

Abstract
Recent studies indicate that metabolic oxidative stress, autophagy, and inflammation are hallmarks of nonalcoholic steatohepatitis (NASH) progression. However, the molecular mechanisms that link these important events in NASH remain unclear. In this study, we investigated the mechanistic role of purinergic receptor X7 (P2X7) in modulating autophagy and resultant inflammation in NASH in response to metabolic oxidative stress. The study uses two rodent models of NASH. In one of them, a CYP2E1 substrate bromodichloromethane is used to induce metabolic oxidative stress and NASH. Methyl choline-deficient diet feeding is used for the other NASH model. CYP2E1 and P2X7 receptor gene-deleted mice are used to establish their roles in regulating metabolic oxidative stress and autophagy. Autophagy gene expression, protein levels, confocal microscopy based-immunolocalization of lysosome-associated membrane protein (LAMP)2A and histopathological analysis were performed. CYP2E1-dependent metabolic oxidative stress induced increases in P2X7 receptor expression and chaperone-mediated autophagy markers LAMP2A and heat shock cognate 70 but caused depletion of light chain 3 isoform B (LC3B) protein levels. P2X7 receptor gene deletion significantly decreased LAMP2A and inflammatory indicators while significantly increasing LC3B protein levels compared with wild-type mice treated with bromodichloromethane. P2X7 receptor-deleted mice were also protected from NASH pathology as evidenced by decreased inflammation and fibrosis. Our studies establish that P2X7 receptor is a key regulator of autophagy induced by metabolic oxidative stress in NASH, thereby modulating hepatic inflammation. Furthermore, our findings presented here form a basis for P2X7 receptor as a potential therapeutic target in the treatment for NASH.
Keywords: 5,5-dimethyl-1-pyrroline N-oxide-nitrone adducts; lipid peroxidation; tyrosine nitration; cytokines; CYP2E1; light chain 3 isoform B; GABA-A receptor-associated protein
nonalcoholic steatohepatitis (NASH) is associated with metabolic oxidative stress, often ascribed to enhanced oxidation of membrane lipids, mitochondrial uncoupling of the electron transport chain, and higher cytochrome P450 enzyme activity (18, 29, 30). Metabolic oxidative stress leads to cellular stress and induces cell death mechanisms, which modulate inflammatory microenvironment in NASH (5, 24).
Cell death mechanisms in disease pathogenesis are synonymous with necrosis, autophagy, and apoptosis; however, much attention is recently focused on the various roles of autophagy in cellular processes, inflammation, energy homeostasis, and immunity (8, 35). Despite increased attention on autophagy, the mechanisms that control autophagy in NASH remain unclear. Autophagy is a central eukaryotic process with many cytoplasmic homeostatic roles (8). It is a critical pathway for degradation of intracellular components by lysosomes and has established roles in hepatic lipid metabolism, insulin sensitivity, and cellular injury (2). Autophagy, dependent on the involvement of the cellular components, has been classified into macro, micro, and chaperone-mediated autophagy. It is crucial, however, that these processes rely on formation of autophagosomes, increased levels of light chain 3 isoform B (LC3B), fusion of autophagosome with the lysosome, and degradation of the cellular cargo by lysosomal enzymes (2, 15, 23, 26). Interestingly, a recent investigation by Lin. et al. (21) reports pharmacological promotion of autophagy-alleviated steatosis and injury in alcoholic and nonalcoholic fatty liver. Given the importance of autophagy in cellular homeostasis and disease pathogenesis, it is also important to study both the inducers and regulators of autophagy in a pathological state like NASH. One of the key components of the inflammatory microenvironment in various disease pathologies is the inotropic purinergic receptors (P2 receptors) that respond to the damage-associated molecular patterns (3, 28). There is an increased focus on a subclass of these receptors (P2X7 receptor), which bind ATP with high affinity and are known to regulate inflammation in early steatohepatitic injury in mice (5). Apart from the binding to ATP, purinergic receptor X7 (P2X7) receptor (P2X7r) has been shown to increase NADPH oxidase activity, increase Kupffer cell- major histocompatibility complex (MHC) class II expression, and regulate inflammation (5). With increased focus on purinergic receptors, inflammasome activation, and autophagy in NASH, we explored the possibility of P2X7r as a key regulator of autophagy and the resultant inflammation in NASH.
The present study utilizes two distinct models of experimental NASH, which have been established to produce pathology of full-blown NASH. The toxin model of NASH utilizes coadministration of high-fat diet and a low-dose environmental toxin bromodichloromethane (BDCM) (7, 31). The diet-induced model exposes mice to methyl-choline-deficient (MCD) diet for 8 wk (36, 37). In a first ever report, we show that in both models of NASH there was an increased metabolic oxidative stress, which caused higher expression of P2X7 receptors. Oxidative stress induced both early and late autophagy proteins and was dependent on the presence of P2X7 receptors. P2X7 receptor modulated LC3B protein depletion, an event that is crucial for autophagy. Further absence of P2X7 receptors significantly reduced downstream inflammasome activation and NASH pathophysiology. The present study also advances our understanding in considering P2X7r-linked autophagy pathways as potential therapeutic targets in NASH.
MATERIALS AND METHODS
Mouse Model
Pathogen-free, custom diet-induced obesity (DIO) adult male mice with a C57BL/6J background (Jackson Laboratories, Bar Harbor, ME) were used as models of toxin-induced NASH. They were fed with a high-fat diet (60% kcal) from 6 wk to 16 wk. All experiments were conducted at the completion of 16 wk. The animals were housed one in each cage before any experimental use. Mice that contained the deleted CYP2E1 gene (cytochrome p450 knockout) (29/Sv-Cyp2e1tm1Gonz/J) (Jackson Laboratories) and mice that contained the deleted P2X7r gene (purinergic receptor X7 knockout) (B6.129P2-P2rx7tm1Gab/J) (Jackson Laboratories) were fed with a high-fat diet and treated identically to DIO mice. The other set of mice were pathogen-free, adult male with a C57BL/6J background fed with MCD diet and were used as models for diet-induced NASH. They were fed with MCD diet from 8 to 16 wk. Mice that contained the disrupted P2X7r gene were also fed with MCD. Mice had ad libitum access to food and water and were housed in a temperature-controlled room at 23–24°C with a 12-h:12-h light/dark cycle. All animals were treated in strict accordance with the NIH Guide for the Humane Care and Use of Laboratory Animals, and the experiments were approved by the institutional review boards at NIEHS, Duke University, and the University of South Carolina at Columbia.
