct interaction between miR-221 and PTEN 3’-UTR site. Firefly luciferase reporter activity in the presence of both pGL3-PTEN vector and pre-miR-221 or anti-miR-221 was compared with that of the controls. Luciferase activity in Caco2 cells cotransfected with pGL3-PTEN vector and pre-miR-221 was decreased markedly compared with the negative control. Luciferase activity in Caco2 cells transfected with anti-miR-221 was increased significantly compared with the negative control, bP < 0.01 vs control group; C: Western blotting showing PTEN protein expression in Caco2 cells transfected with pre-miR-221 or anti-miR-221. β-actin was used as a housekeeping gene to normalize PTEN protein expression. The relative PTEN protein levels normalized against β-actin are shown at the top of each panel; D: Real-time RT-PCR analysis showing PTEN mRNA expression in Caco2 cells transfected with pre-miR-221 or anti-miR-221. All the results are representative of three independent experiments.
To check whether miR-221 actually affects PTEN expression in CRC cells, we analyzed the consequence of the ectopic expression of miR-221. We transfected the pre-miR-221 or anti-miR-221 into Caco2 cells, and we searched for changes in PTEN protein levels by Western blotting. Introduction of miR-221 caused a significant increase of miR-221 value and decreased PTEN protein levels. Conversely, anti-miR-221 caused a significant decrease of miR-221 value and increased PTEN protein amounts (Figure 2C). No significant changes in the PTEN mRNA levels were observed in the cells either transfected with miR-221 or anti-miR-221 (Figure 2D). This result strongly validates a post-transcriptional regulation of PTEN protein by miR-221, and also excludes its role in PTEN mRNA degradation.
miR-221 modulates Caco2 cell radiosensitivity
To determine whether miR-221 and PTEN were involved in the cellular response to radiotherapy in CRC, Caco2 cells were exposed to different doses of X-rays to observe the regular pattern of miR-221 and PTEN. The radiation dose had a significant effect on the expression of miR-221 and PTEN protein in human Caco2 cells in a dose-dependent manner. The miR-221 expression level improved gradually with the increase in radiation dose, while the PTEN protein expression level reduced gradually (Figure 3A and B). However, no significant changes in the PTEN mRNA levels were observed in the cells exposed to irradiation (Figure 3C). All these results gave us a first hint that the expression of miR-221 might be one of the mechanisms acting to regulate radiosensitivity in CRC cells negatively.
Figure 3.
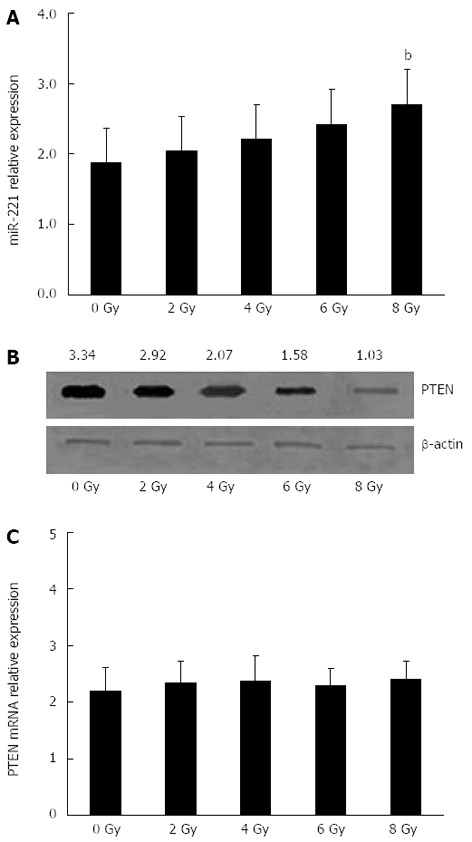
Effects of X-ray dose on miR-221 and phosphatase and tensin homolog deleted on chromosome 10 expression in colorectal carcinoma-derived cells. A: Expression of miR-221 was detected by real-time RT-PCR. The miR-221 expression level improved in a dose-dependent manner with the increase in radiation dose, bP < 0.01 vs control group; B: Expression of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) protein was detected by Western blotting, β-actin was used as a housekeeping gene to normalize PTEN protein expression. The relative PTEN protein levels normalized against β-actin are shown at the top of each panel. The expression of PTEN protein was decreased in a dose-dependent manner with the increase in radiation dose; C: Expression of PTEN mRNA was detected by real-time RT-PCR. No significant changes in the PTEN mRNA levels were observed in the cells exposed to irradiation. The results are representative of three independent experiments.
