

Abstract
We showed that stop of flow triggers a mechanosignaling cascade that leads to the generation of reactive oxygen species (ROS); however, a mechanosensor coupled to the cytoskeleton that could potentially transduce flow stimulus has not been identified. We showed a role for KATP channel, caveolae (caveolin-1), and NADPH oxidase 2 (NOX2) in ROS production with stop of flow. Based on reports of a mechanosensory complex that includes platelet endothelial cell adhesion molecule-1 (PECAM-1) and initiates signaling with mechanical force, we hypothesized that PECAM-1 could serve as a mechanosensor in sensing disruption of flow. Using lungs in situ, we observed that ROS production with stop of flow was significantly reduced in PECAM-1−/− lungs compared with lungs from wild-type (WT) mice. Lack of PECAM-1 did not affect NOX2 activation machinery or the caveolin-1 expression or caveolae number in the pulmonary endothelium. Stop of flow in vitro triggered an increase in angiogenic potential of WT pulmonary microvascular endothelial cells (PMVEC) but not of PECAM-1−/− PMVEC. Obstruction of flow in lungs in vivo showed that the neutrophil infiltration as observed in WT mice was significantly lowered in PECAM-1−/− mice. With stop of flow, WT lungs showed higher expression of the angiogenic marker VEGF compared with untreated (sham) and PECAM-1−/− lungs. Thus PECAM-1 (and caveolae) are parts of the mechanosensing machinery that generates superoxide with loss of shear; the resultant ROS potentially drives neutrophil influx and acts as an angiogenic signal.
Keywords: mechanotransduction, stop of flow, pulmonary endothelium, PECAM, Katp (Kir6.2) channel, NOX2, angiogenic potential
cells sense the physical stimulus in their environment and translate these physical forces into biochemical signals (20, 37). Sensing and responding to a physical force require specialized structures and machinery that can engage in signal transduction (12, 23, 42).
In the vascular system, with a highly distributed network of blood vessels, mechanical forces arising from blood flow initiate signaling that helps maintain vascular structure and function. Indeed, shear associated with blood flow is sensed by the endothelium and the resultant signaling regulates normal vascular physiology (such as embryonic morphogenesis and organization of the vascular tree) while irregular or abnormal shear can lead to vascular dysfunction and disease (19, 27). Thus the mechanosignaling that accompanies various shear profiles and patterns, regular or aberrant, governs susceptibility to atherosclerosis, by inducing athero-protective or athero-prone phenotypes in endothelial cells (10, 22). It thus becomes important to understand the link among the mechanical force, the shear sensing machinery and biochemical signaling within the cell. Despite recent advances in this field, relatively little is known about the sensing apparatus or machinery in cells that transduces the physical forces associated with changes in blood flow into signaling moieties.
We have previously studied endothelial mechanotransduction using in situ (isolated lungs and aorta) as well as in vitro (flow-adapted cells) systems. We showed that endothelial cells respond to abrupt cessation of flow with a signaling cascade characterized by the generation of reactive oxygen species (ROS; Refs. 1, 5, 8, 31, 46). The response to loss of shear required a preceding period of exposure to flow to reach a flow adapted state. Statically cultured cells kept for short periods under flow did not show any response to stop of flow (5, 8, 35). Lack or removal of flow in this model did not compromise oxygen delivery to the lung tissues or cells as normal oxygen tension was maintained during stop of flow in vivo (ventilated lung) and in vitro (continuous flow of medium through side ports) (1, 5, 30, 46). We identified that the earliest change that occurred with cessation of flow was endothelial cell membrane depolarization [via deactivation of a K+ (KATP) channel] leading to NADPH oxidase 2 (NOX2) assembly.
What are the elements on the endothelium that sense the altered flow stimulus and activate a signaling cascade that leads to K+ channel closure and NADPH oxidase activation? We have observed that statically cultured endothelial cells demonstrate low KATP channel expression and activity that are increased upon exposure to prolonged periods of flow (5). This indicated that the channel is induced by flow, pointing to the presence of an upstream shear sensor or a primary transducer on the endothelium that “senses” flow and leads to increased channel expression. A cell surface entity would ideally be well suited to be a primary transducer. Based on previous studies on transduction of mechanical forces including shear stress by a mechanosensory complex comprising of platelet endothelial cell adhesion molecule 1 (PECAM-1; Ref. 42) and on reports of PECAM-1 activation (tyrosine phosphorylation) upon flow (9), we investigated whether PECAM-1, by virtue of its junctional location and cytoskeletal linkage, might serve as a mechanosensor for the loss of blood flow. Our earlier studies on mechanotransduction showed that lack of caveolin-1 (and thus caveolae) prevented the pulmonary endothelial ROS production with cessation of flow (33, 45). We thus investigated the role of PECAM-1 in mechanosensing and the association (if any) of PECAM-1 and caveolin-1 (and thus caveolae) in the pulmonary endothelium.
Based on our results, we conclude that PECAM-1 is an important component of the mechanosensing machinery in the pulmonary endothelium and conversely that the biochemical and cellular responses to altered flow are compromised in the absence of PECAM-1.