Induction of Liver Injury in Mice
DIO mice or high-fat-fed gene-specific knockout mice at 16 wk were administered BDCM (2.0 mmol/kg, diluted in olive oil) through the intraperitoneal route. However, DIO mice treated with olive oil (diluent of BDCM) were used as control. After completion of the treatment, mice of all study groups were killed for liver tissue and serum for the further experiments. MCD-diet-fed wild-type or gene-specific knockout mice were killed at 16 wk for liver tissue for experiments. However, methylcholine-sufficient (MCS) diet-fed mice were used as control.
ELISA
Immunoreactivity for 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-nitrone adduct was detected in liver homogenate using standard ELISA (4).
Histopathology.
Liver tissue sections from each mouse were fixed in 10% neutral buffered formalin. Formalin-fixed liver sections were stained with hematoxylin and eosin and observed under the light microscope. Collagen content in liver tissue was evaluated using Sirius red-stained liver sections. Each liver section was stained with Picro-Sirius red (Sigma-Aldrich, St. Louis, MO) and counterstained with fast green (Sigma-Aldrich).
Serum ALT, alkaline phosphatase, and albumin levels.
Blood from DIO, DIO+BDCM, and P2X7 receptor-deleted mice were collected by cardiac puncture, and serum was separated by standard techniques. The serum alanine aminotransferase (ALT) and alkaline phosphatase were measured at the clinical chemistry core. Albumin was measured using the commercially available kit (Alpha Diagnostic, San Antonio, TX).
Immunohistochemistry.
Formalin-fixed, paraffin-embedded liver tissue from all the mice groups were cut into 5-μm-thick tissue sections. Each section was deparaffinized using standard protocol. Briefly, sections were incubated with xylene twice for 3 min, washed with xylene:ethanol (1:1) for 3 min, and rehydrated through a series of ethanol (twice with 100%, 95%, 70%, 50%), twice with distilled water, and finally rinsed twice with PBS (Sigma-Aldrich). Epitope retrieval of deparaffinized sections was carried out using epitope retrieval solution and steamer (IHC-World, Woodstock, MD) following the manufacturer's protocol. The primary antibodies were 1) anti-4-hydroxynonenal, 2) anti-3-nitrotyrosine, 3) anti-P2X7r, 4) anti- GABA-A receptor-associated protein (GABARAP), 5) anti-lysosome-associated membrane protein (LAMP)-2A, 6) anti-MHC II, 7) anti-IL-1β, 8) anti-IFN-γ, and 9) anti-LC3B. Primary antibodies were purchased from Abcam (Cambridge, MA), Millipore (Temecula, CA), and R&D Systems (Minneapolis, MN) and used in 1:250 dilutions. Antigen-specific immunohistochemistry were performed using Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) following manufacturer's protocols. 3,3' Diaminobenzidine (Sigma-Aldrich) was used as a chromogen substrate. Sections were counter-stained by Mayer's hematoxylin (Sigma-Aldrich). Washing with PBS (Sigma-Aldrich) was performed thrice between the steps. Sections were mounted in Simpo mount (GBI Laboratories, Mukilteo, WA) and observed under a ×20 oil objective. Morphometric analysis was done using CellSens Software from Olympus America.
Western Blotting
Tissue (30 mg) from each liver sample was homogenized in 100 μl of RIPA buffer (Sigma-Aldrich) with protease inhibitor (1×) (Pierce, Rockford, IL) using dounce homogenizer. The homogenate was centrifuged, and the supernatant was diluted 1:5 and used for SDS PAGE and subjected to Western blotting. Novex (Invitrogen, Carlsbad, CA) 4–12% bis-tris gradient gel was used for SDS PAGE. Proteins were transferred to nitrocellulose membrane using precut nitrocellulose/filter paper sandwiches (Bio-Rad Laboratories, Hercules, CA) and Trans-Blot Turbo transfer system (Bio-Rad) in case of low molecular weight proteins and using wet transfer module from Invitrogen in case of high molecular weight proteins. A solution of 5% non-fat milk was used for blocking. Primary antibodies against heat shock cognate 70 (Hsc 70), GABARAP, LC3B, LAMP-2A, Caspase-1, β-actin (all from Abcam), high-motility group box protein (HMGB)-1 (Millipore) at recommended dilutions, and compatible horseradish peroxidase-conjugated secondary antibodies were used. Pierce ECL Western Blotting substrate (Thermo Fisher Scientific, Rockford, IL) was used. The blot was developed using BioMax MS Films and cassettes (with intensifying screen, Kodak). The images were subjected to densitometry analysis using Lab Image 2006 Professional 1D gel analysis software from KAPLEAN Bioimaging Solutions (Liepzig, Germany).
Quantitative RT-PCR
Gene expression levels in tissue samples were measured by two-step qRT-PCR. Total RNA was isolated from liver tissue by homogenization in TRIzol reagent (Invitrogen) according to the manufacturer's instructions and purified with the use of RNeasy mini kit columns (Qiagen, Valencia, CA). Purified RNA (1 μg) was converted to cDNA using iScript cDNA synthesis kit (Bio-Rad) following the manufacturer's standard protocol. qRT-PCR was performed with the gene-specific primers using SsoAdvanced SYBR Green supermix (Bio-Rad) and CFX96 thermal cycler (Bio-Rad). Threshold Cycle (Ct) values for the selected genes were normalized against 18S (internal control) values in the same sample. Each reaction was carried out in triplicate for each gene and for each tissue sample. DIO mouse liver sample was used as the control for comparison with all other liver samples in the toxin-induced NASH group, and MCS-diet-fed mouse liver sample was used as control for comparison with all other liver samples of the diet-induced NASH group. The relative fold change was calculated by the 2−ΔΔCt method. The sequences for the primers used for real-time PCR are provided in Table 1.