To determine whether miR-221 affected Caco2 cell radiosensitivity, cells were transfected with pre-miR-221 or anti-miR-221 and colony formation was assessed following 0-8 Gy radiation. Transfection of Caco2 cells with pre-miR-221 significantly increased survival following 0-8 Gy radiation compared to blank and negative controls. Conversely, transfection of Caco2 cells with anti-miR-221 significantly decreased survival following radiation exposure (Figure 4). The D0 value, the radiation dose required to reduce the level of cell survival from 100% to 37%, which is considered a measure of the intrinsic radiosensitivity of the cell, was calculated following genetic manipulation of miR-221. The quasi-threshold dose (Dq) value represents the sublethal damage repair capacity of the cells, which is also a sensitive indicator for the evaluation of cell radiosensitivity. Blank control cells, cells transfected with negative control or pre-miR-221 or anti-miR-221 oligonucleotides exhibited D0 values of 1.681, 1.666, 2.208 and 1.068 Gy, respectively. The sensitization enhancement ratio (SER), calculated by determining the ratio of the D0 of the control group vs treated cells, was 1.009, 0.761 and 1.574 for negative control-, pre-miR-221-, or anti-miR-221-treated cells, respectively (Table 1), indicating a radiosensitization potential for targeting miR-221.
Figure 4.
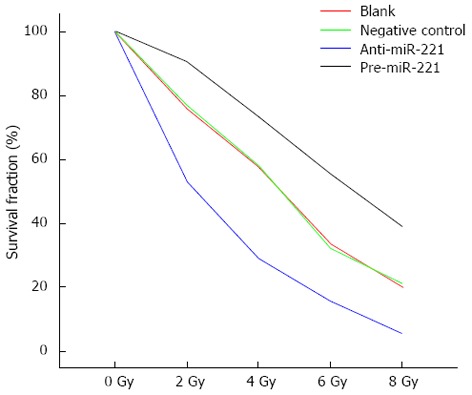
miR-221 modulates colorectal carcinoma cell radiosensitivity. Caco2 cells transfected with negative control, pre-miR-221 or anti-miR-221 oligonucleotides were exposed to 0-8 Gy radiation and incubated for 14 d prior to fixation, staining and assessment of colony formation. The colony formation assays were performed in triplicate.
Table 1.
Impact of miR-221 expression on Caco2 cell radiosensitivity
| Group | D0 | Dq | SF4 | SER |
| Blank control + irradiation | 1.681 | 5.630 | 0.5966 | |
| Negative control + irradiation | 1.666 | 5.813 | 0.5858 | 1.009 |
| Pre-miR-221 + irradiation | 2.208 | 7.828 | 0.7453 | 0.761 |
| Anti-miR-221 + irradiation | 1.068 | 4.655 | 0.2984 | 1.574 |
Caco2 cells were transfected with negative control, pre-miR-221 or anti-miR-221 oligonucleotides. D0 and Dq were determined by standardized software, and the sensitization enhancement ratio (SER) was calculated by determining the ratio of the D0 of the control group vs treated cells. SF4: Surviving fraction at 4 Gy.
To study the mechanism of miR-221 knockdown-induced radiosensitization, we measured irradiation-induced cell death in cells transfected with negative control or anti-miR-221 by flow cytometry. We found that in unirradiated cells transfected with anti-miR-221, there was an increase in necrosis and apoptosis compared to that in the controls. This is consistent with our previous observations that miR-221 knockdown leads to inhibition of cell growth[10]. More interestingly, in irradiated cells, anti-miR-221 transfection enhanced cell death (Figure 5), demonstrating a synergistic effect of miR-221 knockdown with irradiation. Collectively, these results provide strong evidence that miR-221 regulates the radiosensitivity of Caco2 cells.
Figure 5.
