

Abstract
Because transmembrane (TM) protein localization, or nonlocalization, in ordered membrane domains (rafts) is a key to understanding membrane domain function, it is important to define the origin of protein-raft interaction. One hypothesis is that a tight noncovalent attachment of TM proteins to lipids that have a strong affinity for ordered domains can be sufficient to induce raft-protein interaction. The sterol-binding protein perfringolysin O (PFO) was used to test this hypothesis. PFO binds both to sterols that tend to localize in ordered domains (e.g., cholesterol), and to those that do not (e.g., coprostanol), but it does not bind to epicholesterol, a raft-promoting 3α-OH sterol. Using a fluorescence resonance energy transfer assay in model membrane vesicles containing coexisting ordered and disordered lipid domains, both TM and non-TM forms of PFO were found to concentrate in ordered domains in vesicles containing high and low-Tm lipids plus cholesterol or 1:1 (mol/mol) cholesterol/epicholesterol, whereas they concentrate in disordered domains in vesicles containing high-Tm and low-Tm lipids plus 1:1 (mol/mol) coprostanol/epicholesterol. Combined with previous studies this behavior indicates that TM protein association with ordered domains is dependent upon both the association of the protein-bound sterol with ordered domains and hydrophobic match between TM segments and rafts.
Introduction
Lipid rafts are tightly packed sphingolipid and cholesterol-rich liquid-ordered (Lo) membrane domains that are believed to coexist in cells with loosely packed disordered domains composed mostly of unsaturated lipids (1,2). Rafts have been proposed to serve as platforms that regulate protein-protein interaction and are believed to serve many functions in signal transduction, protein trafficking, and pathogenesis (1,2). Proteins that reside on the plasma membrane have different affinities for raft domains (3–5). Several mechanisms have been proposed as possible driving forces for transmembrane (TM) protein association with lipid rafts. One idea is that TM proteins might be dragged into rafts by interacting with raft components, such as saturated fatty acids, lipids with saturated acyl chains (e.g., sphingolipids), or raft-associating sterols (e.g., cholesterol). However, how these interactions impart raft affinity to TM proteins, and/or whether these interactions are sufficient to drive TM protein insertion into lipid rafts has not been confirmed.
Perfringolysin O (PFO) is an ideal protein to study the interaction between membrane proteins and lipid rafts. PFO belongs to the family of cholesterol-dependent cytolysins (CDCs), and forms large homooligomeric pore complexes comprising up to 50 subunits in cholesterol-containing membranes (6). The ability of CDCs to specifically recognize cholesterol-rich regions of cell membranes has led to the hypothesis that CDCs are targeted to and bind lipid rafts (7). For example, PFO, listeriolysin O, and aerolysin have been reported to associate with raft domains, which may accelerate clustering (oligomerization) of membrane-bound toxin monomers (8–10). Both intact PFO and derivatives of PFO membrane binding domain (domain 4) have been used as markers of cholesterol-rich regions of cell membranes (10–12). Although PFO is not a typical TM protein, as it has a TM β-barrel, like TM residues in TM α-helical proteins, TM residues in PFO should also be unable to pack tightly with the linear acyl chains of lipids. Therefore, the principles determining PFO interaction with lipid rafts should be similar to those for α-helical membrane proteins.
Previous reports indicated that membrane cholesterol serves as the cellular receptor for PFO and some other CDCs, and is necessary to induce the conformational changes for PFO membrane insertion and pore formation (13). However, although PFO was found to colocalize with raft markers, such as flotillin and Src family kinases (10–12), to date, there is no experimental evidence for the necessity of lipid rafts for PFO activity in vitro (14–16). Recent work in model membranes has shown that PFO binds to and forms pores more readily in vesicles composed of unsaturated lipids that do not form ordered domains, while tightly packing phospholipids, which interact strongly with sterols and form ordered domains, have been shown to interfere with the interaction of PFO with membranes, in the sense that in membranes composed of tightly packed lipids, a higher concentration of sterol is needed for PFO to interact with membranes (14–16). This led to the hypothesis that PFO may insert into disordered domains and then move into ordered domains subsequent to insertion (16). Even so, it is not clear whether PFO raft affinity is directly related to the raft affinity of cholesterol, or due to other the properties of the protein upon binding to cholesterol/membranes.
Here, we used two sterols in addition to cholesterol to separate the roles of cholesterol in PFO binding to membranes and raft association. One is coprostanol, which contains a 3β-hydroxyl group that can interact with PFO, but which disrupts ordered domain formation (16,17). The other is epicholesterol, which contains a 3α-hydroxyl group and can promote ordered domain formation, but which does not interact with PFO (16,17). Using various combinations of these sterols the affinity of PFO for ordered domains on lipid vesicles with coexisting ordered and disordered domains was measured by fluorescence resonance energy transfer (FRET). The results show that PFO has a high affinity for ordered domains in vesicles containing cholesterol or 1:1 cholesterol/epicholesterol, but has a high affinity for disordered domains in vesicles containing 1:1 coprostanol/epicholesterol. This indicates that PFO association with ordered domains is dependent upon the raft-associating properties of PFO-bound sterols.
Materials and Methods
Materials
1,2-dimyristoleoyl-sn-glycero-3-phosphocholine (DMoPC), 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol (ovine wool), ganglioside M1 (GM1), sphingomyelin (SM; egg), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-pyrenesulfonyl (pyrene-DPPE), 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DPhPE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). Lipids were stored in ethanol or chloroform at −20°C. Concentrations were determined by dry weight or by absorbance, using an ε of 35,000 cm−1 M−1 at 350 nm for pyrene-DPPE in methanol, 95,000 cm−1 M−1 at 560 nm for Rho-DOPE in methanol, and 21,000 cm−1 M−1 at 463 nm for NBD-DPhPE in methanol (18). The sterols, 5-cholesten-3α-ol (epicholesterol) and 5β-cholestan-3β-ol (coprostanol), were purchased from Steraloids (Newport, RI). The labeling reagents N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl)iodoacetamide (BODIPY-FL) and 6-acryloyl-2-dimethylaminonaphthalene (acrylodan) were purchased from Invitrogen (Grand Island, NY). Acetyl-K2W2L8AL8W2K2-amide (LW peptide) was purchased from Anaspec (San Jose, CA) and used without further purification. Cholera toxin B (CT-B) subunit was purchased from EMD Chemicals (Gibbstown, NJ). All other chemicals were reagent grade.