Table 1.
Sequences for the primers used for real-time PCR in 5′ to 3′ orientation
| Gene | Primer Sequence |
|---|---|
| CYP2E1 | Sense: GGCGCATCGTGGTCCTGCAT |
| Antisense: CCGCACGTCCTTCCATGTGGG | |
| P2X7r | Sense: GGGACGCTGAAGAACACCTT |
| Antisense: CCTAACTTCGTCACCCCACC | |
| GABARAP | Sense: AGAGGAGCATCCGTTCGAGA |
| Antisense: CGAGCTTTGGGGGCTTTTTC | |
| Atg-2A | Sense: AACATCCAACGTGCTAGGGG |
| Antisense: GGACAGTGGGTGGATCTTGG | |
| LC3B | Sense: GCTGTGAGGACAACAGCAAC |
| Antisense: AGTGAGTGAGTGACCAGGGA | |
| MHC II | Sense: TGATTCTGGGGGTCCTCGCCC |
| Antisense: CGTGGTCGGCCTCAATGTCGT | |
| LAMP2A | Sense: GTACCTGACAAGGCGACACA |
| Antisense: CCCAAGAGACAGCGAATCCA | |
| TGF-β | Sense: CTCACCGCGACTCCTGCTGC |
| Antisense: TCGGAGAGCGGGAACCCTCG | |
| COL-1-α-1 | Sense: TGGACGATGGGGCTGGGGAG |
| Antisense: GGGTGCCCAGTGGTCGCTTC | |
| α-SMA | Sense: GGAGAAGCCCAGCCAGTCGC |
| Antisense: ACCATTGTCGCACACCAGGGC | |
| Hsc 70 | Sense: AGTTGGCATTGATCTCGGCA |
| Antisense: GTGTTGGTGGGGTTCATTGC | |
| Caspase-1 | Sense: GGACCCTCAAGTTTTGCCCT |
| Antisense: AACTTGAGCTCCAACCCTCG | |
| IL-1β | Sense: CCTCGGCCAAGACAGGTCGC |
| Antisense: TGCCCATCAGAGGCAAGGAGGA | |
| TNF-α | Sense: CAACGCCCTCCTGGCCAACG |
| Antisense: TCGGGGCAGCCTTGTCCCTT | |
| IFN-γ | Sense: TGCGGGGTTGTATCTGGGGGT |
| Antisense: GCGCTGGCCCGGAGTGTAGA | |
| HMGB-1 | Sense: GGACTCTCCTTTAACCGCCA |
| Antisense: TTGTGATAGCCTTCGCTGGG |
P2X7, purinergic receptor X7; GABARAP, GABA-A receptor-associated protein; Atg-2A, autophagy-related protein 2A; LC3B, light chain 3 isoform B; MHCII, histocompatibility complex class II; LAMP2A, lysosome-associated membrane protein 2A; SMA, smooth muscle actin; Hsc 70; heat shock cognate 70; HMGB-1, high-motility group box protein 1.
Confocal Laser Scanning Microscopy
Frozen tissue sections after formalin fixation were analyzed by confocal microscopy using Zeiss LSM 510-UV Meta (Carl Zeiss, Oberkochen, Germany) and a Plan-NeoFluor ×40/1.3 objective with different zoom levels. The primary antibody for LAMP-2A was obtained from Abcam; the Cy3-conjugated affinitipure secondary antibody was obtained from Jackson ImmunoResearch (West Grove, PA). The BODIPY dye and DAPI used were supplied by Molecular Probes (Eugene, OR).
Statistical Analyses
All in vivo experiments were repeated three times with three mice per group (N = 3; data from each group of 3 mice were pooled). All in vitro experiments were repeated three times, and the statistical analysis was carried out by ANOVA followed by the Bonferroni post hoc correction for intergroup comparisons. Quantitative data from Western blots as depicted by the relative intensity of the bands were analyzed by performing a Student's t-test. P < 0.05 was considered statistically significant.
RESULTS
Steatohepatitic Injury in Obese Mice is Associated with Increased Metabolic Oxidative Stress
Metabolic oxidative stress in obesity-induced steatohepatitic injury is caused following increased CYP2E1 activity, mitochondrial uncoupling of the electron transport chain, or toxin metabolism following administration of hepatotoxic drugs (1, 5, 11, 22). We and others (5, 31, 32) have shown that toxin-induced steatohepatitis models produce metabolic oxidative stress. The toxin- and diet-induced steatohepatitic injury models that produce metabolic oxidative stress are relevant to clinical outcomes because patients with NASH show oxidative stress and have been hypothesized to augment disease progression (27). Our results demonstrate that CYP2E1 increases lipid peroxidation and oxidative and nitrosative stress upon steatohepatitic injury. 4-Hydroxynonenal (4-HNE), a marker for lipid peroxidation, was found to be increased in liver tissues from both the toxin (DIO+BDCM) and diet models (MCD-diet-fed mice) (Fig. 1, A, i–ii and iv–v, and C) compared with the respective controls (DIO and MCS diet). DIO mice with deletion of the CYP2E1 gene and administered BDCM showed decreased 4-hydroxynonenal adducts, compared with DIO+BDCM only group (Fig. 1, A, ii–iii, and C). Tyrosine nitration, a marker of oxidative and nitrosative stress, as evidenced by 3-nitrotyrosine immunoreactivity was found to be increased in liver tissues from both the toxin (DIO+BDCM) and diet models (MCD-diet-fed mice) (Fig. 1, B, i–ii and iv–v, and D) compared with the respective controls (DIO and MCS diet). DIO mice with deletion of the CYP2E1 gene and administered BDCM showed decreased 3-nitrotyrosine immunoreactivity, compared with DIO+BDCM only group (Fig. 1, B, ii–iii, and D). To estimate the extent of protein radical formation and corresponding oxidation, DMPO-nitrone adducts were measured in the liver homogenates of the toxin model of steatohepatitic injury. Results indicated that DMPO-nitrone adducts were significantly increased in DIO+BDCM group compared with DIO only group (P < 0.05) (Fig. 1E). CYP2E1-deleted mice fed with high-fat diet and coadministered BDCM had a significant decrease in the nitrone adducts (Fig. 1E, P < 0.05). Hepatic CYP2E1 mRNA expressions showed an increase in DIO+BDCM- (not statistically significant) and MCD-diet-treated mice compared with DIO- or MCS-diet-fed mice (Fig. 1, F and G; P < 0.05), suggesting that increased CYP2E1 expression might play a prominent role in the metabolic oxidative stress generation in steatohepatitic injury. The