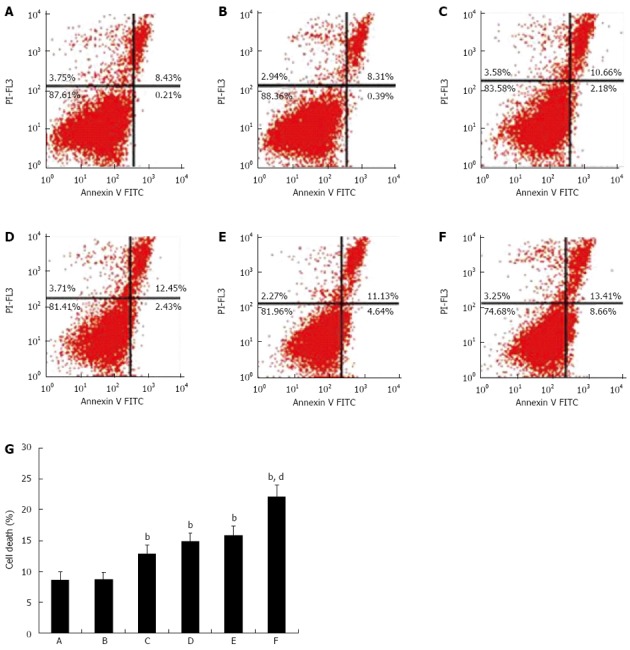
Effects of anti-miR-221 on irradiation-induced death in Caco2 cells. The percentages of necrotic and apoptotic cells were measured by calculating the ratio of the number of corresponding cells to the number of total cells. A: Blank control; B: Caco2 cells transfected with negative control; C: Caco2 cells transfected with anti-miR-221; D: Caco2 cells under irradiation; E: Caco2 cells transfected with negative control under irradiation; F: Caco2 cells transfected with anti-miR-221 under irradiation. The right upper quadrant (FITC+/PI+) shown as necrotic cells. The right lower quadrant (FITC+/PI-) shown as apoptotic cells; G: The status of cell death was determined by flow cytometry. Groups A-F are described as above. n = 3, mean ± SD; bP < 0.01 vs blank or negative control group. dP < 0.01 vs sole anti-miR-221 or irradiation group.
Enhancement of anti-miR-221 on CRC cell radiosensitivity is mediated by PTEN
PTEN is a tumor suppressor protein, thus, we hypothesized that anti-miR-221 might sensitize Caco2 cells to radiotherapy by upregulating PTEN protein expression. To study further downstream pathways of miR-221, we conducted Western blotting to look at phosphorylation of AKT, a downstream target of PTEN. In Caco2 cells transfected with anti-miR-221, there was a significant increase in PTEN expression accompanied by downregulation of AKT phosphorylation (Figure 6A).
Figure 6.
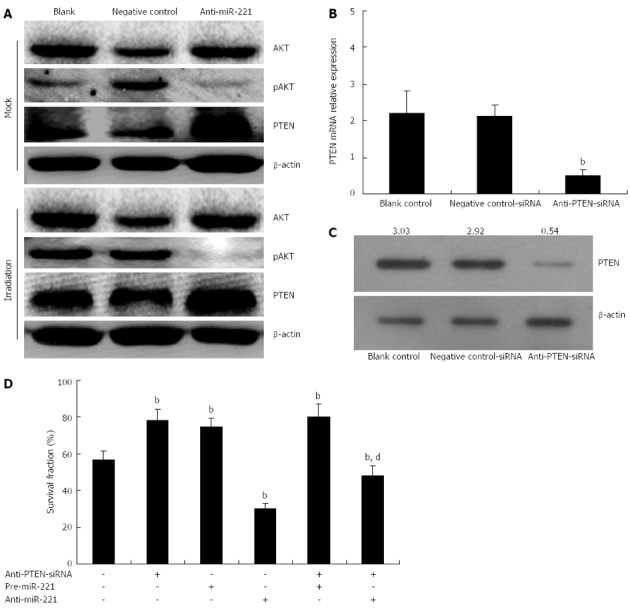
Ectopic expression of miR-221 affects the radiosensitivity of colorectal carcinoma cells by targeting phosphatase and tensin homolog deleted on chromosome 10. A: miR-221 regulates the phosphatase and tensin homolog deleted on chromosome 10 (PTEN)/AKT pathway. Caco2 cells transfected with negative control or anti-miR-221 were mock treated or irradiated (4 Gy). Total cell lysates were obtained for Western blotting using the indicated antibodies. Caco2 cells were pretreated with or without anti-PTEN-siRNA (80 nmol/L) for 24 h prior to the addition of pre-miR-221 (50 nmol/L) or anti-miR-221 (50 nmol/L); B, C: Real-time RT-PCR and Western blotting showing PTEN mRNA and protein reduced markedly after transfection with anti-PTEN-siRNA (bP < 0.01 vs blank control and negative control group); D: The status of cell radiosensitivity was determined by colony formation assay. The suppression of Caco2 cell survival fraction at 4 Gy by anti-miR-221 was partially, but not completely, abrogated by anti-PTEN-siRNA (SF4 from 29.77% to 47.88%). n = 3, mean ± SD; bP < 0.01 vs negative control group; dP < 0.01 vs sole anti-miR-221 group.