Purification and fluorescent labeling of PFO
A functional cysteine-less derivative of wild-type PFO (PFO C459A) and a prepore mutant (PFO C459A Y181A), which cannot insert to form TM segments, were gifts of A. Heuck, U. Mass. Amherst. PFO variants of these proteins with an Ala to Cys substitution at residue 215 were generated by the QuickChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) as previously described (19). We refer to PFO C459A A215C as WT in this report because it has wild-type length TM segments, and inserts into membrane identically to PFO C459A, which is a fully active form of PFO. We refer to PFO C459A Y181A A215C as prepore or prepore mutant PFO. PFO was expressed in Escherichia coli BL21(DE3)pLysS and purified similarly as described previously (16). The purified PFO was stored in phosphate-buffered saline (PBS, 1.8 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, and 2.7 mM KCl at pH 7.4) at a concentration of ∼20–30 μM at −20°C. Labeling of PFO at Cys-215 with BODIPY-FL or acrylodan was carried out similarly as described previously (19). The fluorescent-labeled PFO was stored in PBS pH 7.4 at a concentration of ∼4–5 μM at −20°C.
Preparation of lipid vesicles
Multilamellar vesicles (MLVs) were prepared with the desired mixtures of lipids. Lipids in solvent were mixed and then dried with N2. They were then redissolved in CHCl3 and redried under N2 and then high vacuum for at least 1 h. The redried lipid mixtures were then dispersed in PBS, pH 5.1, at 70°C to give the desired final concentration and agitated at 55°C for 15 min using a VWR multitube vortexer (Westchester, PA) placed within a convection oven (GCA Corp., Precision Scientific, Chicago, IL). Samples were cooled to room temperature (∼23°C) before use.
Fluorescence intensity measurements
Fluorescence emission intensity was measured (unless otherwise noted) at room temperature (∼23°C) on a SPEX Fluorolog 3 spectrofluorimeter (Jobin-Yvon, Edison, NJ). For fixed wavelength measurements, excitation and emission wavelength sets used (in nm) were (280, 340) for tryptophan, (350,448) for acrylodan, (490,510) for BODIPY, and (350, 379) for pyrene-DPPE. Unless otherwise noted, fluorescence intensity in single background samples lacking fluorophore was subtracted. For acrylodan-labeled PFO emission spectra, samples and backgrounds were excited at 350 nm, and emission was acquired from 420 to 560 nm.
Measurement of vesicle binding by tryptophan fluorescence
The ability of WT PFO to associate with lipid vesicles was assessed by measuring the increase in intrinsic Trp emission intensity, which occurs when Trp residues located at the tip of domain 4 come into contact with membrane (20). A small aliquot of PFO was added to 1 ml of an MLV suspension containing 500 μM total lipids in PBS pH 5.1. The final PFO concentration was 55 nM. After 1 h incubation at room temperature, Trp emission intensity was measured as described previously. (Note that PFO interaction with membranes is enhanced at low pH. This may reflect a physiological role for PFO pore formation in acidic organelles (16,21,22).)
Measurement of vesicle insertion by acrylodan fluorescence
PFO-membrane interaction was also monitored by the changes in the acrylodan fluorescence intensity of WT PFO labeled with acrylodan on Cys-215. Residue 215 becomes buried in the bilayer when PFO inserts into the bilayer and forms TM β-hairpins (23). To do this, a small aliquot of acrylodan-labeled PFO was added to 1 ml of an MLV suspension containing 500 μM total lipids in PBS pH 5.1. The final acrylodan-labeled PFO concentration was 25 nM. After 1 h incubation at room temperature, acrylodan emission spectra were measured as described above.
Measurement of vesicle association by centrifugation
The ability of WT and prepore PFO to associate with vesicles was assessed by ultracentrifugation. MLVs (500 μM total lipid) composed of different lipid species with 40 or 45 mol % sterol were prepared in 1 ml of PBS pH 5.1, and incubated with a small aliquot of BODIPY-labeled PFO for 1 h at room temperature. The final BODIPY-labeled PFO concentration was 25 nM. Samples were then spun for 30 min in a Beckman L8-85 ultracentrifuge (Indianapolis, IN) at 84,000 × g at 4°C. This was sufficient to pellet all of the vesicles. After spinning, supernatants containing the unbound PFO were removed, and pellets containing the MLVs and bound PFO were resuspended in 1 ml of PBS, pH 5.1. BODIPY fluorescence was then measured for both the supernatant and the pellet.
Detection of domain formation by FRET
FRET measurements were made in 1 ml MLV samples dispersed in PBS pH 5.1 prepared as described previously (19,24). Samples contained 500 μM total lipids with 0.05 mol % pyrene-DPPE as a donor and 2 mol % Rho-DOPE as an acceptor in F samples. Fo samples contained only donor. The fluorescence of pyrene-DPPE was measured from 15 to 60°C as described previously (24). Fluorescence measurements were taken every 5°C. The ratio of donor fluorescence intensity in the presence of acceptor to its absence (F/Fo) was calculated. Background fluorescence was negligible.
FRET assay of raft affinity
FRET experiments were carried out to assess WT and prepore PFO affinity for raft domains similarly to as described by Nelson et al. (18) and Lin et al. (19), with 2 mol % NBD-DPhPE used as the FRET acceptor. Briefly, 100 μl of MLVs prepared in PBS, pH 5.1 at a total lipid concentration of 5 mM were incubated with 3 μg PFO. Two types of samples were prepared. F sample vesicles contained FRET acceptor (2 mol % NBD-DPhPE of total lipid). Fo sample vesicles lacked FRET acceptor. To measure CT-B raft affinity, the same procedure was followed as for PFO, but vesicles contained an additional 2 mol % ganglioside GM1, and 5 μg CT-B chain. For samples with LW peptide, peptide was added from an ethanolic stock solution to lipids before preparation of the MLVs at a concentration of 0.25 mol % of total lipids. After incubation at room temperature for at least 1 h, samples were diluted with PBS pH 5.1 to 1 ml and Trp emission intensity was measured as described above. The ratio of the local concentration of acceptor around the donor in a membrane containing both liquid ordered and liquid disordered domains (CLoLd) relative to that in a homogeneous membrane lacking domains (CLd) is given by the equation: CLoLd/CLd = ln (F/Fo)LoLd/ln (F/Fo)Ld (18). Uncorrected F/Fo is the ratio of measured donor fluorescence (after subtraction of background fluorescence) in the presence of acceptor to that in its absence (= Fmeasured/Fomeasured). Corrected F/Fo = (Fmeasured – xFunbound)/(Fomeasured – xFunbound), where Funbound is the Trp fluorescence of the same amount of PFO in the samples would have if it was fully unbound to vesicles, and x = fraction of unbound protein derived from the centrifugation assay.