protein levels of CYP2E1 showed a small increase in DIO+BDCM group compared with DIO group (not statistically significant) but was unchanged in the MCS and MCD groups (Fig. 1H). Given the importance of P2X7 receptor in innate immunity, inflammation, cell death, and disease pathogenesis in NASH following its activation, we investigated the role of metabolic oxidative stress in modulating the expression of hepatic P2X7 receptors, those that might amplify inflammation. Gene expression was measured by qRT-PCR. Results indicated that there was a significant increase in P2X7 receptor mRNA expression in DIO+BDCM group and MCD-diet-fed group compared with DIO- and MCS-diet groups (P < 0.05) (Fig. 2, A and B). Deletion of the CYP2E1 gene in mice fed with high-fat diet and exposed to BDCM had significant decrease in the P2X7 receptor mRNA expression compared with DIO+BDCM group (P < 0.05) (Fig. 2A). Similar observations were found in the P2X7 receptor immunoreactivity in hepatic tissue. DIO+BDCM group and MCD-diet-fed group had higher P2X7 receptor levels compared with DIO-only and MCS-diet groups (Fig. 2C, i–ii and iv–v). Mice that had the deletion of CYP2E1 gene but were coadministered with high-fat diet and BDCM had decreased levels of P2X7 receptors (Fig. 2C, iii). Hepatocytes, Kupffer cells, and liver sinusoidal endothelial cells had increased expression of P2X7 receptor mRNA in the toxin model compared with untreated samples.
Fig. 1.

A: 4-Hydroxynonenal (4-HNE), (a marker of lipid peroxidation) immunoreactivity in liver slices from diet-induced obesity (DIO), DIO + bromodichloromethane (BDCM), CYP2E1 knockout (KO), methylcholine-sufficient (MCS)-diet-fed mice, and methylcholine-deficient (MCD)-diet-fed mice (i–v). B: nitrotyrosine immunoreactivity as shown by immunohistochemistry in liver slices (i–v; DIO, DIO+BDCM, CYP2E1 knockout, MCS, and MCD groups). C: morphometry (percentage) of 4-hydroxynonenal-reactive cells (*P < 0.05). D: morphometry (percentage) of nitrotyrosine (NTyr)-immunoreactive cells (*P < 0.05). E: 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-nitrone adducts, a marker of protein free radicals in toxin model of nonalcoholic steatohepatitis (NASH) as assessed by ELISA with anti-DMPO antibody (*P < 0.05). F and G: CYP2E1 mRNA expression as measured by quantitative real-time PCR (P < 0.05) in toxin-induced and MCD models of NASH. H: Western blot analysis of CYP2E1 protein levels. Lanes 1–5 represent DIO, DIO+BDCM, CYP2E1 Knockout, MCS, and MCD, respectively. I: band quantification of the immunoblots using normalization against β-actin. The Western analysis was done using liver homogenates from the respective groups. Representative immunoblot is shown.
Fig. 2.

A and B: mRNA expression of purinergic receptor X7 (P2X7) receptor as assessed by quantitative real-time PCR, normalized against DIO and MCS groups. CYP2E1 KO represents liver mRNA expression from CYP2E1 gene-depleted mice fed with high-fat diet (*P < 0.05). C: immunohistochemistry of P2X7 receptor protein from liver slices of DIO, DIO+BDCM, CYP2E1 KO, MCS, and MCD groups. The localization of the receptor-positive staining is shown by arrows.
CYP2E1-Mediated Metabolic Oxidative Stress in Steatohepatitic Injury Induces Macroautophagy and Causes Increased Expression of Chaperone-Mediated Autophagy-Related Proteins
To study the effect of metabolic oxidative stress on the induction of autophagy-related proteins in steatohepatitic injury, markers of macro and chaperone-mediated autophagy were analyzed. Results indicated that mRNA expressions of early autophagy genes, GABARAP, autophagy-related protein 2A (Atg2A), and LC3B, and components of the lysosomal translocation complex, LAMP2A and Hsc 70, were significantly elevated in DIO+BDCM group and MCD-diet-fed group compared with DIO- and MCS-diet fed group, respectively (Fig. 3, A and B) (P < 0.05). Mice that had a deletion of CYP2E1 gene and were coexposed to high-fat diet and BDCM had a significant decrease in early autophagy markers GABARAP, Atg2A, and LC3B compared with DIO+BDCM group, whereas there was no significant change in the expression pattern of Hsc 70 (Fig. 3A). LAMP2A mRNA levels were decreased in CYP2E1-deleted mice, but the change was not significant (Fig. 3A). Immunoreactivity of early autophagy marker GABARAP (initiator of the autophagy process) and LAMP2A, an important constituent of the lysosomal translocation complex, was increased in livers of steatohepatitic mice in both models of injury (Fig. 3, C and D, i–ii and iv–v). Absence of the CYP2E1 gene caused a decrease in the immunoreactivity of both GABARAP and LAMP2A, suggesting that metabolic oxidative stress and related oxidized proteins caused due to the involvement of CYP2E1 has led to the initiation and sustenance of the autophagy process (Figs. 3, C, iii, and D, iii). Hsc 70, which is responsible for recognizing and mediating substrate targeting for chaperone-mediated autophagy, was not significantly altered in both DIO+BDCM group and MCD-diet group (Fig. 3, E and F) (P < 0.05); however, the deletion of the CYP2E1 gene had significantly decreased levels of Hsc 70 in the toxin model (Fig. 3, E and F) (P < 0.05), suggesting a possible role of CYP2E1 in regulating the Hsc 70 expressions in this model of steatohepatitic injury.
