

Abstract
Hepatic stellate cells (HSC) and liver endothelial cells (LEC) migrate to sites of injury and perpetuate alcohol-induced liver injury. High-mobility group box 1 (HMGB1) is a protein released from the nucleus of injured cells that has been implicated as a proinflammatory mediator. We hypothesized that HMGB1 may be released from ethanol-stimulated liver parenchymal cells and contribute to HSC and LEC recruitment. Ethanol stimulation of rat hepatocytes and HepG2 cells resulted in translocation of HMGB1 from the nucleus as assessed by Western blot. HMGB1 protein levels were increased in the supernatant of ethanol-treated hepatocytes compared with vehicle-treated cells. Migration of both HSC and LEC was increased in response to conditioned medium for ethanol-stimulated hepatocytes (CMEtOH) compared with vehicle-stimulated hepatocytes (CMVEH) (P < 0.05). However, the effect of CMEtOH on migration was almost entirely reversed by treatment with HMGB1-neutralizing antibody or when HepG2 cells were pretransfected with HMGB1-siRNA compared with control siRNA-transfected HepG2 cells (P < 0.05). Recombinant HMGB1 (100 ng/ml) also stimulated migration of HSC and LEC compared with vehicle stimulation (P < 0.05 for both HSC and LEC). HMGB1 stimulation of HSC increased the phosphorylation of Src and Erk and HMGB1-induced HSC migration was blocked by the Src inhibitor PP2 and the Erk inhibitor U0126. Hepatocytes release HMGB1 in response to ethanol with subsequent recruitment of HSC and LEC. This pathway has implications for HSC and LEC recruitment to sites of ethanol-induced liver injury.
Keywords: HMGB1; ethanol; hepatocyte; hepatic stellate cells (HSC), liver endothelial cells (LEC)
alcoholic liver disease is one of the most common causes of liver cirrhosis and portal hypertension worldwide; however, the mechanisms by which ethanol leads to liver injury and fibrosis are not fully understood (14, 18, 35, 44). Amongst various injury pathways, ethanol and/or its by-products certainly directly injure hepatocytes (4, 6, 15, 22, 31). Hepatic stellate cells (HSC) and liver endothelial cells (LEC) are integral to the subsequent steps by which parenchymal cell injury leads to liver fibrosis. Both cell types reside in a quiescent state in the hepatic sinusoids; however, injury and the ensuing wound healing response recruits both these cell types to sites of injury. HSC and LEC then act in tandem for development of fibrosis and its associated angiogenesis (39). The process by which injured cells release molecules that recruit inflammatory cells in absence of infection has been referred to as “sterile inflammation.” Although prior studies have characterized this process for recruitment of inflammatory cells, the mechanisms by which LEC and HSC participate are less understood.
Owing to its unique anatomic location downstream from the gut along with its fenestrated and permeable sinusoidal characteristics, the liver is continuously exposed to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharides (LPS) or endogenous danger-associated molecular patterns (DAMPs) (47). For example, activation of Kupffer cells by LPS derived from intestinal bacteria is viewed as a pivotal step in the injury process (27, 29). PAMPs and DAMPs are recognized by specific receptors, which in turn activate inflammasome signaling pathways that promote migration of innate immune and vascular cells to sites of injury. In response to continuous injury, this may lead to chronic inflammation and fibrosis. Recognition of DAMPs by Toll-like receptor 4 (TLR4) activates NF-κB and leads to sterile inflammation (19). In this process, TLR4 activated cells release molecules through the inflammasome pathway, to recruit effector cells to sites of injury, although how this occurs in ethanol-induced injury is not fully understood.
High-mobility group box 1 (HMGB1) is an intracellular DNA-binding protein expressed by all mammalian cells. It interacts with various receptors including TLR2, TLR4, TLR9, and RAGE (receptor for advanced glycation end products) (21). HMGB1 resides in the nucleus as a histone protein (20, 28, 30, 34, 38), but in the sterile inflammation response, it is released from necrotic cells (8, 11, 30) and functions as an inflammatory mediator. In this sterile inflammation response, HMGB1 is known to function as a TLR4 ligand (16). Although recent studies implicated HMGB1 in ischemic liver injury (23, 40, 41) and in acute liver failure by viral hepatitis (26, 46), it is not clear whether and how HMGB1 contributes to alcoholic liver injury. In this study, we test the concept that HMGB1 may be released from ethanol-treated liver parenchymal cells and contribute to HSC and LEC recruitment.
METHODS
Cell culture and preparation.
Human HSCs, human LECs, primary rat hepatocytes, and HepG2 cells were used in this study. HSCs and LECs were purchased from ScienCell Research Laboratories (San Diego, CA) (3). Rat hepatocytes (3×106) were isolated by standard techniques as previously described (37) and plated onto 10-cm gelatin-coated tissue culture dishes. Hepatocytes were allowed to attach to tissue culture plasticware overnight prior to use. HSCs, rat hepatocytes, and HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO) with 10% fetal bovine serum and 1% penicillin/streptomycin (ScienCell) under standard tissue culture conditions (a humidified 5% carbon dioxide incubator at 37°C). LECs were cultured in medium containing 5% fetal bovine serum, 2% endothelial cell growth supplement, and 1% penicillin-streptomycin. Cells at passages 3–8 were studied. Cells were serum starved overnight prior to experiments. LPS-free HMGB1 and HMGB1-neutralizing antibody were purchased from HMGBiotech (Milan, Italy). All cell culture and transfection reagents were purchased from GIBCO (Rockville, MD).