Laser scanning microscopy
Giant unilamellar vesicles (GUVs) were prepared using the electroformation method as previously described (19,25). The inclusion in the GUVs of the fluorescently labeled lipid Rho-DOPE (0.02 mol %), a marker for disordered domains (26), allowed the optical visualization of domains. After electroformation, GUVs were observed under confocal laser scanning microscopy and 3D representations constructed (19). All of the measurements were performed at room temperature.
Results
PFO interacting with membranes containing different sterols
The interaction of WT PFO with model membrane vesicles having compositions appropriate for domain association studies was monitored by the increase of domain 4 Trp emission intensity observed when PFO associates with membranes (20). These studies were necessary to define what lipid mixtures would tightly bind to PFO, and thus be suitable for FRET experiments defining PFO association with ordered and disordered domains.
We started with DSPC/DMoPC mixtures with cholesterol. We previously found a 1:1 (mol/mol) mixture of DSPC/DMoPC could form coexisting ordered and disordered domains with cholesterol (18,19). In agreement with previous observations (16,20), a cholesterol-dependent increase in Trp fluorescence was detected when PFO interacts with vesicles composed of DSPC/DMoPC and sufficient cholesterol (Fig. 1 A), with the binding threshold (which we are defining here as the sterol concentration giving a half-maximal fluorescence increase) at ∼35 mol % cholesterol. A similar binding curve and threshold sterol concentration for PFO binding was observed when PFO was incubated with DSPC/DMoPC vesicles containing coprostanol, indicating that coprostanol could mediate PFO-membrane association, in agreement with previous results in DOPC vesicles (16). In contrast, PFO did not interact with DSPC/DMoPC vesicles containing epicholesterol. This lack of interaction also agrees with previous studies in DOPC vesicles with epicholesterol (16).
Figure 1.
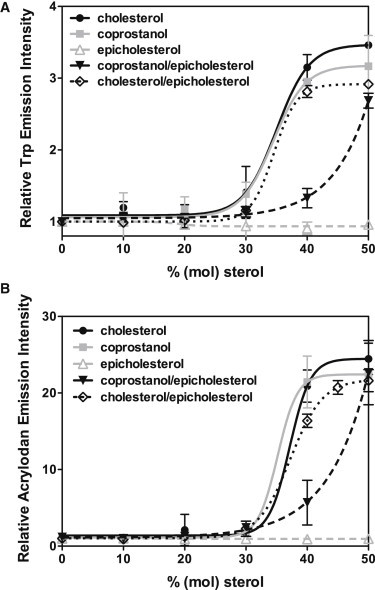
PFO interaction with vesicles composed of DSPC/DMoPC/sterol. Trp fluorescence (A) of 55 nM PFO and acrylodan fluorescence (B) of 25 nM acrylodan-labeled PFO interacting with MLVs (500 μM total lipid) composed of 1:1 DSPC/DMoPC with different amounts of cholesterol (●), coprostanol ( ), epicholesterol (
), epicholesterol (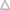 ), 1:1 cholesterol/epicholesterol (▾), or 1:1 coprostanol/epicholesterol (♢) were measured in PBS pH 5.1. Average (mean) values and SD values from triplicates are shown. Error bars are not shown where they are too small to illustrate.
), 1:1 cholesterol/epicholesterol (▾), or 1:1 coprostanol/epicholesterol (♢) were measured in PBS pH 5.1. Average (mean) values and SD values from triplicates are shown. Error bars are not shown where they are too small to illustrate.
Binding of PFO to vesicles also was observed in vesicles containing 1:1 (mol/mol) cholesterol/epicholesterol or coprostanol/epicholesterol mixtures (Fig. 1 A). Interestingly, the threshold concentration of cholesterol or coprostanol needed to bind PFO decreased by up to twofold in the presence of epicholesterol (in 1:1 3β-sterol/epicholesterol mixtures a 40 mol % total sterol threshold for binding is equivalent to 20 mol % of the PFO binding sterol). This may be due to competition of epicholesterol with cholesterol or coprostanol for interaction with phospholipids, which can increase cholesterol or coprostanol exposure to water and thus reactivity with PFO (14,27,28).
Next, the membrane insertion of PFO TM β-barrel was detected by the fluorescence intensity of a fluorescent probe, acrylodan attached to Cys-215, a residue located within the TM hairpin that forms when PFO undergoes TM insertion (23). There was a steep sterol-dependent increase of acrylodan fluorescence observed when PFO interacts with model membranes containing an increasing amount of sterols. The increase in acrylodan fluorescence occurred at the same sterol concentration as the increase of Trp fluorescence (Fig. 1 B), indicating that in these lipid mixtures, once membrane-bound, PFO efficiently unravels its domain 3 α-helical bundles and inserts the residues from these segments into the lipid bilayer. Overall, these studies indicated that DSPC/DMoPC vesicles with high amounts of sterol (>40 mol %) would be suitable for FRET studies.
PFO interaction and insertion into membranes was also studied on vesicles composed of 1:1 SM/DMoPC, DSPC/DPhPC, and SM/DPhPC, all with various sterols at various concentrations. Similar Trp and acrylodan fluorescence changes were seen in all the lipid mixtures examined (Fig. S1 in the Supporting Material), indicating that these mixtures would also be suitable for FRET studies when a high sterol concentration was used.