Fig. 3.

Metabolic oxidative stress influences autophagy protein profiles in NASH. A and B: mRNA expressions of early and late autophagy-related proteins in toxin and diet models of NASH (*P < 0.05) as measured by quantitative real-time PCR. GABARAP: GABA-A receptor-associated protein; Atg-2A, autophagy-related protein 2A; LC3B, light chain 3 isoform B; Hsc 70, heat shock cognate 70; LAMP2A, lysosome-associated membrane protein. Immunoreactivity of GABARAP (C) and LAMP2A (D) as measured by immunohistochemistry is shown. i–v: DIO, DIO+BDCM, CYP2E1 KO, MCS, and MCD, respectively. E and F: Western blot image for Hsc 70 and its corresponding band quantification analysis (*P < 0.05). The normalization was carried out against the levels of β-actin in the liver homogenates.
P2X7 Receptor Modulates Autophagy Induced by Metabolic Oxidative Stress in Steatohepatitic Injury
Based on our present findings that show a significant increase in P2X7 receptor gene expression upon increased metabolic oxidative stress in NASH models, we investigated its role in mediating autophagy and modulating proteins in lysosomal translocation complex. mRNA expression of early autophagy-inducing genes, GABARAP, Atg2A, and LC3B, were significantly downregulated in P2X7 receptor gene-deleted mice coadministered with high-fat diet+BDCM and in MCD-diet-fed mice compared with DIO+BDCM- and MCD-diet-fed groups, respectively (Fig. 4, A and B). Lysosomal translocation complex proteins Hsc 70 and LAMP2A, which are significant players in chaperone-mediated autophagy, were downregulated in P2X7 receptor-deleted mice, either treated with toxin BDCM or fed with MCD diet, compared with wild-type counterparts (P < 0.05) (Fig. 4, A and B). Western blot analysis of GABARAP revealed a significant decrease in the band intensity, signifying decreased GABARAP protein levels in P2X7 gene-deleted mice treated with both the toxin and MCD diet compared with the wild-type counterparts (P < 0.05) (Fig. 4, C and D). Interestingly, P2X7 receptor-deleted mice from both DIO+BDCM- and MCD-diet groups had significantly increased levels of LC3B compared with their wild-type counterparts (P < 0.05) (Fig. 4, C and D). In contrary to the P2X7 receptor gene-deleted mice, protein levels of LC3B were decreased in DIO+BDCM- and MCD-diet-fed groups compared with DIO- and MCS-diet-fed groups, suggesting LC3B depletion in NASH (Fig. 4E). Immunohistochemistry of LC3B also showed increased LC3B protein immunoreactivity in P2X7 receptor-deleted liver compared with DIO+BDCM only group (Fig. 4F). LAMP2A protein levels, as studied by immunohistochemistry, showed a decreased immunoreactivity in P2X7 receptor-deleted mice, having been exposed to toxin or MCD diet compared with the wild-type counterparts (Fig. 5A). Because Hsc 70 plays a crucial role in recruiting peptides for the lysosomal translocation process and is a key mediator of late autophagy, Western blot analysis of Hsc 70 was carried out (6, 20). Results showed that P2X7 receptor gene-deleted mice had a significant decrease in Hsc 70 protein levels compared with their wild-type counterparts (P < 0.05) (Fig. 5, B and C). The results demonstrate that P2X7 receptor modulates both the early autophagy and the late chaperone-mediated lysosomal translocation processes in steatohepatitic injury.
Fig. 4.

P2X7 receptor deletion modulates autophagy in NASH. A and B: mRNA expressions of early and late autophagy-related proteins in toxin and diet models of NASH (*P < 0.05) as measured by quantitative real-time PCR in DIO+BDCM and P2X7 receptor gene-deleted mice, coexposed to high-fat diet and toxin, as well as MCD and P2X7 receptor gene-deleted mice fed with MCD diet. C: Western blot analysis of GABARAP and LC3B and their corresponding band quantification analysis (D) (*P < 0.05). E: Western blot analysis of LC3B protein in DIO, DIO+BDCM, MCS, and MCD liver homogenates. Bands are normalized against β-actin. F: immunohistochemistry of LC3B protein in liver slices of DIO, DIO+BDCM, and P2X7 receptor knockout mice coexposed to high-fat diet and BDCM showing perivenular areas.
Fig. 5.
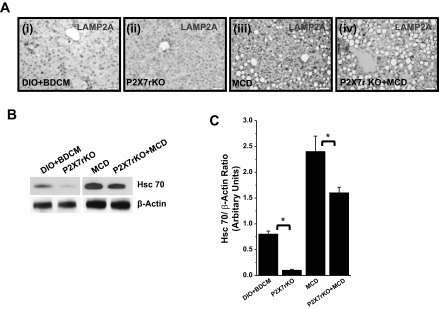
A: immunoreactivity of LAMP2A as analyzed by immunohistochemistry. i–iv: DIO+BDCM, P2X7 receptor-deleted group coexposed to high-fat diet and BDCM (toxin), as well as MCD and P2X7 receptor knockout group exposed to MCD diet, respectively. B and C: Western blot analysis of Hsc 70 in DIO+BDCM, P2X7 receptor-deleted group coexposed to high-fat diet and BDCM (toxin), as well as MCD and P2X7 receptor-deleted group (MCD) (*P < 0.05). C represents the band quantification analysis.
P2X7 Receptor Is a Key Regulator of LAMP2A Association with the Lysosomal Membrane During Increased Metabolic Oxidative Stress in Steatohepatitic Injury
To show the extent of lysosomal membrane association of LAMP2A, confocal laser scanning microscopy was performed. Results showed that there was an increase (3-fold) in the number of events of lysosomal membrane (green) associations of LAMP2A (red), as evidenced by colocalization spots (yellow) in DIO+BDCM group compared with DIO only group (Fig. 6A, v–viii). P2X7 receptor-deleted mice that were coadministered with high-fat diet and BDCM had no visible colocalizations in the same numbers of field analyzed (Fig. 6A, ix–xii). MCD mice liver had more LAMP-2A membrane fusions compared with MCS mice as evidenced by confocal microscopy (Fig. 6B). These results suggested that the absence of P2X7 receptor gene significantly affected LAMP2A association with the lysosomal membrane.