Transfection.
siRNA targeting human HMGB1 and a scrambled control were purchased from Qiagen (Valencia, CA). Cells were seeded in 60-mm dishes and transfected with siRNA by using oligofectamine (Invitrogen, Carlsbad, CA) as described previously. Transfected cells were incubated in medium containing 10% FBS for 3 days prior to experiments.
Preparation of conditioned medium.
Rat hepatocytes or HepG2 cells were cultured in a 100 × 15 mm dish and treated with 0–100 mM of ethanol or vehicle for 24 h. Supernatant were collected after centrifugation at 500 g for 15 min to remove necrotic cells or cell debris. The resultant supernatants were collected to use as conditioned media (CMEtOH for ethanol-stimulated hepatocytes or CMVEH for vehicle-stimulated hepatocytes). For separate experiments, HepG2 cells were cultured in a 100 × 15 mm dish containing basal DMEM with 50 and 100 mM of ethanol for 24 h. In parallel, basal DMEM with 50 and 100 mM of ethanol were prepared through same incubation time. The resultant supernatants were collected to use as EtOH CM for conditioned medium from ethanol-stimulated HepG2 cells and EtOH DMEM for basal DMEM containing ethanol.
Isolation of nuclear and cytoplasmic proteins and Western blotting.
Cells were washed twice with ice-cold PBS and homogenized in a cell lysis buffer at 4°C for 20 min. After centrifugation, the protein concentration in the lysates was measured by a Bradford assay. In some experiments nuclear and cytoplasmic cell lysates from HepG2 cells and rat hepatocytes were collected for Western blot analysis by using previously validated protocols (3). Lysates containing 30–50 μg of proteins were heated for 3 min at 100°C. Protein lysates were separated on a 12 or 15% acrylamide gel and transferred to polyvinylidene difluoride membranes (GE Healthcare, Buckinghamshire, UK). After 60-min incubation with 5% nonfat dry milk (Bio-Rad) or 5% albumin from bovine serum (Sigma-Aldrich) at room temperature to block the nonspecific binding, membranes were incubated at 4°C overnight with specific primary antibodies and then, for 2 h with secondary antibodies conjugated to horseradish peroxidase at 4°C. Membranes were washed and protein bands were detected with an enhanced chemiluminescence detection system (ECL Plus, Santa Cruz Biotechnology) according to the manufacturer's instructions. When necessary, membranes were stripped and reprobed with an anti-GAPDH antibody (1:105). Digitalization of films was performed with a scanner (Epson V750, Nagano, Japan). Quantification of band density was performed by use of Image J 1.40G (NIH, Bethesda).
HMGB1 ELISA.
HMGB1 concentrations were measured by enzyme-linked immunosorbent assay (ELISA) (IBL, Toronto, Ontario, Canada) that detects rat and mouse HMGB1 according to the manufacturer's instructions.
Real-time PCR.
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Invitrogen Life Technologies). The reverse-transcription reaction was performed by using 1 μg total RNA that was reverse-transcribed into the first-strand cDNA by Superscript II reverse transcriptase with random primers (Invitrogen Life Technologies). PCR mixture was prepared with SYBR Green PCR Master Mix (PE Applied Biosystems, Foster City, CA) by using the primers as shown in Table 1. Thermal cycling conditions were 10 min at 95°C followed by 40 cycles of 95°C for 15 s and 60°C for 1 min on an ABI PRISM 7000 Sequence Detection System (PE Applied Biosystems). Gene expression was normalized with rat 14S mRNA or mouse β-actin mRNA content.
Table 1.
Primer sequence
| Sequence | Size | |
|---|---|---|
| Rat | ||
| HMGB1 | ||
| Sense | CCGGATGCTTCTGTCAACTT | 216 |
| Antisense | TTGATTTTTGGGCGGTACTC | |
| 14S | ||
| Sense | GGGGAAGGACAAGAAGGAAG | 162 |
| Antisense | CCACCAGTTACTCGGCAAAT | |
| Mouse | ||
| HMGB1 | ||
| Sense | CCGGATGCTTCTGTCAACTT | 208 |
| Antisense | TTGATTTTTGGGCGGTACTC | |
| β-Actin | ||
| Sense | AGAGGGAAATCGTGCGTGAC | 138 |
| Antisense | CAATAGTGATGACCTGGCCGT |
Migration assay.