Vesicles containing mixtures of coprostanol and epicholesterol form ordered domains
To study PFO raft affinity, lipid compositions forming coexisting ordered and disordered domains are required. To confirm that the compositions studied previously (all of which contain a mixture of a high Tm-lipid, which tends to form ordered domains, and a low Tm-lipid, which tends to form disordered domains) would actually form coexisting ordered and disordered domains in the presence of a high concentration of sterol, a FRET method was used (24). In this method, when a membrane has coexisting ordered and disordered domains, the segregation of donor-labeled and acceptor-labeled lipids with different affinities for ordered and disordered domains results in a decrease in FRET, which is detected as an increase in the normalized donor fluorescence (F/Fo). To measure FRET, the acceptor used was Rho-DOPE, which partitions strongly into Ld domains, whereas the donor used, pyrene-DPPE, has a significant affinity for Lo domains.
As shown in Fig. 2, at room temperature weaker pyrene to rhodamine FRET (higher F/Fo) was observed in 1:1 DSPC/DMoPC vesicles containing cholesterol, a 1:1 mixture of cholesterol/epicholesterol, or a 1:1 mixture of coprostanol/epicholesterol than that measured in homogeneous (domain-lacking) DMoPC vesicles with the same sterols. This is indicative of coexisting ordered and disordered domain formation in the DSPC/DMoPC-containing samples. (We did not use pure coprostanol because it tends to inhibit domain formation (see Discussion).) A sigmoidal increase of FRET upon increasing temperature was observed, such that at high temperature FRET was similar in samples containing DSPC/DMoPC and those just containing DMoPC. This shows that the ordered domains melted (i.e., transitioned from ordered to disordered) at high temperature, consistent with previous results (24).
Figure 2.
Detection of domain formation in vesicles composed of DSPC/DMoPC/sterol by FRET. MLVs (500 μM total lipid) composed of DMoPC with 45 mol % cholesterol ( ), DMoPC with 45 mol % 1:1 coprostanol/epicholesterol (
), DMoPC with 45 mol % 1:1 coprostanol/epicholesterol ( ), 1:1 DSPC/DMoPC with 45 mol % cholesterol (▪), 1:1 DSPC/DMoPC with 45 mol % 1:1 cholesterol/epicholesterol (
), 1:1 DSPC/DMoPC with 45 mol % cholesterol (▪), 1:1 DSPC/DMoPC with 45 mol % 1:1 cholesterol/epicholesterol ( ), or 1:1 DSPC/DMoPC with 45 mol % 1:1 coprostanol/epicholesterol (♦) were prepared in PBS pH 5.1. F samples contained both FRET donor (0.05 mol % pyrene-DPPE) and FRET acceptor (2 mol % Rho-DOPE). Fo samples only contained FRET donor (0.05 mol % pyrene-DPPE). The ratio of donor fluorescence in the presence of acceptor to that in its absence (F/Fo) is graphed. Average (mean) values and SD values from triplicates are shown. Abbreviations: chol = cholesterol; epichol = epicholesterol; and cop = coprostanol.
), or 1:1 DSPC/DMoPC with 45 mol % 1:1 coprostanol/epicholesterol (♦) were prepared in PBS pH 5.1. F samples contained both FRET donor (0.05 mol % pyrene-DPPE) and FRET acceptor (2 mol % Rho-DOPE). Fo samples only contained FRET donor (0.05 mol % pyrene-DPPE). The ratio of donor fluorescence in the presence of acceptor to that in its absence (F/Fo) is graphed. Average (mean) values and SD values from triplicates are shown. Abbreviations: chol = cholesterol; epichol = epicholesterol; and cop = coprostanol.
Somewhat stronger FRET (lower F/Fo) at low temperatures was observed in DSPC/DMoPC vesicles with a 1:1 coprostanol/epicholesterol mixture than that observed in DSPC/DMoPC vesicles containing cholesterol or a 1:1 cholesterol/epicholesterol mixture, suggesting that smaller or fewer domains might be formed in the vesicles containing 1:1 coprostanol/epicholesterol (24). This could be explained by the fact that coprostanol disrupts domain formation (17).
Domain formation similar to that in vesicles with DSPC/DMoPC was detected in liposomes composed of SM/DMoPC, DSPC/DPhPC, and SM/DPhPC in the presence of cholesterol or 1:1 mixtures of coprostanol/epicholesterol (Fig. S2).
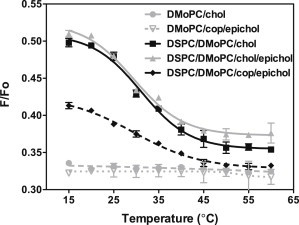
The FRET assay does not define what type of ordered domains are present (gel or liquid ordered) in samples containing lipid mixtures with epicholesterol or coprostanol/epicholesterol. To define this, microscopy studies on GUVs containing various sterols were carried out. Lo domains are rounded, whereas gel domains have jagged, highly irregular shapes (29–31). Fig. 3 A shows that mixtures of high-Tm and low-Tm lipids with epicholesterol form Lo domains. We also examined domains in GUVs containing epicholesterol/coprostanol mixtures. For mixtures of high Tm and low Tm lipids with 1:1 coprostanol/epicholesterol, domains were too small to see (even though easily detected by FRET) at any concentration with mixtures of lipids containing DMoPC or containing SM, and could only be seen at very low epicholesterol concentrations (maximum 12.5 mol % epicholesterol) for mixtures of DSPC/DPhPC. However, Lo domains were observed in DSPC/DPhPC at 20 mol % epicholesterol in 1:2 coprostanol/epicholesterol (Fig. 3 B).
Figure 3.
Fluorescence micrographs of GUVs show formation of coexisting Lo and Ld domains in the presence of epicholesterol and epicholesterol plus coprostanol. (A) GUVs composed of high-Tm lipid, low-Tm lipid, and epicholesterol: (a) 1:1 DSPC/DMoPC with 22.5 mol % epicholesterol; (b) 1:1 SM/DMoPC with 22.5 mol % epicholesterol; (c) 1:1 DSPC/DPhPC with 20 mol % epicholesterol; and (d) 1:1 SM/DPhPC with 20 mol % epicholesterol. The scale bar is 5 μm. Dashed white line is added to help visualize vesicle perimeter. (B) Two representative micrographs of GUVs composed of 1:1 DSPC/DPhPC with 10 mol % coprostanol and 20 mol % epicholesterol. The scale bar for the micrograph on the left is 10 μm and on the right is 5 μm. Abbreviation: epichol = epicholesterol. Vesicles in (A) and (B) were labeled with 0.02 mol % Ld-marker Rho-DOPE. To see this figure in color, go online.