Fig. 6.

A: LAMP2A colocalization with lysosomal membrane (×60 zoom 3). Confocal laser scanning image of LAMP2A and membrane lipid (Bodipy stain) from liver sections of DIO, DIO+BDCM, and P2X7 receptor-deleted group coexposed to high-fat diet and BDCM. B: LAMP2A colocalization with lysosomal membrane (×60). Confocal laser scanning image of LAMP2A and membrane lipid (Bodipy stain) from liver sections of MCS, MCD, and P2X7 receptor-deleted group fed with MCD diet. Gray arrows show LAMP2A localizations, whereas white arrows depict colocalizations of LAMP2A with lysosomal membrane depicted by bodipy staining. Number of colocalizing events as shown by overlay of images were counted to reveal the extent of LAMP2A attachment to the lysosomal membrane.
P2X7 Receptor Modulates Autophagy-Linked Inflammation in NASH
To explore the regulation of P2X7 receptor and its downstream effectors in autophagy-linked inflammation in steatohepatitic injury, mRNA expression of caspase-1, IL-1β, TNF-α, and IFN-γ were studied. Results indicated that there was a significant increase (more than 17-fold) of caspase-1, IL-1β (4-fold), TNF-α, and IFN-γ in both DIO+BDCM group and MCD-diet-fed group, compared with DIO- or MCS-diet-fed group (Fig. 7, A and B). Cleaved caspase-1, which is an indicator of active caspase-1, was significantly decreased in P2X7 receptor gene-deleted mice from both DIO+BDCM-and MCD-treated group compared with the wild-type counterparts (Fig. 7, C and D). Immunohistochemistry of IL-1β and IFN-γ showed an increase in the immunoreactivity of both these proteins in livers of DIO+BDCM- and MCD-diet-fed groups compared with only DIO- or MCS-diet-fed groups (Fig. 7, E and F, i–ii and iv–v), whereas a decrease was observed in livers of mice that contained the deletion of the P2X7 receptor gene (Fig. 7, E and F, iii and vi) (16, 19). Results also indicated that P2X7 gene-deleted mice from both DIO+BDCM-coexposed group had decreased amounts of HMGB-1 protein, a damage-associated molecular pattern linked to inflammation, as analyzed by Western blot (Fig. 8, A and B), compared with their wild-type controls (P < 0.05) although the HMGB-1 protein level was not significantly different in MCD-treated group (Fig. 8, A and B). The decreased expression of HMGB-1, IL-1β, IFN-γ, and cleaved caspase-1 in P2X7 receptor-deleted mice shows that metabolic oxidative stress-induced autophagy and corresponding inflammation are regulated by the P2X7 receptors in steatohepatitic injury.
Fig. 7.

P2X7 receptor modulates inflammation in NASH. A and B: mRNA expression of proinflammatory mediators, as measured by quantitative real-time PCR in DIO, DIO+BDCM, P2X7 receptor gene-deleted mice, coexposed to high-fat diet and BDCM (toxin), MCS, MCD, and P2X7 receptor gene-deleted mice fed with MCD diet. The expression levels were normalized against DIO in toxin model and MCS in diet model. C: Western blot analysis of cleaved caspase-1 (active caspase-1) and its corresponding band quantification analysis (D) (*P < 0.05). Individual lanes representing DIO+BDCM and P2X7 receptor gene-deleted groups were separated from the parent blot to ensure better visibility. E and F: immunoreactivity of IL-1β and IFN-γ as analyzed by immunohistochemistry. i–vi: DIO, DIO+BDCM, P2X7 receptor-deleted group coexposed to high-fat diet and BDCM (toxin), MCS, MCD, and P2X7 receptor-deleted group exposed to MCD diet, respectively.
Fig. 8.

A: Western blot analysis of high-motility group box protein (HMGB)-1 from liver homogenates of DIO+BDCM, P2X7 receptor-deleted group coexposed to high-fat diet and BDCM (toxin), MCD, and P2X7 receptor-deleted group exposed to MCD diet, respectively. B: band quantification analysis following normalization against β-actin (*P < 0.05). C: quantitative real-time PCR analysis of the mRNA expression profiles of fibrosis markers in DIO+BDCM, P2X7 receptor-deleted group coexposed to high-fat diet and BDCM (toxin), MCD, and P2X7 receptor-deleted group exposed to MCD diet, respectively (*P < 0.05). SMA, smooth muscle actin. D: hematoxylin and eosin staining of liver sections from DIO+BDCM group and P2X7 gene-deleted group coexposed to BDCM and high-fat diet. Areas of necrosis are shown in demarcated gray dashed lines and arrow heads. E: Picro-Sirius red staining for fibrosis in liver sections from DIO+BDCM group and P2X7 gene-deleted group coexposed to BDCM and high-fat diet.
P2X7 Receptor Gene-Deleted Mice Are Protected from Steatohepatitic Injury and Fibrosis
To ensure that the genetic deletion of P2X7 receptor in mice exposed with either high-fat diet+BDCM or MCD-diet-fed group were protected from symptoms of steatohepatitic injury, mRNA expressions of fibrotic markers α-smooth muscle actin (SMA), TGF-β, and Col-1-α1 were analyzed. Results showed that α-SMA, TGF-β, and Col-1-α1 were significantly decreased in P2X7 receptor gene-deleted mice compared with wild-type mice either fed with high-fat diet and BDCM or MCD (Fig. 8C, P < 0.05). Hematoxylin and eosin stainings of the liver slices of P2X7 receptor-deleted mice administered the toxin and fed with high-fat diet showed decreased hepatocyte necrosis compared with wild-type mice fed with high-fat diet (Fig. 8D). There was a decrease in Picro-Sirius red staining in livers of P2X7 receptor gene-deleted mice compared with the wild-type mice fed with high-fat diet and BDCM, suggesting decreased fibrosis (Fig. 8E). DIO+BDCM mice also had increased serum ALT, alkaline phosphatase, and albumin levels compared with DIO and P2X7 receptor gene-deleted mice.