Additionally, if anti-miR-221 enhancement of CRC cell radiosensitivity was indeed mediated by PTEN, we would expect that the PTEN-specific and irreversible antagonist, anti-PTEN-siRNA, would abolish this effect. To test this hypothesis, we measured the cell radiosensitivity variations induced by pre-miR-221 or anti-miR-221 in CRC cells previously transfected with anti-PTEN-siRNA. The aim of this experiment was to study if and how the PTEN-depleted cellular environment responds to pre-miR-221 or anti-miR-221 addition and irradiation. Caco2 cells were pretreated with or without anti-PTEN-siRNA (80 nmol/L) for 24 h prior to the addition of pre-miR-221 (50 nmol/L) or anti-miR-221 (50 nmol/L) and the status of cell radiosensitivity was determined by colony formation assay following different doses of X-ray exposure. The data showed that a reduction of PTEN dosage by means different from miR-221 overexpression led to analogous outcomes: when we transfected Caco2 cells with anti-PTEN-siRNA, which was able to reduce both PTEN mRNA and protein by about 80% (Figure 6B and C), we observed a sharp increase in cell survival following radiation as compared with the negative controls (Figure 6D). Thus, reducing PTEN levels in CRC cells, either by miR-221 overexpression or by anti-PTEN-siRNA transduction, is sufficient to induce a comparable cell survival increase.
When pre-miR-221 was transfected into Caco2 cells previously treated with anti-PTEN-siRNA, we observed that anti-PTEN-siRNA and miR-221 seemed to cooperate to increase the survival rate (Figure 6D). However, when anti-miR-221 was transfected into Caco2 cells previously treated with anti-PTEN-siRNA, we observed that the suppression of cell survival by anti-miR-221 was partially abrogated by anti-PTEN-siRNA (Figure 6D, Table 2). These results indicated that the inhibitory effect of anti-miR-221 on CRC cell survival following irradiation was partially, but not completely, mediated by PTEN, suggesting that anti-miR-221 could also activate some PTEN-independent signaling pathways to repress CRC cell growth in addition to the up-regulation of PTEN.
Table 2.
Impact of PTEN on miR-221-mediated Caco2 cell radiosensitivity
| Group | D0 | Dq | SF4 | SER |
| Blank control + irradiation | 1.677 | 5.590 | 0.5667 | |
| Anti-PTEN-siRNA + irradiation | 2.327 | 7.788 | 0.7569 | 0.718 |
| Anti-miR-221 + irradiation | 1.082 | 4.702 | 0.2977 | 1.550 |
| Anti-miR-221 and anti-PTEN-siRNA + irradiation | 1.511 | 5.010 | 0.4788 | 1.110 |
Caco2 cells were pretreated with anti-PTEN-siRNA for 24 h prior to the addition of anti-miR-221. D0 and Dq were determined by standardized software, and the sensitization enhancement ratio (SER) was calculated by determining the ratio of the D0 of the control group vs treated cells. SF4: Surviving fraction at 4 Gy.
DISCUSSION
Radiotherapy is one of the most important treatment methods for CRC. However, only one-third of CRC patients are highly responsive to radiation. The other patients are relatively resistant to radiation, and they tend to progress even after high-dose treatment[15-17]. Factors leading to CRC radioresistance include location, size, and microenvironment such as inadequate vascular supply[18,19]. More importantly, cellular and genetic factors that are related to radiation responses may explain radiation-resistant cellular phenotypes[20,21].