FRET assay reveals sterol structure controls PFO raft affinity
Using the lipid compositions studied above, a different FRET assay we developed previously was used to study the effect of sterols on PFO affinity for ordered domains (rafts) (18,19). FRET from PFO Trp (donor) to the acceptor probe lipid NBD-DPhPE, which has bulky acyl chains and partitions favorably into Ld domains (19), was measured to evaluate PFO association with rafts. The basis of the assay is that in membranes with coexisting Lo and liquid-disordered (Ld) domains, FRET is weak when PFO associates with acceptor-depleted Lo domains and strong when it associates with acceptor-rich Ld domains. To compare FRET values for proteins with different inherent FRET efficiencies, CLoLd/CLd, which describes the effective local acceptor concentration around protein molecules in domain-containing membranes (CLoLd) relative to that in the homogeneous Ld membranes (CLd), was used (18,19). The effective local (i.e., within FRET range) acceptor concentration is high when a protein partitions into a domain with a high concentration of acceptor (i.e., Ld domains in these samples) and low when a protein partitions into domains with a low concentration of acceptor (i.e., Lo domains in these samples). High CLoLd/CLd values indicate that a protein has a high affinity for Ld domains, whereas low CLoLd/CLd indicates a protein has a high affinity for Lo domains.
To calibrate FRET, we compared the raft affinity of PFO to that of two standard marker proteins: LW peptide, a TM-helix peptide previously shown to have a high affinity for Ld domains (32), and CT-B, a protein that binds to the raft-associating lipid GM1 and has a very high affinity for Lo domains (18,33). The expected FRET pattern was observed for LW peptide and CT-B in vesicles composed of 1:1 DSPC/DMoPC with 45 mol % cholesterol, 1:1 cholesterol/epicholesterol, or 1:1 coprostanol/epicholesterol. In all cases, CLoLd/CLd was high for LW peptide and low for CT-B (Fig. 4).
Figure 4.
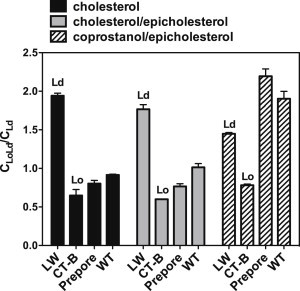
FRET-detected raft affinity of PFO in DSPC/DMoPC/sterol vesicles forming coexisting Lo and Ld domains. Raft affinity of LW peptide, CT-B, and PFO in MLVs (500 μM total lipid) composed of 1:1 DSPC/DMoPC with 45 mol % cholesterol (black bars), 45 mol % 1:1 cholesterol/epicholesterol (gray bars), or 45 mol % 1:1 coprostanol/epicholesterol (striped bars) was measured in PBS pH 5.1. 2 mol % NBD-DPhPE was used as the FRET acceptor. The CLoLd/CLd ratio represents the average local acceptor concentration of acceptor around the donor (protein) in vesicles containing Lo and Ld domains (CLoLd) relative to that in a homogenous bilayer (DMoPC with 45 mol % sterol) lacking domains (CLd). CLoLd/CLd is high for a protein in Ld domains and low for a protein in Lo domains. The actual FRET (F/Fo) data are shown in Fig. S3. For vesicles containing a mixture of coprostanol and epicholesterol, in which PFO binding is not complete, corrected CLoLd/CLd values are shown (see Fig. S4 and Materials and Methods). Uncorrected F/Fo and CLoLd/CLd data are shown in Fig. S5. Average (mean) values and SD values from triplicates are shown.
Next, raft affinity was measured both for TM (WT) and non-TM (prepore mutant) PFO using FRET. In vesicles containing 1:1 DSPC/DMoPC with 45 mol % cholesterol, the CLoLd/CLd ratio for both WT and prepore PFO was between that of LW and CT-B but closer to CT-B, indicating a considerable extent of association with (i.e., partition into) DSPC- and cholesterol-rich Lo domains (Fig. 4). The association of prepore PFO with Lo domains was slightly stronger than that of WT PFO. This was also true in vesicles containing 1:1 DSPC/DMoPC with 45 mol % 1:1 cholesterol/epicholesterol.
In contrast, in vesicles containing 1:1 DSPC/DMoPC with 45 mol % 1:1 coprostanol/epicholesterol (Fig. 4) or with 45 mol % 1:2 coprostanol/epicholesterol (Fig. S6), the CLoLd/CLd ratio for both WT and prepore PFO was very high, in fact generally even higher than that of LW peptide, indicating a very strong association with DMoPC- and coprostanol-rich Ld domains. In this case WT PFO associated somewhat more with the ordered domains (likely to be Lo domains) formed in this mixture than did the prepore PFO. Because PFO does not bind epicholesterol, the sterol that does bind to PFO, coprostanol, must largely determine PFO location (see Discussion).
We previously showed that hydrophobic mismatch between the TM segments of PFO and membrane domain widths could influence the domains to which PFO binds (19). Mismatch cannot explain the low raft affinity observed with coprostanol-containing vesicles as the prepore protein, which lacks TM domains and so cannot respond to hydrophobic mismatch, shows a raft affinity very close to that of the WT protein.
Using this FRET assay a similar pattern of PFO domain association with ordered and disordered domains was also observed in the other lipid mixtures examined (Fig. 5), confirming that the localization of sterols to which PFO binds determined the domain localization of PFO. It is noteworthy that in cholesterol-containing vesicles, prepore PFO usually had a slightly higher association with Lo domains than WT PFO, but this pattern reversed in vesicles containing coprostanol and epicholesterol (see Discussion).
Figure 5.