Migration of HSC and LEC was evaluated by Boyden chamber assay in response to conditioned medium or recombinant HMGB1. Boyden assay was performed as previously described (7). In brief, modified Boyden chambers (Becton Dickinson, Heidelberg, Germany) were used with filters (8 μm pores, Neuro Probe, Gaithersburg, MD) coated with collagen type I (50 μg/ml). HMGB1 (100 ng/ml) or conditioned medium was added to the lower chamber and 3,000 cells in 50 μl of serum-free DMEM were added to the upper chamber. In some experiments, HMGB1-neutralizing antibody was added in lower wells of Boyden chambers. For inhibition of RAGE or TLR4 receptors, HSC and LEC were preincubated for 30 min at 4°C with 40 μg/ml anti RAGE (R&D Systems) or 5 μg/ml anti-TLR4 (Biolegend) or murine control IgG (Biolegend) (5). After 4 h incubation at 37°C, cells remaining on the upper surface of filters were scraped off with a cotton swab and cells on the lower surface were fixed with ethanol and stained with DAPI. Cells were counted by use of the ImagePro program. Results are the means ± SE of the number of cells counted in ×4 low-power fields per filter.
Cell viability and caspase 3/7 assay.
To evaluate the cytotoxicity of ethanol toward hepatocytes, cell viability assay was performed. In brief, after incubation of hepatocytes with ethanol (0, 10, 50, and 100 mM) for 24 h, the cells were exposed to a 0.4% Trypan blue solution for 3 min and viewed under a light microscope. Cell viability was defined as the ratio of unstained cells to the total number of cells. HepG2 cells (1 × 104/well) were used for MTS (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation assay, Promega) and caspase 3/7 assay (Apo-One Homogeneous Caspase-3/7 Assay, Promega) with incubation time 4 h or 30 min, respectively. Camptothecin (10 and 20 μM) was used as a positive control for apoptosis.
Immunocytochemistry.
HepG2 cells were treated with basal DMEM containing 100 mM ethanol for 24 h. The cells were fixed with 2.5% paraformaldehyde for 5 min and washed three times with PBS. For permeability, the cells were exposed to 0.1% Triton X-100 in PBS for 5 min. After blocking with 10% FBS in PBS, the cells were incubated with the HMGB1 antibody overnight at +4°C, and then secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG) was applied for 1 h at room temperature. Phalloidin and ToTo3 were used to stain cytoplasm and nucleus for 20 min, respectively.
Immunohistochemistry.
Paraffin-embedded sections of mouse liver were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed in sodium citrate buffer pH 6. After blocking endogenous peroxidase for 15 min, the sections were blocked with 10% FBS in PBS for 1 h at room temperature. The sections were incubated with the HMGB1 antibody overnight at +4°C, and then biotinylated secondary antibody (Vector Laboratories) was applied for 1 h at room temperature. ABC reagent (Vector Laboratories) was used to develop the stain, and brown staining in cytoplasm was quantitated in randomly chosen sections.
In vivo ethanol feeding studies in mice.
C57B6 mice were fed with Lieber-DeCarli liquid diet (32% calories from ethanol, 5% volume) or nonalcohol isocaloric control liquid diet for 4 wk as described (24). All animal work was performed under Mayo IACUC oversight.
Statistical analysis.
Data expressed were as means ± SE of data obtained from at least three independent experiments and compared by Mann-Whitney U-test or ANOVA for multiple comparison parametric data. A P value less than 0.05 was considered statistically significant.
RESULTS
Ethanol induces HMGB1 release from hepatocytes.
To examine whether ethanol stimulates HMGB1 release from hepatocytes, rat hepatocytes were treated with 0, 10, 50, and 100 mM of ethanol for 24 h and supernatants were collected and assayed for HMGB1 release by Western blot analysis and ELISA. HMGB1 expression by Western blot analysis (Fig. 1A) and HMGB1 concentration by HMGB1 ELISA (Fig. 1B) were significantly increased in the supernatant of cells incubated with ethanol in a concentration-dependent manner. There was no significant change in HMGB1 mRNA expression after ethanol treatment (Fig. 1C), suggesting that the increased release of HMGB1 does not involve increased transcription. Because HMGB1 can be passively released from necrotic cells, viability of hepatocytes after ethanol treatment was also evaluated. Cell viability did not differ between hepatocytes treated with ethanol and vehicle for 24 h (Fig. 1D). This result was supported by data using HepG2 cells, in which ethanol dosing showed neither necrosis nor apoptosis (Fig. 1D). Since cytoplasmic HMGB1 translocation correlates with eventual cellular release (12), Western blot analysis was performed with nuclear and cytoplasmic protein lysates of ethanol-treated rat hepatocytes to determine whether ethanol treatment induces the translocation of HMGB1 from nucleus to cytoplasm in hepatocytes. Nuclear HMGB1 protein levels decreased while cytoplasmic levels increased in response to ethanol treatment, with levels in the total lysates remaining unchanged (Fig. 1E; top and bottom). Similar results were observed in ethanol-treated HepG2 cells (Fig. 1F). This was further corroborated by immunocytochemistry studies showing cytoplasmic translocation of HMGB1 in HepG2 cells treated with ethanol (Fig. 1G). Collectively, these data indicate that ethanol actively stimulates HMGB1 release from hepatocytes.
Fig. 1.