Activation of oncogenes and inactivation of tumor suppressor genes lead to aberrant activity of signal transduction pathways, and radioresistance can be a result of abnormal functioning of these signaling pathways[22-24]. PTEN functions as a tumor suppressor gene, specifically by negatively regulating the AKT/PKB signaling pathway[25]. Previous studies have shown that PTEN is a dual-specificity phosphatase possessing both lipid and protein phosphatase activities. Activated PTEN affects a dephosphorylation of PIP3, generates PIP2, and decreases the phosphorylation level of AKT, which result in cell growth arrest and apoptosis[26]. Genetic inactivation of PTEN is a hallmark of many cancers, including CRC, and reduced expression occurs in many other tumor types. Deficiency of PTEN in the intestine has been reported to induce precancerous polyps, via the induction of formation and fission of crypts, structures located at the base of the intestine containing a rapidly dividing pool of intestinal stem cells[27,28]. Moreover, restoring PTEN expression in PTEN-deficient tumor cells has been shown to enhance radiosensitivity; however, little is known regarding the impact of miRNAs on PTEN expression in CRC[29,30].
Zhang et al[31] studied the miR-221 expression in a gastric cancer-derived cell line and demonstrated that PTEN was the target of miR-221. miR-221 is a newly discovered miRNA which is upregulated in multiple malignant tumors such as hepatocellular carcinoma, bladder cancer, and pancreatic cancer, and facilitates tumors entering S phase from G0/G1 phase by inhibiting the expression of cyclin dependent kinase inhibitor (CDKI)[32]. miR-221 therefore represents an attractive candidate for selective treatment with miR-221-specific inhibitor. miR-221 expression is abnormally increased in CRC and can promote CRC development, which has been confirmed by our previous studies[10]. However, the mechanism by which miR-221 modulates the malignant phenotype, including radioresistance, within CRC remains unknown. Here, we observed miR-221 upregulation in several human CRC-derived lines compared with HUVEC, corroborating the findings of our previous studies[10]. In this study, we predicted that PTEN would be a target gene of the miR-221 by computer-aided algorithm. Moreover, we found binding sites for human miR-221 in the PTEN 3′-UTR by using luciferase activity assay, suggesting that miR-221 might affect PTEN expression. Indeed, we demonstrated that introduction of miR-221 caused a significant increase of miR-221 value and decreased PTEN protein levels. Conversely, anti-miR-221 caused a significant decrease in miR-221 value and increased PTEN protein. Based upon these findings, we hypothesized PTEN as a target of miR-221 in CRC to regulate cell radiosensitivity.
As PTEN is a target of miR-221, and has been described previously as an important regulator of radiation sensitivity, these results suggest that increasing PTEN expression by silencing miR-221 could enhance the radiosensitivity of CRC cells. In this study, transfection of Caco2 cells with pre-miR-221 significantly increased survival following X-ray exposure compared to blank and negative controls. Conversely, transfection of Caco2 cells with anti-miR-221 significantly decreased survival following irradiation. Indeed, we proved that the Caco2 cells were sensitized to radiation by knockdown of miR-221; however, whether PTEN was the sole or main target for miR-221 regulation of radiosensitivity remains unknown. Thus, by using the PTEN-specific antagonist, anti-PTEN-siRNA, we demonstrated that the regulatory effect of anti-miR-221 on CRC cell radiosensitivity was partially, but not completely, mediated by PTEN, suggesting that miR-221 could regulate other PTEN-independent signaling pathways to enhance CRC radiosensitivity. It has been previously shown that CDKN1B/p27 and CDKN1C/p57 are also the target of miR-221 and consistently, CDKN1B/p27 and CDKN1C/p57 expressions inversely correlate in most cancers with miR-221 overexpression, which suggests that the inhibitory effect of anti-miR-221 on CRC cell radiosensitivity is only partially abrogated by anti-PTEN-siRNA[33]. We think that our results, which identify PTEN as a target for miR-221 in the context of CRC cell lines, fit well within a dynamic view of the miRNA-mediated regulation of gene expression: it is well known and widely predicted that the relationship between miRNAs and target mRNAs is not a “one to one” connection, because the same mRNA can be regulated by more than one miRNA, and that the choice of how many and which miRNAs target one 3’-UTR is strongly determined by the specific cellular environment[34-36]. An miRNA that regulates targets playing opposite roles in the control of cell proliferation may act as a tumor suppressor in some cancers and as an oncogene in others, depending on which targets are driving tumorigenesis in that specific cellular milieu.