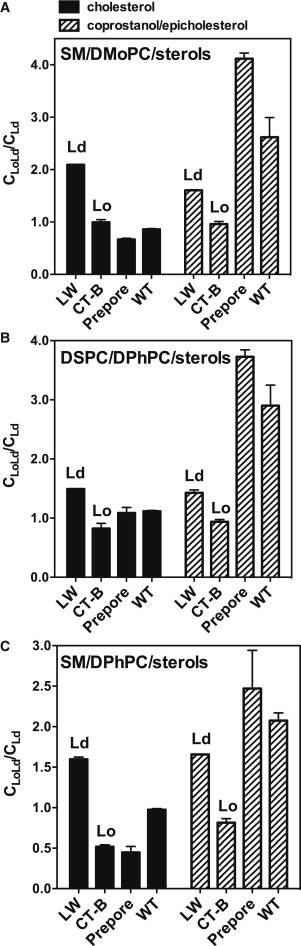
FRET-detected raft affinity of PFO in vesicles containing various lipid compositions forming coexisting Lo and Ld domains. (A) Raft affinity of LW peptide, CT-B, and PFO in MLVs (500 μM total lipid) composed of 1:1 SM/DMoPC with 45 mol % cholesterol (black bars) or 45 mol % 1:1 coprostanol/epicholesterol (striped bars). (B) Raft affinity of LW peptide, CT-B, and PFO in MLVs (500 μM total lipid) composed of 1:1 DSPC/DPhPC with 40 mol % cholesterol (black bars) or 40 mol % 1:1 coprostanol/epicholesterol (striped bars). (C) Raft affinity of LW peptide, CT-B, and PFO in MLVs (500 μM total lipid) composed of 1:1 SM/DPhPC with 40 mol % cholesterol (black bars) or 40 mol % 1:1 coprostanol/epicholesterol (striped bars). Samples were prepared in PBS pH 5.1. 2 mol % NBD-DPhPE was used as the FRET acceptor. The CLoLd/CLd ratio represents the average local acceptor concentration of acceptor around the donor (protein) in vesicles containing Lo and Ld domains (CLoLd) relative to that in a homogenous bilayer (DMoPC with 40 mol % or 45 mol % sterol) lacking domains (CLd). (We could not use DPhPC/sterol for the experiments in (B) and (C) because we could not make MLVs of DPhPC with a high sterol concentration.) The actual FRET (F/Fo) data are shown in Fig. S3. For vesicles containing a mixture of coprostanol and epicholesterol, in which PFO binding is not complete, corrected CLoLd/CLd values are shown (see Fig. S4 and Materials and Methods). Uncorrected F/Fo and CLoLd/CLd data are shown in Fig. S5. Average (mean) values and SD values from triplicates are shown.
Discussion
Effect of lipid structure and sterol structure on PFO localization
Rafts are well-established contributors to the action of many bacterial toxins, such as PFO. Raft domains have been proposed to promote toxin binding, oligomerization, and oligomer insertion into membranes to form pores (35,36). However, in vitro studies using artificial lipid vesicles do not provide evidence supporting the idea that rafts are required for PFO assembly and action. In fact, at a fixed cholesterol concentration PFO can more readily associate and assemble in vesicles with a lipid composition forming the loosely packed Ld state than one forming the Lo state (15,16). This is not definitive, because in membranes with coexisting ordered and disordered domains the cholesterol concentration should be higher in the ordered domains than in the disordered domains. Thus, the question of whether and why PFO associates with ordered domains must be studied in model membranes with coexisting ordered and disordered domains.
To study this we prepared such model membrane vesicles and then determined where PFO located. Our previous studies showed that both sterol binding and hydrophobic match between TM segment length and Lo domain bilayer width can promote raft affinity (18,19). However, remaining unanswered is the the question of whether PFO raft affinity is directly related to the raft affinity of cholesterol. To answer this question we examined the effect of sterol structure upon PFO raft interaction. Different sterols have different tendencies to participate in ordered domains formation (17,37,38). Coprostanol, which has a large bend between the A and B rings (Fig. S7), interferes with tight packing found in ordered domains and inhibits ordered domain formation (17,38). This also implies that coprostanol binds to Ld domains much better than to ordered domains. Because coprostanol does not tend to form ordered domains with saturated lipids, to study the effect of coprostanol on PFO raft affinity, we needed to use vesicles with a mixture of coprostanol and epicholesterol. Epicholesterol, which has a 3α-OH in place of the 3β-OH of cholesterol, does not bind to PFO but does promote ordered domain formation. Based on this, we reasoned, and experimentally confirmed, that vesicles with a mixture of coprostanol and epicholesterol would form coexisting ordered and disordered domains. Using vesicles containing a mixture of coprostanol and epicholesterol, FRET showed that, compared to cholesterol-containing vesicles, PFO preferentially partitioned into Ld domains. As only coprostanol could bind to PFO in this sterol mixture, the localization of PFO must have been mainly determined by the properties of PFO-bound coprostanol. It should be noted that although direct PFO-epicholesterol interactions would not form, the epicholesterol could influence PFO association with rafts by affecting the extent of exclusion of coprostanol from ordered domains. PFO-bound coprostanol should be similar to unbound coprostanol in tending to localize in Ld domains because PFO only seems to bind to the -OH end of sterol molecules. The membrane-exposed portion of PFO-bound sterol should pack with membrane lipids similar to unbound sterol (except in cases in which the exposure of the OH group influences raft affinity, such as in ceramide-containing membranes, see below). Thus, we conclude that when PFO is bound to a sterol that associates with rafts, its raft association is promoted, and when PFO is bound to a sterol that does not associate well with rafts, its association with Ld domains is promoted.
Relationship between sterol domain localization and PFO domain localization
It is important to note that the localization of PFO in ordered or disordered domains does not reflect the location of the PFO-binding sterol before binding to PFO, but rather the equilibrium location of PFO after it binds to membrane and assembles into the TM oligomer (or prepore if that is the final conformation). This is confirmed by the fact that TM segment length affects domain location of PFO (18,19). Because PFO initially binds to membranes in the prepore form, PFO should initially bind to domains independent of the TM segment length. Thus, if the initial domain binding site determined final PFO domain localization, PFO domain localization would not be affected by TM segment length. This is contradicted by experimental observations using PFO with different TM segment lengths (19).