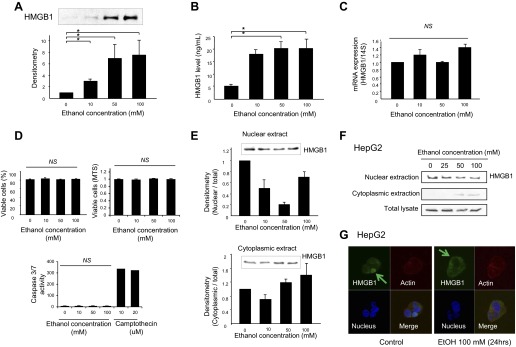
Ethanol induces high-mobility group box 1 (HMGB1) cytosolic translocation and release from hepatocytes. Rat hepatocytes were treated with ethanol (0–100 mM) for 24 h and supernatants were collected for Western blot assay and HMGB1 ELISA. Western blot assay shows significant increase of HMGB1 protein levels in the supernatants of ethanol-treated cells (A). HMGB1 concentration was increased in the supernatants of ethanol-treated hepatocytes by HMGB1 ELISA (B). Rat hepatocytes were treated with ethanol (0–100 mM) for 24 h and mRNA was prepared for real-time PCR. There was no significant change in HMGB1 mRNA levels after ethanol treatment (C). Ethanol- or vehicle-treated hepatocytes were collected and stained with Trypan blue. There was no significant difference in the proportion of viable cells between vehicle-treated cells and ethanol-treated cells. In MTS and caspase 3/7 assay performed with HepG2 cells, neither necrosis nor apoptosis was observed in response to ethanol (D). Rat hepatocytes (E) and HepG2 cells (F) were treated with ethanol (0–100 mM) for 24 h and nuclear and cytoplasmic fractions were isolated and prepared for SDS-PAGE. HMGB1 expression was significantly decreased in nuclear fractions and increased in cytoplasmic fraction after ethanol treatment by Western blot analysis. In immunocytochemistry study, HMGB1 was more visible in the cytoplasm of ethanol-treated HepG2 cells compared with control (G). HMGB, green; blue, nucleus; red, cytoplasm (original magnification ×65). Results were obtained from at least 3 independent experiments. Data in bar graphs represent means ± SE. *P < 0.05; NS, not significant.
Recombinant and ethanol-stimulated hepatocyte HMGB1 induce migration of hepatic stellate cells and liver sinusoidal endothelial cells.
The effect of HMGB1 on migration of HSC and LEC was evaluated by modified Boyden chamber assay. Recombinant human HMGB1 induced migration of human HSCs (Fig. 2A) and human LECs (Fig. 2B) in a concentration-dependent manner. Similarly, significantly greater numbers of HSC (Fig. 2C) and LEC (Fig. 2D) migrated in the Boyden assay in response to CMEtOH compared with CMVEH. Interestingly, the migration of cells decreased in CMe100 (conditioned medium from 100 mM ethanol-stimulated hepatocytes) compared with CMe50 (conditioned medium from 50 mM ethanol-stimulated hepatocytes). This is consistent with prior studies in which increasing HMGB1 levels were less effective in stimulating migration of endothelial progenitor cells, possibly due to changes in protein conformation or receptor-ligand binding kinetics (5). To exclude an effect of residual ethanol from CMEtOH on HSC migration, EtOH CM and EtOH DMEM were subjected to Boyden chamber assay and their effect on cell migration were compared in both HSC and LEC. The results showed significantly higher migration in EtOH CM compared with EtOH DMEM, excluding the possibility that increased migration in response to EtOH CM is due to residual EtOH in the conditioned medium (Fig. 2, E and F). Next, to confirm that HMGB1 was causative in CMEtOH-induced migration, experiments were performed with HMGB1-neutralizing antibody. The migratory effect of CMEtOH was blocked by HMGB1-neutralizing antibody in both HSC (Fig. 3A) and LEC (Fig. 3B). To further evaluate the effect of HMGB1 on the cell migration, CMEtOH and CMVEH were prepared from HepG2 cells after HMGB1-siRNA transfection. HepG2 cells were used in this assay because they are more amenable to transfection than primary hepatocytes. HMGB1 protein levels were significantly decreased in HMGB1-siRNA-transfected cells compared with control-siRNA-transfected cells and nuclear levels were reduced in response to ethanol (Fig. 3C). More cells migrated in response to CMEtOH of control-siRNA-transfected cells, compared with CMEtOH of HMGB1-siRNA-transfected cells for both HSCs (Fig. 3D) and LECs (Fig. 3E). In total, these data indicate that HMGB1 stimulates migration of both HSC and LEC.
Fig. 2.

HMGB1 induces migration of human (h) hepatic stellate cells (HSC) and liver endothelial cells (LEC). HSC and LEC (3,000 cells/well) were loaded into the upper wells of Boyden assay chamber and recombinant HMGB1 (0–100 ng/ml) or conditioned medium for ethanol-stimulated hepatocytes (CMEtOH) compared with vehicle-stimulated hepatocytes (CMVEH) were loaded into the lower wells and incubated for 4 h at 37°C. CMEtOH was collected from the media of rat hepatocytes treated with ethanol (10, 50, and 100 mM) for 24 h. Migration of HSC (A) and LEC (B) was significantly increased in response to recombinant HMGB1. Similar findings were noted in response to CMEtOH (C with HSC and D with LEC). In separate experiment evaluating the effect of ethanol on cell migration, migration of HSC (E) and LEC (F) was significantly increased in EtOH CM compared with EtOH DMEM. (*P < 0.05). CMe100, conditioned medium from 100 mM ethanol-stimulated hepatocytes; CMe50, conditioned medium from 50 mM ethanol-stimulated hepatocytes; EtOH CM, conditioned medium from ethanol-stimulated HepG2 cells; EtOH DMEM, basal DMEM containing ethanol. Results depicted are compiled from at least 3 experiments. Data represents means ± SE. *P < 0.05.