DISCUSSION
To date, nonalcoholic fatty liver disease and its associated steatohepatitic injury are considered an emerging epidemic in light of the dramatic increase in obesity rates (9, 12). With the progressive nature of NASH and its rising prevalence, there is a significant need for specific and targeted treatments. This is complicated by the fact that there are not many validated therapies for nonalcoholic fatty liver disease other than weight loss, which is well known to have a poor long-term success rate (10). Cellular death pathways and inflammation play crucial roles in NASH pathophysiology, and it is essential that we identify new mediators of NASH pathophysiology that are important regulators of autophagy and inflammatory pathways. Impairment of autophagy that is correlated with hepatic lipid accumulation and obesity has been found to have a significant impact on progression of NASH (2). There is also a perception that agents that augment autophagy can have therapeutic potential in NASH (21). However, no significant research has been in place that links metabolic oxidative stress, autophagy, and P2X7 receptors and their crosstalk in modulating disease progression in NASH. Targeting a key regulator of each of these components can have benefits in therapy of NASH. This study shows that P2X7 receptor, which is upregulated by metabolic oxidative stress (Fig. 2), serves as a key regulator in modulating the oxidative stress induced-autophagy process (Fig. 3). P2X7 downregulation in CYP2E1 knockout mice, coupled with an earlier report by this group that showed extracellular ATP (ligand for P2X7) release from necrosed hepatocytes (those that had higher 4-HNE staining) in a CCl4-mediated NASH model, correlates oxidative stress, upregulation of P2X7 receptor, and its downstream events (5). The P2X7 receptor might modulate the autophagy process by allowing depletion of LC3B, which is supposedly an early autophagy marker (Fig. 4E), while increasing, albeit in small proportions, Hsc 70 and LAMP2A mRNA levels (Fig. 3, A and B) and allowing LAMP2A association with the lysosomal membrane (Fig. 6). Interestingly, LC3B mRNA levels in NASH models showed a significant increase, whereas the protein levels decreased (Figs. 3, A and B, and 4E). The contrasting result might point to a translational level regulation and can be explored further. These events can be crucial mediators of chaperone-mediated autophagy in the hepatic lobe, thus increasing inflammation, in a manner perhaps similar to P2X7 receptor-mediated release of autophagolysosomes/phagolysosomes into the extracellular matrix, causing increase in inflammation (33). The above mechanism of release of phagolysosomes to the extracellular matrix might be speculative for NASH at this point, but this study certainly proves the dependence of depleted LC3B and increased levels of LAMP2A and Hsc 70 on P2X7 receptor in both models of experimental NASH.
A previous study by our group showed that P2X7 receptor was crucial for causing Kupffer cell activation and inflammation following release of ATP from necrosed hepatocytes in CCl4-mediated early steatohepatitic injury (5). In the present study, we show that metabolic oxidative stress, which is associated with NASH, caused an upregulation of P2X7 receptors (Fig. 2). Metabolic oxidative stress also, mainly characterized by oxidatively modified proteins, has been known to induce chaperone-mediated autophagy, increased substrate translocation by Hsc 70 toward the lysosomal membrane, and increased LAMP2A levels (17). Furthermore, P2X7 receptors have been associated with autophagy, disruption of normal lysosomal functions, and release of autophagolysosomes to the extracellular matrix in the microglial cells, causing an increase in inflammation (33). Our present study shows that there was a significant downregulation of both early and late autophagy proteins, including LAMP2A in P2X7 receptor gene-deleted mice. P2X7 receptor-deleted mice also showed decreased release of IL-1β, IFN-γ, and HMGB-1 (damage-associated molecular pattern that contributes to inflammation, Ref. 34), observations that were dependent on significantly lower cleaved caspase-1 protein levels, implying the possible involvement of P2X7 receptors. This study, however, falls short on exploring the exact molecular mechanism of P2X7 receptor involvement and on having a clear answer whether it is the increased calcium ion-mediated change in lysosomal pH, lysosomal functional impairment, or expulsion of the lysophagosome to the extracellular matrix that caused an increase in the inflammation. The present study also establishes a direct correlation between the decreased numbers of LAMP2A-lysosomal membrane associations, reduced inflammation, and decreased NASH pathophysiology in P2X7 receptor gene-deleted mice (Figs. 6, 7, and 8). These observations assume significance because LAMP2A levels and its association with the lysosomal membrane are an important step for chaperone-mediated autophagy, which occurs late in the autophagy process. Our studies showed a depletion of LC3B, a autophagosome protein in NASH pathophysiology (Fig. 4E), but not in LAMP2A, which is contrary to a study by Fortunato et al. (13) that showed decreased LAMP2A levels linked to decreased fusion of the autophagosome with the lysosome, resulting in necrotic cell death and inflammation. This may be due to a different mechanism of a dysfunctional lysosomal function compared with our model that primarily evidenced LC3B depletion. Recent studies also link P2X7 receptor to a dysfunctional lysosome and autophagy protein LC3B (25, 33). In both cases, the fate of the cell and its link to immune activation remain a dysregulated lysosomal compartment and are regulated by ATP-binding P2X7 receptors in the latter study (25). Interestingly, our study indicates significantly decreased LC3B protein levels in DIO+BDCM- and MCD-diet-fed livers compared with DIO- and MCS-diet-fed groups, whereas absence of P2X7 receptor gene elevates the LC3B protein levels (Fig. 4, C–E). This result is important because it has been shown that depletion of autophagic protein LC3B enhances caspase-1 activation and increases in inflammatory microenvironment (25). We have also found higher caspase-1 activation and levels of inflammatory indicators IL-1β, TNF-α, IFN-γ, and HMGB-1 in DIO+BDCM- and MCD-diet-fed groups, whereas a decrease in these indicators was seen in P2X7 receptor gene-deleted mice (Fig. 7). Taken together, the present study provides the first evidence that P2X7 receptor deletion results in downregulation of autophagy-related proteins, inflammation, and disease pathophysiology in NASH (Figs. 7 and 8). Having significant evidence that P2X7 receptor is a key regulator of autophagy in NASH, it may be presumed that targeting the P2X7 receptor or the autophagy process in NASH can emerge as a good therapeutic option for the treatment of NASH. It is increasingly becoming clear that there are a series of studies that promote autophagy as a beneficial mechanism in NASH and an equal number of studies that conclude autophagy is central in causing NASH; the present study only predicts the role of P2X7 receptor-mediated defective autophagy as a cause for inflammation in NASH (14). The translational impact of this study will be enhanced with more mechanistic studies in the future involving cell-specific P2X7 receptor knockdowns and functional roles of different autophagy proteins in the presence or absence of the P2X7 receptor.