Another set of observations that shows final PFO location is not the same as the location to which PFO binds come from experiments using vesicle containing a mixture of high-Tm lipid, low-Tm lipid, sterol and ceramide (18). Ceramide displaces cholesterol from ordered domains, and yet PFO in such membranes does locate in the ordered domains (27). Presumably, the PFO first binds to the cholesterol-containing Ld domains and then moves into the ceramide-rich ordered domains, together with bound sterol (18).
An interesting question is whether other proteins would show behavior similar to that of PFO upon their binding of a ligand/lipid that has a very strong or very weak affinity for rafts. The effect of binding might be modest for a monomeric protein, and should be much stronger for an oligomeric one. This may be a very important factor in altering raft affinity during clustering of membrane proteins into complexes. In addition, even a modest change in raft affinity, of a few-fold or less, might have important functional consequences.
Another question is the extent to which the physical properties of ordered domains influence PFO localization. We believe the exclusion of PFO from ordered domains in the samples containing mixtures of coprostanol and epicholesterol cannot be explained by tight/highly ordered lipid acyl chain packing excluding TM PFO segments. This is because prepore PFO is a peripheral protein, lacking TM segments, and it is also excluded from ordered domains in membranes containing coprostanol. Instead, this is very likely due to coprostanol being largely localized to disordered domains. It is also possible that other structural parameters that differ in epicholesterol-rich and cholesterol-rich Lo domains have some influence on PFO localization. Because the WT PFO associates with ordered domains better than does the prepore PFO in coprostanol-containing samples, its TM segments must promote association with ordered domains in these samples. (It should be noted that binding of various sterols by WT and prepore PFO is almost identical, so that cannot explain the WT/prepore differences (18).) How can this be if packing disfavors association of TM segments with ordered domains? An explanation is that, as we have previously shown, hydrophobic mismatch is an important factor that controls PFO localization in domains (19). Thus, in the samples containing epicholesterol and coprostanol, the mismatch between the length spanned by the TM segments of PFO and Ld domain bilayer width no doubt favors entry into the ordered domains, and overcomes any effects due to poor packing between ordered state lipids and amino acid side chains.
Interpretation of FRET data
In interpreting FRET results, it is important to note that the domains formed by coprostanol and epicholesterol were different from those formed by cholesterol. First, relative to cholesterol, stronger FRET between the donor (which has some affinity for ordered domains) and the acceptor (which partitions strongly into disordered domains) indicated that lipids with a mixture of coprostanol and epicholesterol formed a lesser amount of ordered domain bilayers, and/or smaller ordered domains, and/or affected the partitioning of the FRET probes in ordered and disordered domains so as to decrease the segregation of donor and acceptor (24). Second, as judged by FRET, ordered domains formed by coprostanol and epicholesterol were less thermally stable than that promoted by cholesterol. The temperature dependence of domain melting indicated that with the same phospholipid compositions, ∼5°C decrease in the melting temperature Tm was observed for vesicles containing 1:1 coprostanol/epicholesterol relative to that for vesicles containing cholesterol and/or 1:1 cholesterol/epicholesterol. This could either mean that the domains disappear at a lower Tm value when coprostanol is present, or that they fall below the size threshold for detection by FRET at a lower temperature (24).
These issues make it important to calibrate the difference in FRET response for PFO in membranes with different sterols using proteins that act as Ld and Lo markers. Even so, it should be noted that the partition of the marker proteins could be affected to some degree by lipid composition, and thus absolute values for raft affinity cannot be defined from the FRET data. An alternative, which in favorable cases can provide absolute values for PFO raft affinity, is microscopy studies in giant vesicles (18,19). However, we found large domains only formed at a concentration of sterol too low to allow PFO binding to the vesicles (data not shown). In any case, it is noteworthy that when FRET and microscopy experiments can be carried out under similar conditions, they have agreed as to PFO localization in ordered and disordered domains (18,19).
Conclusions
The affinity of PFO for ordered domains appears to be determined by a number of properties. One is binding to sterol. The raft affinity of prepore PFO, which does not penetrate bilayers, is totally dependent on the raft affinity of the sterol to which it is bound. If the sterol packs well into ordered domains, prepore PFO has a high affinity for ordered domains, whereas if the sterol has a low affinity for ordered domains, the prepore PFO has a low affinity for them as well. The same properties must apply to WT PFO in its transmembraneous pore-forming state. Whether the protein-bound sterol is exposed to water is an additional factor that must be considered. Prior studies have shown that free cholesterol has weak affinity for ceramide-rich domains in vesicles that contain SM and low-Tm lipids because ceramide competes with cholesterol for interaction with SM that minimizes the aqueous exposure of hydrophobic sites on cholesterol or ceramide (27). This is not an issue for sterol bound to PFO, because the PFO shields the sterol from water. As a result, as prior studies have shown, PFO has a strong affinity for ordered domains in ceramide-rich vesicles (18). A third property that is important for PFO in a TM state is hydrophobic mismatch between bilayer width and TM segment length. Mismatch in which the width of a domain is less than the length of PFO TM segments disfavors association with that domain due to unfavorable exposure of hydrophobic residues on PFO to water (19). This has been shown to be true both when the mismatch involves Ld domains and when it involves Lo domains. The effect of mismatch should be greatest for oligomeric protein complexes or oligomeric proteins such as PFO, because they cannot tilt to lessen mismatch (19). It should be noted that although these factors influencing Lo affinity would have a cumulative effect, it would not necessarily involve them being strictly additive. It should also be noted that these principles should apply to α-helical proteins as well as to β-barrel proteins.
Acknowledgments
This work was supported by National Institutes of Health (NIH) GM 099892.