Fig. 3.

HMGB1 is essential for CMEtOH-induced migration of HSC and LEC. In Boyden chamber assay, CMEtOH-induced cell migration was blocked by HMGB1-neutralizing antibody (1 μg/ml) in HSC (A) and LEC (B). HepG2 cells were transfected with HMGB1-siRNA. HMGB1 protein expression was markedly decreased by HMGB1-siRNA transfection, and nuclear (N) levels were reduced in response to ethanol based on Western blot assay. GAPDH blot is shown from nuclear and cytosolic (C) preparations (C). CMEtOH from HMGB1-siRNA-transfected cells did not induce migration of HSC (D) or LEC (E), whereas the number of migrated cells was significantly increased by CMEtOH from control-siRNA-transfected cells. Results depicted are compiled from at least 3 experiments. Data represents means ± SE. *P < 0.05.
HMGB1 requires Src/Erk pathway for HSC migration but not for LEC migration.
We next sought to identify signaling mechanisms that mediate HSC and LEC migration in response to HMGB1. Since Src and Erk are implicated in liver nonparencyhmal cell migration (3, 48), we evaluated the changes of the activation of Src and Erk in response to recombinant HMGB1. After treatment with HMGB1 (100 ng/ml) for varying durations (0–90 min), cells were collected for Western blot assay. Both Src and Erk were activated after 15-min stimulation with recombinant HMGB1 in HSC (Fig. 4A). This response was not observed in LEC, in which only small and nonsignificant changes were observed in p-Src after extended duration of incubation (Fig. 4B).
Fig. 4.
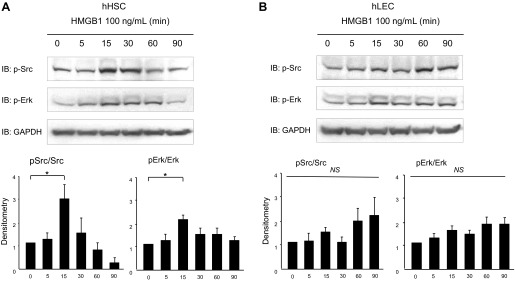
HMGB1 induces Src and Erk phosphorylation in HSC. Serum-starved HSC and LEC were treated with recombinant HMGB1 (100 ng/ml) for different durations (0–90 min). Total cell lysates were used for Western blot to detect phosphorylated Src and Erk. Src and Erk phosphorylation were significantly increased in HSC in response to 15-min treatment with HMGB1 (A) but not in LEC (B). *P < 0.05. Blots are representative of at least 3 replicates. Graph depicts data compiled from experiments and replicates in total. IB, immunoblotting.
To further explore the role of Src in HMGB1-induced migration, Boyden chamber assay was performed after blocking Src by two different approaches. First, HSCs and LECs were pretreated with 10 μM of the Src inhibitor PP2 for 30 min and Boyden chamber assay was performed. PP2 attenuates HMGB1-induced migration of HSC (Fig. 5A). However, PP2 did not affect HMGB1-induced migration of LEC (Fig. 5B). Next, cells were transfected with a Src dominant-negative mutant retroviral construct (Src-Y419F) that attenuates Src function or a Src-wild-type (Src-WT) retroviral construct (48). In Boyden chamber assay, cell migration was increased in response to recombinant HMGB1 protein in Src-WT HSCs, but not Src-Y419F mutant-transfected HSCs (Fig. 5C). However, transfection of Src-Y419F mutant construction did not affect HMGB1-induced migration of LEC (Fig. 5D). These results suggested that Src is required for the migratory effect of HMGB1 on HSC but not LEC.
Fig. 5.

Src contributes to HMGB1-induced HSC migration. Boyden chamber assay was performed on HSC and LEC exposed to recombinant HMGB1 (100 ng/ml). PP2 (10 μM) pretreatment for 30 min blocked HMGB1-induced migration of HSC (A) but did not affect HMGB1 migration of LEC (B). HSC and LEC were transfected with Src mutant retroviral construct (Src-Y419F) or Src-wild type (Src-WT) retrovirus. In Boyden chamber assay, cell migration was increased in response to recombinant HMGB1 in Src-WT HSCs, but not Src-Y419F mutant HSCs (C). Src-Y419F mutant construct transfection did not affect HMGB1 migration of LEC (D). Results were obtained from at least 3 independent experiments. Data in bar graphs represent means ± SE and are expressed as number of cells in the filter. *P < 0.05.