In summary, we demonstrated that miR-221 could regulate CRC cell radiosensitivity by targeting PTEN. Our data suggest that upregulation of PTEN expression by transfection of anti-miR-221 has important biological effects on the radiosensitivity of CRC cells. These results identify anti-miR-221 as a potential therapeutic approach for CRC via upregulation of PTEN. However, it is noteworthy that the results in this study are based on only one cultured CRC cell line that might not necessarily comprehensively reflect other lines and the in vivo situation. Therefore, further experiments are required to elucidate the antitumor mechanisms of anti-miR-221 in in vivo systems.
COMMENTS
Background
miRNAs regulate gene expression by mainly binding to the 3’-untranslated region (UTR) of target mRNAs, leading to mRNA degradation or translation inhibition. miRNAs are aberrantly expressed in various cancers, suggesting that they play a vital role as a novel class of oncogenes or tumor suppressor genes, depending on the targets they regulate.
Research frontiers
Colorectal carcinoma (CRC) is one of the most dangerous malignancies in China. Previous studies have shown that miR-221 expression is elevated in radioresistant CRC cell lines; however, it is not known whether and how miR-221 controls the cellular response to irradiation. In this study, the authors investigated the alterations of miR-221 and phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene expression in CRC cells after X-ray irradiation, and the mechanisms underlying the enhancement of radiosensitivity to irradiation in CRC cells transfected with anti-miR-221.
Innovations and breakthroughs
Some human miRNAs are consistently deregulated in human cancer, suggesting a role for these genes in tumorigenesis. This study showed that knocking down miR-221 by antisense oligonucleotides upregulated PTEN expression and PTEN was identified as a direct target of miR-221 in CRC. Moreover, upregulated PTEN expression suppressed AKT activity and increased radiation-induced cell death, resulting in enhancement of radiosensitivity in CRC cells.
Applications
This study indicated that anti-miR-221 enhanced the radiosensitivity of CRC cells by upregulating PTEN, and miR-221 might be a novel potential strategy for CRC treatment.
Peer review
Anti-miR-221 could enhance the radiosensitivity of CRC cells by upregulating PTEN. This study provides evidence for the antioncogenic activity of anti-miR-221 in the irradiation of CRC and this may be a useful biomarker or therapeutic target in CRC.
Footnotes
Supported by National Natural Science Foundation of China, No. 81101896; and the National Research Foundation for Doctoral Program of Higher Education of China, No. 20124433110010
P- Reviewers: Mihm S, Pyo H, Xie K S- Editor: Qi Y L- Editor: Logan S E- Editor: Zhang DN
References
- 1.Puente Gutiérrez JJ, Marín Moreno MA, Domínguez Jiménez JL, Bernal Blanco E, Díaz Iglesias JM. Effectiveness of a colonoscopic screening programme in first-degree relatives of patients with colorectal cancer. Colorectal Dis. 2011;13:e145–e153. doi: 10.1111/j.1463-1318.2011.02577.x. [DOI] [PubMed] [Google Scholar]
- 2.Rundle AG, Lebwohl B, Vogel R, Levine S, Neugut AI. Colonoscopic screening in average-risk individuals ages 40 to 49 vs 50 to 59 years. Gastroenterology. 2008;134:1311–1315. doi: 10.1053/j.gastro.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Q, Huang CM, Chen N, Li L, Wang XD, Zhang W, Bi F, Tang QL, Li ZP, Wang W. Chemotherapy and radiotherapy downregulate the activity and expression of DNA methyltransferase and enhance Bcl-2/E1B-19-kDa interacting protein-3-induced apoptosis in human colorectal cancer cells. Chemotherapy. 2012;58:445–453. doi: 10.1159/000345916. [DOI] [PubMed] [Google Scholar]