Supporting Material
References
- 1.Brown D.A., London E. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 1998;164:103–114. doi: 10.1007/s002329900397. [DOI] [PubMed] [Google Scholar]
- 2.Simons K., Sampaio J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levental I., Lingwood D., Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. USA. 2010;107:22050–22054. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bijlmakers M.J. Protein acylation and localization in T cell signaling (Review) Mol. Membr. Biol. 2009;26:93–103. doi: 10.1080/09687680802650481. (Review) [DOI] [PubMed] [Google Scholar]
- 5.Shogomori H., Hammond A.T., Brown D.A. Palmitoylation and intracellular domain interactions both contribute to raft targeting of linker for activation of T cells. J. Biol. Chem. 2005;280:18931–18942. doi: 10.1074/jbc.M500247200. [DOI] [PubMed] [Google Scholar]
- 6.Rossjohn J., Feil S.C., Parker M.W. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 7.Tweten R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gekara N.O., Weiss S. Lipid rafts clustering and signalling by listeriolysin O. Biochem. Soc. Trans. 2004;32:712–714. doi: 10.1042/BST0320712. [DOI] [PubMed] [Google Scholar]
- 9.Abrami L., van Der Goot F.G. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J. Cell Biol. 1999;147:175–184. doi: 10.1083/jcb.147.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waheed A.A., Shimada Y., Ohno-Iwashita Y. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts) Proc. Natl. Acad. Sci. USA. 2001;98:4926–4931. doi: 10.1073/pnas.091090798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohno-Iwashita Y., Shimada Y., Iwashita S. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe. 2004;10:125–134. doi: 10.1016/j.anaerobe.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Shimada Y., Maruya M., Iwashita S., Ohno-Iwashita Y. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Euro. J. Biochem. 2002;269:6195–6203. doi: 10.1046/j.1432-1033.2002.03338.x. [DOI] [PubMed] [Google Scholar]
- 13.Giddings K.S., Johnson A.E., Tweten R.K. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. USA. 2003;100:11315–11320. doi: 10.1073/pnas.2033520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sokolov A., Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 2010;285:29480–29490. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flanagan J.J., Tweten R.K., Heuck A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry. 2009;48:3977–3987. doi: 10.1021/bi9002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson L.D., Johnson A.E., London E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 2008;283:4632–4642. doi: 10.1074/jbc.M709483200. [DOI] [PubMed] [Google Scholar]
- 17.Xu X., London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry. 2000;39:843–849. doi: 10.1021/bi992543v. [DOI] [PubMed] [Google Scholar]
- 18.Nelson L.D., Chiantia S., London E. Perfringolysin O association with ordered lipid domains: implications for transmembrane protein raft affinity. Biophys. J. 2010;99:3255–3263. doi: 10.1016/j.bpj.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Q., London E. Altering hydrophobic sequence lengths shows that hydrophobic mismatch controls affinity for ordered lipid domains (rafts) in the multitransmembrane strand protein perfringolysin O. J. Biol. Chem. 2013;288:1340–1352. doi: 10.1074/jbc.M112.415596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura M., Sekino N., Ohno-Iwashita Y. Interaction of theta-toxin (perfringolysin O), a cholesterol-binding cytolysin, with liposomal membranes: change in the aromatic side chains upon binding and insertion. Biochemistry. 1995;34:6513–6520. doi: 10.1021/bi00019a032. [DOI] [PubMed] [Google Scholar]
- 21.Portnoy D.A., Tweten R.K., Bielecki J. Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect. Immun. 1992;60:2710–2717. doi: 10.1128/iai.60.7.2710-2717.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Brien D.K., Melville S.B. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect. Immun. 2004;72:5204–5215. doi: 10.1128/IAI.72.9.5204-5215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran R., Tweten R.K., Johnson A.E. The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc. Natl. Acad. Sci. USA. 2005;102:7139–7144. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pathak P., London E. Measurement of lipid nanodomain (raft) formation and size in sphingomyelin/POPC/cholesterol vesicles shows TX-100 and transmembrane helices increase domain size by coalescing preexisting nanodomains but do not induce domain formation. Biophys. J. 2011;101:2417–2425. doi: 10.1016/j.bpj.2011.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiantia S., Kahya N., Schwille P. Raft domain reorganization driven by short- and long-chain ceramide: a combined AFM and FCS study. Langmuir. 2007;23:7659–7665. doi: 10.1021/la7010919. [DOI] [PubMed] [Google Scholar]
- 26.Chiantia S., Schwille P., London E. Asymmetric GUVs prepared by MβCD-mediated lipid exchange: an FCS study. Biophys. J. 2011;100:L1–L3. doi: 10.1016/j.bpj.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Megha, London E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 28.Alanko S.M., Halling K.K., Ramstedt B. Displacement of sterols from sterol/sphingomyelin domains in fluid bilayer membranes by competing molecules. Biochim. Biophys. Acta. 2005;1715:111–121. doi: 10.1016/j.bbamem.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Feigenson G.W. Phase behavior of lipid mixtures. Nat. Chem. Biol. 2006;2:560–563. doi: 10.1038/nchembio1106-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feigenson G.W. Phase boundaries and biological membranes. Annu. Rev. Biophys. Biomol. Struct. 2007;36:63–77. doi: 10.1146/annurev.biophys.36.040306.132721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J., Wu J., Feigenson G.W. Phase studies of model biomembranes: complex behavior of DSPC/DOPC/cholesterol. Biochim. Biophys. Acta. 2007;1768:2764–2776. doi: 10.1016/j.bbamem.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fastenberg M.E., Shogomori H., London E. Exclusion of a transmembrane-type peptide from ordered-lipid domains (rafts) detected by fluorescence quenching: extension of quenching analysis to account for the effects of domain size and domain boundaries. Biochemistry. 2003;42:12376–12390. doi: 10.1021/bi034718d. [DOI] [PubMed] [Google Scholar]
- 33.Lencer W.I., Saslowsky D. Raft trafficking of AB5 subunit bacterial toxins. Biochim. Biophys. Acta. 2005;1746:314–321. doi: 10.1016/j.bbamcr.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Gekara N.O., Jacobs T., Weiss S. The cholesterol-dependent cytolysin listeriolysin O aggregates rafts via oligomerization. Cell. Microbiol. 2005;7:1345–1356. doi: 10.1111/j.1462-5822.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- 36.Mañes S., del Real G., Martínez-A C. Pathogens: raft hijackers. Nat. Rev. Immunol. 2003;3:557–568. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- 37.Xu X.L., Bittman R., London E. Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts). Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide. J. Biol. Chem. 2001;276:33540–33546. doi: 10.1074/jbc.M104776200. [DOI] [PubMed] [Google Scholar]
- 38.Beattie M.E., Veatch S.L., Keller S.L. Sterol structure determines miscibility versus melting transitions in lipid vesicles. Biophys. J. 2005;89:1760–1768. doi: 10.1529/biophysj.104.049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.