In Western blot assay, PP2 pretreatment also blocked HMGB1-induced Erk phosphorylation in HSC (Fig. 6A), whereas this response was not significant in LEC (Fig. 6B). These results suggest that Erk may be downstream of Src in HMGB1 migration signaling in HSC. Therefore, we further explored role of Erk in HMGB1-induced migration. Boyden chamber assay was performed after pretreatment of cells with the Erk inhibitor U0126. HMGB1-induced migration of HSC was blocked by 30-min pretreatment with U0126 (5 μM) (Fig. 6C), whereas U0126 did not affect HMGB1-induced migration of LEC (Fig. 6D). Thus the Src/Erk pathway plays a pivotal role in HMGB1-induced HSC migration but not for HMGB1-induced LEC migration. In LEC, HMGB1 (100 ng/ml) increased Akt activation by Western blot assay (Fig. 7A). Additionally, HMGB1-induced migration of LEC was blocked by 30-min pretreatment with Akt inhibitor (Fig. 7B). These data suggest that Akt may be involved in LEC migration in response to HMGB1.
Fig. 6.
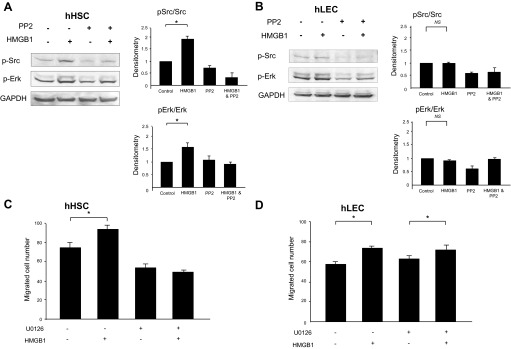
Erk contributes to HMGB1-induced HSC migration. HSC and LEC were treated with recombinant HMGB1 (100 ng/ml) or PP2 or U0126 for 15 min then prepared for Western blot assay or for cell migration studies. PP2 pretreatment blocked the activation of Erk in HSC (A) but not in LEC (B). HMGB1-induced cell migration was blocked by 30-min pretreatment with the Erk inhibitor U0126 (5 μM) in HSC (C) but not in LEC (D). Results were obtained from at least 3 independent experiments. Data in bar graphs represent means ± SE and are expressed as number of cells in the filter. *P < 0.05.
Fig. 7.

Akt may contribute to HMGB1-induced LEC migration. Western blotting and Boyden chamber assay were performed on LEC treated with recombinant HMGB1 (100 ng/ml). Serum-starved LEC was treated with recombinant HMGB1 for different time course (0–90 min). Total cell lysates were used for Western blotting. Akt phosphorylation was increased in time 60 and 90 min (A). HMGB1-induced cell migration was attenuated by 30-min pretreatment with Akt inhibitor (B). Results were obtained from at least 3 independent experiments. Data in bar graphs represent means ± SE and are expressed as number of cells in the filter. *P < 0.05.
HMGB1 induced cell migration via TLR4 and RAGE in HSC.
We performed experiments with inhibitory antibodies against two major HMGB1 receptors, TLR4 and RAGE, to evaluate their role in HMGB1-induced cell migration in HSC and LEC. Neutralizing anti-TLR4 and anti-RAGE antibodies significantly blocked the HMGB1-induced cell migration in HSC (Fig. 8A). However, neither anti-TLR4 nor anti-RAGE antibodies blocked the HMGB1-induced migration in LEC (Fig. 8B). These data indicate that HMGB1 induces cell migration through TLR4 and RAGE in HSC.
Fig. 8.

HMGB1 induced cell migration via Toll-like receptor 4 (TLR4) or RAGE (receptor for advanced glycation end products). HSC and LEC were preincubated with neutralizing anti-TLR4 or anti-RAGE antibodies or control IgG or PBS before migration assay. HSC and LEC (3,000 cells/well) were loaded into the upper wells and recombinant human HMGB1 (100 ng/ml) or vehicle was loaded into the lower wells of Boyden chamber. HMGB1-induced cell migration of HSC (A) was significantly blocked by anti-TLR4 or anti-RAGE antibodies. However, these antibodies did not affect HMGB1-induced cell migration of LEC (B). Results were obtained from at least 3 independent experiments. Data in bar graphs represent means ± SE and are expressed as number of cells in the filter. *P < 0.05.
HMGB1 in alcohol-induced liver injury in vivo.
To evaluate the role of HMGB1 in alcohol-induced liver injury in vivo, mice were fed alcohol (24). Steatosis was documented in liver tissue of alcohol-fed mice as previously described (Fig. 9A). HMGB1 mRNA levels from total liver were significantly increased in alcohol-fed mice (Fig. 9B). Increased HMGB1 mRNA tissue levels in vivo are likely a result of infiltrating inflammatory cells that also generate HMGB1 since HMGB1 mRNA levels from hepatocytes in vitro were not elevated in response to ethanol (Fig. 1C). HMGB1 cytoplasmic translocation was also detected in hepatocytes in alcohol-fed mice as assessed by immunohistochemistry (Fig. 9C). Although serum HMGB1 levels were increased by 30%, this change did not reach statistical significance (Fig. 9D). These data suggest that HMGB1 translocation occurs in alcohol-induced liver injury in vivo. In vivo studies with more inflammatory and/or fibrotic alcohol feeding models will be useful to explore this further.
Fig. 9.