- 4.Colibaseanu DT, Mathis KL, Abdelsattar ZM, Larson DW, Haddock MG, Dozois EJ. Is curative resection and long-term survival possible for locally re-recurrent colorectal cancer in the pelvis? Dis Colon Rectum. 2013;56:14–19. doi: 10.1097/DCR.0b013e3182741929. [DOI] [PubMed] [Google Scholar]
- 5.Sun K, Deng HJ, Lei ST, Dong JQ, Li GX. miRNA-338-3p suppresses cell growth of human colorectal carcinoma by targeting smoothened. World J Gastroenterol. 2013;19:2197–2207. doi: 10.3748/wjg.v19.i14.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anindo MI, Yaqinuddin A. Insights into the potential use of microRNAs as biomarker in cancer. Int J Surg. 2012;10:443–449. doi: 10.1016/j.ijsu.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 8.Fabbri M. miRNAs as molecular biomarkers of cancer. Expert Rev Mol Diagn. 2010;10:435–444. doi: 10.1586/erm.10.27. [DOI] [PubMed] [Google Scholar]
- 9.Zhou YL, Liu C, Dai XX, Zhang XH, Wang OC. Overexpression of miR-221 is associated with aggressive clinicopathologic characteristics and the BRAF mutation in papillary thyroid carcinomas. Med Oncol. 2012;29:3360–3366. doi: 10.1007/s12032-012-0315-8. [DOI] [PubMed] [Google Scholar]
- 10.Sun K, Wang W, Zeng JJ, Wu CT, Lei ST, Li GX. MicroRNA-221 inhibits CDKN1C/p57 expression in human colorectal carcinoma. Acta Pharmacol Sin. 2011;32:375–384. doi: 10.1038/aps.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagan I, Fatehullah A, Deevi RK, Bingham V, Campbell FC. Rescue of glandular dysmorphogenesis in PTEN-deficient colorectal cancer epithelium by PPARγ-targeted therapy. Oncogene. 2013;32:1305–1315. doi: 10.1038/onc.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong B, Cheng Y, Ma L, Zhang C. MiR-21 regulates biological behavior through the PTEN/PI-3 K/Akt signaling pathway in human colorectal cancer cells. Int J Oncol. 2013;42:219–228. doi: 10.3892/ijo.2012.1707. [DOI] [PubMed] [Google Scholar]
- 13.Zhao G, Cai C, Yang T, Qiu X, Liao B, Li W, Ji Z, Zhao J, Zhao H, Guo M, et al. MicroRNA-221 induces cell survival and cisplatin resistance through PI3K/Akt pathway in human osteosarcoma. PLoS One. 2013;8:e53906. doi: 10.1371/journal.pone.0053906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang C, Kang C, Wang P, Cao Y, Lv Z, Yu S, Wang G, Zhang A, Jia Z, Han L, et al. MicroRNA-221 and -222 regulate radiation sensitivity by targeting the PTEN pathway. Int J Radiat Oncol Biol Phys. 2011;80:240–248. doi: 10.1016/j.ijrobp.2010.12.049. [DOI] [PubMed] [Google Scholar]
- 15.Mirnezami AH, Sagar PM, Kavanagh D, Witherspoon P, Lee P, Winter D. Clinical algorithms for the surgical management of locally recurrent rectal cancer. Dis Colon Rectum. 2010;53:1248–1257. doi: 10.1007/DCR.0b013e3181e10b0e. [DOI] [PubMed] [Google Scholar]
- 16.Dellas K, Reese T, Richter M, Arnold D, Dunst J. Concurrent chemoradiation of metastases with capecitabine and oxaliplatin and 3D-CRT in patients with oligometastatic colorectal cancer: results of a phase I study. Radiat Oncol. 2012;7:83. doi: 10.1186/1748-717X-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caravatta L, Padula GD, Picardi V, Macchia G, Deodato F, Massaccesi M, Sofo L, Pacelli F, Rotondi F, Cecere G, et al. Concomitant boost radiotherapy and multidrug chemotherapy in the neoadjuvant treatment of locally advanced rectal cancer: results of a phase II study. Acta Oncol. 2011;50:1151–1157. doi: 10.3109/0284186X.2011.582880. [DOI] [PubMed] [Google Scholar]
- 18.Ferenschild FT, Vermaas M, Verhoef C, Ansink AC, Kirkels WJ, Eggermont AM, de Wilt JH. Total pelvic exenteration for primary and recurrent malignancies. World J Surg. 2009;33:1502–1508. doi: 10.1007/s00268-009-0066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You YN, Roses RE, Chang GJ, Rodriguez-Bigas MA, Feig BW, Slack R, Nguyen S, Skibber JM. Multimodality salvage of recurrent disease after local excision for rectal cancer. Dis Colon Rectum. 2012;55:1213–1219. doi: 10.1097/DCR.0b013e318270837f. [DOI] [PubMed] [Google Scholar]