GRANTS
This work has been supported by NIH pathway to Independence Award (4R00ES019875-02 to S. Chatterjee), NIH R01 (R01DK053792 to A. M. Diehl), and the Intramural Research Program of the National Institutes of Health and the National Institute of Environmental Health Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.D., R.K.S., A.K., G.M., and S.C. performed experiments; S.D., R.K.S., A.K., G.M., and S.C. prepared figures; R.K.S., A.K., M.B.K., G.M., A.M.D., and S.C. analyzed data; A.K., M.B.K., G.M., A.M.D., and S.C. interpreted results of experiments; M.B.K., G.M., A.M.D., and S.C. edited and revised manuscript; S.C. conception and design of research; S.C. drafted manuscript; S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the technical services of Benny Davidson at the Instrumentation Resource Facility (IRF), University of South Carolina, School of Medicine, and Jeoffrey Hurlburt and Ralph Wilson at NIEHS. We thank Dr. Anindya Chanda for the internal review of this manuscript. We also thank Dr. James Carson, Department of Exercise Science and the IRF at the University of South Carolina, School of Medicine for equipment usage and consulting services.
REFERENCES
- 1.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol 57: 860–866, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol 5: 159–166, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 20: 897–915, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee S, Ehrenshaft M, Bhattacharjee S, Deterding LJ, Bonini MG, Corbett J, Kadiiska MB, Tomer KB, Mason RP. Immuno-spin trapping of a post-translational carboxypeptidase B1 radical formed by a dual role of xanthine oxidase and endothelial nitric oxide synthase in acute septic mice. Free Radic Biol Med 46: 454–461, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee S, Rana R, Corbett J, Kadiiska MB, Goldstein J, Mason RP. P2X7 receptor-NADPH oxidase axis mediates protein radical formation and Kupffer cell activation in carbon tetrachloride-mediated steatohepatitis in obese mice. Free Radic Biol Med 52: 1666–1679, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246: 382–385, 1989 [DOI] [PubMed] [Google Scholar]
- 7.Das S, Kumar A, Seth RK, Tokar EJ, Kadiiska MB, Waalkes MP, Mason RP, Chatterjee S. Proinflammatory adipokine leptin mediates disinfection byproduct bromodichloromethane-induced early steatohepatitic injury in obesity. Toxicol Appl Pharmacol 269: 297–306, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol 189: 15–20, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diehl AM. Nonalcoholic steatohepatitis. Semin Liver Dis 19: 221–229, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Durazzo M, Belci P, Collo A, Grisoglio E, Bo S. Focus on therapeutic strategies of nonalcoholic Fatty liver disease. Int J Hepatol 2012: 464706, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eccleston HB, Andringa KK, Betancourt AM, King AL, Mantena SK, Swain TM, Tinsley HN, Nolte RN, Nagy TR, Abrams GA, Bailey SM. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid Redox Signal 15: 447–459, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver 6: 149–171, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137: 350–360; e351–355, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Friedman SL. Focus. J Hepatol 58: 845–846, 2013 [DOI] [PubMed] [Google Scholar]
- 15.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22: 407–417, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawano A, Tsukimoto M, Mori D, Noguchi T, Harada H, Takenouchi T, Kitani H, Kojima S. Regulation of P2X7-dependent inflammatory functions by P2X4 receptor in mouse macrophages. Biochem Biophys Res Commun 420: 102–107, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15: 4829–4840, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol 58: 395–398, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Li G, Liang X, Lotze MT. HMGB1: the central cytokine for all lymphoid cells. Front Immunol 4: 68, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci 68: 749–763, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 58: 993–999, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, Doeller JE, Kraus DW, Darley-Usmar VM, Bailey SM. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J 417: 183–193, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol 298: C776–C785, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Mendelson KG, Contois LR, Tevosian SG, Davis RJ, Paulson KE. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc Natl Acad Sci USA 93: 12908–12913, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orenstein SJ, Cuervo AM. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21: 719–726, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacana T, Sanyal AJ. Vitamin E and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care 15: 641–648, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu Y, Dubyak GR. P2X7 receptors regulate multiple types of membrane trafficking responses and non-classical secretion pathways. Purinergic Signal 5: 163–173, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol 37: 56–62, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, Chatterjee S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol Sci 134: 291–303, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shertzer HG, Kendig EL, Nasrallah HA, Johansson E, Genter MB. Protection from olanzapine-induced metabolic toxicity in mice by acetaminophen and tetrahydroindenoindole. Int J Obes 34: 970–979, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takenouchi T, Nakai M, Iwamaru Y, Sugama S, Tsukimoto M, Fujita M, Wei J, Sekigawa A, Sato M, Kojima S, Kitani H, Hashimoto M. The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J Immunol 182: 2051–2062, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209: 1519–1528, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Qin ZH. Coordination of autophagy with other cellular activities. Acta Pharmacol Sin 34: 585–594, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, Omenetti A, Jung Y, Teaberry V, Choi SS, Guy CD, Pollard J, Charlton P, Diehl AM. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 50: 1421–1430, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45: 1366–1374, 2007 [DOI] [PubMed] [Google Scholar]