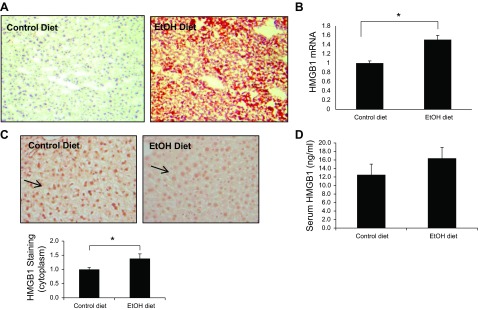
HMGB1 cytoplasmic translocation in alcohol-induced liver injury in vivo. C57B6 mice were fed with alcohol diet or control diet for 4 wk. After mice were euthanized, Oil Red O staining (A), HMGB1 mRNA (B), and immunohistochemistry for HMGB1 (C) were assessed in liver tissue. Serum HMGB1 levels also were measured (D). Original magnification ×20; 5 mice in each group; *P < 0.05.
DISCUSSION
In this study, we examined the role of HMGB1 in the hepatocyte response to ethanol. Our results indicate that ethanol induces HMGB1 secretion by hepatocytes. In addition, secreted HMGB1 induces migration of HSC and LEC. We also uncover specific signaling mechanisms by which this is achieved. For example, migration of HSC requires activation of the nonreceptor tyrosine kinase Src as well as activation of the kinase Erk. In total, the work identifies a pathway that may contribute to alcohol injury responses in liver.
Previous studies have demonstrated that intranuclear HMGB1 can be actively secreted or passively released into extracellular space in response to pathogens or in response to necrotic cell injury (1, 2, 25, 32, 34, 41, 43). Extracellular HMGB1 in turn acts as a recruitment signal to induce an inflammatory reaction (41, 43). This process has been best characterized in liver in context of ischemia-reperfusion injury (9). Our results indicate that ethanol also activates the HMGB1 injury pathway. In our experiments, HMGB1 release was likely active since cell viability was intact in response to ethanol and thus release was not a result of the passive release of intracellular contents by necrotic cells. The steps by which HMGB1 is actively released from cells remain under active investigation, although our initial data show that active release via exosomes seems to be less likely than a traditional secretory pathway (data not shown).
Prior studies have shown that HMGB1 acts as a chemokine for various cell types (10, 33, 45). However, the molecular steps by which HMGB1 stimulates cell migration are not fully understood especially in HSC and LEC (8, 13, 42). In the present studies we found that HMGB1 induces migration of both HSC and LEC; however, the mechanisms of cell motility were different in the two cell types. We used a number of complementary approaches to explore the role of the Src and Erk intracellular mediators in cell migration. These studies revealed that although HMGB1 utilizes both Src and Erk for HSC migration, these mediators are not required for HMGB1-induced migration of LEC. Further studies will be required to elucidate how HMGB1 induces LEC migration; however, Akt may be a likely mediator based on the present studies and prior literature (48).
LPS is increasingly recognized as an instigator of diverse liver injury processes. In alcohol-induced injury, there is an increase in gut permeability that increases LPS delivery to the liver through the portal vein. LPS acts on a multitude of liver cells that express TLR4 to induce responses that include cell activation and migration (9, 17, 36). Furthermore, recent studies indicate that sterile inflammation, that is, injury in the absence of infection, can induce similar responses (28). These nonmicrobial endogenous danger signals are termed DAMPs and include HMGB1, heat-shock protein, uric acid, and double-stranded genomic DNA (20, 28, 34). Interestingly, HMGB1 has been implicated not only as a potential TLR4 agonist but also as a molecule that is produced and released in response to TLR4 activation (16). Thus HMGB1 could contribute to sterile inflammation through both of these mechanisms. These responses are important for wound healing reactions to tissue injury (20), and our work implicates HMGB1 in these processes in HSC.
Prior studies have shown that HMGB1 can induce cell migration (10, 33) and proinflammatory cytokine production (34, 43). Our study supports a potential role for this molecule in ethanol-induced liver injury. Ethanol induces translocation and secretion of HMGB1 from the hepatocytes. HMGB1 in turn induces migration of HSCs and LECs to the injured sites via Src/Erk and Akt pathways, respectively. The results support the concept that ethanol-induced HMGB1 release may play an important role in the pathogenesis of chronic inflammation and recruitment of cells contributing to the wounding response in alcoholic liver disease.
GRANTS
Work was supported by NIH AA021171 and AA021788 (V. Shah).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.S.S., J.H.K., and V.H.S. conception and design of research; Y.S.S., J.H.K., and V.K.V. performed experiments; Y.S.S. and J.H.K. interpreted results of experiments; Y.S.S. and J.H.K. drafted manuscript; J.H.K. and U.Y. analyzed data; J.H.K. and V.K.V. prepared figures; J.H.K., L.Y., T.M.d.A., D.A.S., and V.K.V. edited and revised manuscript; J.H.K. and V.H.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Eugene Chang, Tim Billiar, and Greg Gores for helpful discussions.