- 20.Daly ME, Kapp DS, Maxim PG, Welton ML, Tran PT, Koong AC, Chang DT. Orthovoltage intraoperative radiotherapy for locally advanced and recurrent colorectal cancer. Dis Colon Rectum. 2012;55:695–702. doi: 10.1097/DCR.0b013e31824d464c. [DOI] [PubMed] [Google Scholar]
- 21.Tanis PJ, Doeksen A, van Lanschot JJ. Intentionally curative treatment of locally recurrent rectal cancer: a systematic review. Can J Surg. 2013;56:135–144. doi: 10.1503/cjs.025911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao C, Zhou J, Yang Z, Huang Y, Wu X, Shen H, Tang J, Chen Q. KRAS, BRAF and PIK3CA mutations and the loss of PTEN expression in Chinese patients with colorectal cancer. PLoS One. 2012;7:e36653. doi: 10.1371/journal.pone.0036653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh LL, Er TK, Chen CC, Hsieh JS, Chang JG, Liu TC. Characteristics and prevalence of KRAS, BRAF, and PIK3CA mutations in colorectal cancer by high-resolution melting analysis in Taiwanese population. Clin Chim Acta. 2012;413:1605–1611. doi: 10.1016/j.cca.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 24.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 25.Deevi R, Fatehullah A, Jagan I, Nagaraju M, Bingham V, Campbell FC. PTEN regulates colorectal epithelial apoptosis through Cdc42 signalling. Br J Cancer. 2011;105:1313–1321. doi: 10.1038/bjc.2011.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, Ball RY, Mitrou PN, McTaggart A, Arends MJ. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer. 2011;11:123. doi: 10.1186/1471-2407-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu CP, Kao TY, Chang WL, Nieh S, Wang HL, Chung YC. Clinical significance of tumor suppressor PTEN in colorectal carcinoma. Eur J Surg Oncol. 2011;37:140–147. doi: 10.1016/j.ejso.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Jang KS, Song YS, Jang SH, Min KW, Na W, Jang SM, Jun YJ, Lee KH, Choi D, Paik SS. Clinicopathological significance of nuclear PTEN expression in colorectal adenocarcinoma. Histopathology. 2010;56:229–239. doi: 10.1111/j.1365-2559.2009.03468.x. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe T, Semba S, Yokozaki H. Regulation of PTEN expression by the SWI/SNF chromatin-remodelling protein BRG1 in human colorectal carcinoma cells. Br J Cancer. 2011;104:146–154. doi: 10.1038/sj.bjc.6606018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song M, Chen D, Lu B, Wang C, Zhang J, Huang L, Wang X, Timmons CL, Hu J, Liu B, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS One. 2013;8:e65821. doi: 10.1371/journal.pone.0065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang CZ, Han L, Zhang AL, Fu YC, Yue X, Wang GX, Jia ZF, Pu PY, Zhang QY, Kang CS. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer. 2010;10:367. doi: 10.1186/1471-2407-10-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spahn M, Kneitz S, Scholz CJ, Stenger N, Rüdiger T, Ströbel P, Riedmiller H, Kneitz B. Expression of microRNA-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int J Cancer. 2010;127:394–403. doi: 10.1002/ijc.24715. [DOI] [PubMed] [Google Scholar]
- 33.Fornari F, Gramantieri L, Ferracin M, Veronese A, Sabbioni S, Calin GA, Grazi GL, Giovannini C, Croce CM, Bolondi L, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27:5651–5661. doi: 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- 34.Rossi S, Calin GA. Bioinformatics, non-coding RNAs and its possible application in personalized medicine. Adv Exp Med Biol. 2013;774:21–37. doi: 10.1007/978-94-007-5590-1_2. [DOI] [PubMed] [Google Scholar]
- 35.Sun K, Guo C, Deng HJ, Dong JQ, Lei ST, Li GX. Construction of lentivirus-based inhibitor of hsa-microRNA-338-3p with specific secondary structure. Acta Pharmacol Sin. 2013;34:167–175. doi: 10.1038/aps.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schetter AJ, Okayama H, Harris CC. The role of microRNAs in colorectal cancer. Cancer J. 2012;18:244–252. doi: 10.1097/PPO.0b013e318258b78f. [DOI] [PMC free article] [PubMed] [Google Scholar]