REFERENCES
- 1.Albayrak A, Uyanik MH, Cerrah S, Altas S, Dursun H, Demir M, Uslu H. Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol 23: 633–638, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81: 1–5, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E, Charlton M, Watts RJ, Mukhopadhyay D, Shah VH. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest 120: 2379–2394, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ceni E, Crabb DW, Foschi M, Mello T, Tarocchi M, Patussi V, Moraldi L, Moretti R, Milani S, Surrenti C, Galli A. Acetaldehyde inhibits PPARgamma via H2O2-mediated c-Abl activation in human hepatic stellate cells. Gastroenterology 131: 1235–1252, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, Zeiher AM, Chavakis T, Dimmeler S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res 100: 204–212, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Charrier AL, Leask A, French SW, Brigstock DR. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J Hepatol 55: 399–406, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das A, Shergill U, Thakur L, Sinha S, Urrutia R, Mukhopadhyay D, Shah VH. Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest Liver Physiol 298: G908–G915, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol 152: 1197–1206, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem 285: 39888–39897, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fages C, Nolo R, Huttunen HJ, Eskelinen E, Rauvala H. Regulation of cell migration by amphoterin. J Cell Sci 113: 611–620, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Falciola L, Spada F, Calogero S, Langst G, Voit R, Grummt I, Bianchi ME. High mobility group 1 protein is not stably associated with the chromosomes of somatic cells. J Cell Biol 137: 19–26, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3: 995–1001, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson WD, Moll RH. Experimental evaluation of relationships among populations resulting from intergradation among cultivars of ZEA MAYS L. Genetics 74: 133–138, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haydon G, Lalor PF, Hubscher SG, Adams DH. Lymphocyte recruitment to the liver in alcoholic liver disease. Alcohol 27: 29–36, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Holmuhamedov EL, Czerny C, Beeson CC, Lemasters JJ. Ethanol suppresses ureagenesis in rat hepatocytes: role of acetaldehyde. J Biol Chem 287: 7692–7700, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoque R, Vodovotz Y, Mehal W. Therapeutic strategies in inflammasome mediated diseases of the liver. J Hepatol 58: 1047–1052, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jagavelu K, Routray C, Shergill U, O'Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 52: 590–601, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karaa A, Kamoun WS, Clemens MG. Chronic ethanol sensitizes the liver to endotoxin via effects on endothelial nitric oxide synthase regulation. Shock 24: 447–454, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167: 5887–5894, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 8: 279–289, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 143: 1158–1172, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Lalor PF, Faint J, Aarbodem Y, Hubscher SG, Adams DH. The role of cytokines and chemokines in the development of steatohepatitis. Semin Liver Dis 27: 173–193, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, Hou YJ, Chang YX, Tu QQ, Feng GS, Shen F, Wu MC, Wang HY. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 54: 1620–1630, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res 6: 523–531, 1982 [DOI] [PubMed] [Google Scholar]
- 25.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Muller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res 97: e73–e83, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Majumdar M, Ratho R, Chawla Y, Singh MP. High levels of circulating HMGB1 as a biomarker of acute liver failure in patients with viral hepatitis E. Liver Int 33: 1341–1348, 2013 [DOI] [PubMed] [Google Scholar]
- 27.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol 50: 1258–1266, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330: 362–366, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res 35: 787–793, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller S, Scaffidi P, Degryse B, Bonaldi T, Ronfani L, Agresti A, Beltrame M, Bianchi ME. New EMBO members' review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J 20: 4337–4340, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto N, Greenwel P, Friedman SL, Zhang F, Dannenberg AJ, Cederbaum AI. Ethanol and arachidonic acid increase α2(I) collagen expression in rat hepatic stellate cells overexpressing cytochrome P450 2E1. Role of H2O2 and cyclooxygenase-2. J Biol Chem 275: 20136–20145, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol 164: 441–449, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauvala H, Huttunen HJ, Fages C, Kaksonen M, Kinnunen T, Imai S, Raulo E, Kilpelainen I. Heparin-binding proteins HB-GAM (pleiotrophin) and amphoterin in the regulation of cell motility. Matrix Biol 19: 377–387, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 371: 838–851, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13: 1324–1332, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Shah V, Chen AF, Cao S, Hendrickson H, Weiler D, Smith L, Yao J, Katusic ZS. Gene transfer of recombinant endothelial nitric oxide synthase to liver in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol 279: G1023–G1030, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Stott K, Tang GS, Lee KB, Thomas JO. Structure of a complex of tandem HMG boxes and DNA. J Mol Biol 360: 90–104, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol 53: 976–980, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med 204: 2913–2923, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201: 1135–1143, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang FP, Li L, Li J, Wang JY, Wang LY, Jiang W. High mobility group box-1 promotes the proliferation and migration of hepatic stellate cells via TLR4-dependent signal pathways of PI3K/Akt and JNK. PLoS One 8: e64373, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Yamagishi Y, Horie Y, Kato S, Kajihara M, Tamai H, Granger DN, Ishii H. Ethanol modulates gut ischemia/reperfusion-induced liver injury in rats. Am J Physiol Gastrointest Liver Physiol 282: G640–G646, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18: 250–259, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Zhou RR, Zhao SS, Zou MX, Zhang P, Zhang BX, Dai XH, Li N, Liu HB, Wang H, Fan XG. HMGB1 cytoplasmic translocation in patients with acute liver failure. BMC Gastroenterol 11: 21, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 18: 1272–1282, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou L, Cao S, Kang N, Huebert RC, Shah VH. Fibronectin induces endothelial cell migration through beta1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem 287: 7190–7202, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]