MATERIALS AND METHODS
Reagents and Antibodies
Dihydroethidium (DHE), carboxy-H2DFFDA, and acetylated low-density lipoprotein (AcLDL) coupled to Alexa 594 and Amplex Red were purchased from Life Technologies (Carlsbad, CA). Acetonitrile (HPLC grade) was purchased from Sigma-Aldrich (St. Louis, MO). Antibodies to PECAM-1 (M20) were from Santa Cruz Biotechnology (Santa Cruz, CA), and antibodies to VEGF and VE-cadherin were from Abcam (Cambridge, MA). Anti-caveolin-1 was either from Abcam (Cambridge, MA) or Transduction Laboratories (San Jose, CA). Polyclonal anti-VE-cadherin-FITC was from eBiosciences (San Diego, CA). Anti-β-actin and nonimmune IgG (mouse and rabbit) were from Sigma-Aldrich. Antibody to PECAM that binds to IgD1 in PECAM was obtained from Centocor (Malvern, PA; Ref. 38a). The phospho-PECAM (p-Tyr713PECAM) antibody was from Assay Biotechnology (Sunnyvale, CA). Anti-flotillin was from was BD Transduction Laboratories (Lexington, KY). Anti-mouse IgG-coated magnetic beads were from Dynal Biotech (Life Technologies, Grand Island, NY). The 25-micrometer green fluorescent microspheres (Fluoro-Max) were purchased from Thermoscientific (Lafayette, CO). The Duolink kit was from Olink (Uppsala, Sweden).
Animal Use
Animal use was reviewed and approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Male C57BL/6, caveolin-1−/−, and NOX2−/− mice weighing ∼20 g were obtained from Jackson Laboratories (Bar Harbor, ME). Breeding pairs of PECAM-1−/− mice originally created by Tak W. Mak (13) were provided to us by Dr. Joseph Madri (Yale University). These mice were backcrossed for over 10 generations into the C57BL/B6 background. All protocols were performed in accordance with National Institutes of Health guidelines and with the approval the University of Pennsylvania Animal Use Committee.
Cell Culture
Isolation of pulmonary microvascular endothelial cells (PMVEC) was carried out as described previously except that we used anti-VE-cadherin (instead of anti-PECAM) to immunoselect for endothelial cells from PECAM-1−/− lungs (8). Briefly, lung tissue was trimmed at the periphery and digested, and VE-cadherin positive cells isolated by immunoselection using anti-VE-cadherin-labeled Dynal magnetic beads (8, 11, 25).
Flow Adaptation of Endothelial Cells and Cessation of Flow In Vitro
The artificial capillary system (FiberCell Systems, Frederick, MD) for flow adapting PMVEC has been described previously (5, 6, 8). Stop of flow was achieved by turning off the peristaltic pump to stop flow of culture medium over the cells; the cells, however, were fed by oxygenated medium from the side ports to ensure that lack of flow did not compromise oxygen and nutrient supply. Cells were subjected to 24 h of flow adaptation followed by stop of flow and removed from the capillary chambers or kept under statically cultured conditions.
Lung Isolation
Mouse lung isolation and perfusion were carried out as described previously (6, 8, 49, 50). Briefly, mice were anesthetized, the trachea was cannulated, and the lungs were ventilated with 5% CO2 in air. The chest was opened, the pulmonary artery was cannulated, and the lungs were cleared of blood by gravity-driven flow of Krebs-Ringer bicarbonate solution (KRB; in mM: 118 NaCl, 4.7 KCl, 1.2 MgSO4·7H2O, 1.2 KH2PO4, and 24.9 NaHCO3) supplemented with 10 mM glucose and 5% wt/vol dextran. The lung was dissected free and hung in an environmental chamber maintained at 37°C for dye loading. Perfusion was maintained at 2 ml/min with a peristaltic pump.
Intravital Microscopy for ROS Detection in the Intact Lung
Imaging of ROS using a difluorofluorescein probe [5-(and-6)-carboxy-2′,7′-difluorodihydrofluorescein diacetate (CMH2DFFDA)] was based on reports previously described (47, 48). After isolation and perfusion of the lung as described above, the lung was allowed to equilibrate in KRB buffer for 5 min. Next, endothelial marker Alexa 594 acetylated low density lipoprotein (Alexa594-AcLDL) was added (final concentration of 0.5 μg/ml) to the perfusate followed by 10 μM H2DFFDA. After an additional 15 min of perfusion to facilitate loading of the dyes in the pulmonary endothelium, the lung was removed from the chamber and immediately positioned on the stage of a confocal microscope while perfusion and ventilation were maintained. Stop of flow was attained by turning off the perfusion pump, but ventilation continued throughout the duration of the lung imaging. H2DFFDA fluorescence was measured with wild-type (WT) and PECAM-1−/− and NOX2−/− lungs under flow and at different periods poststop of flow.
Superoxide Production as Monitored by HPLC Detection of 2-Hydroxyethidium
Detection of 2-hydroxyethidium (2-OHE), the superoxide-specific oxidation product of DHE, was adapted from previous reports (51, 52). Lung isolation and perfusion were carried out as above, and the lung was perfused for 30 min with 50 μM DHE. Experiments were carried out in a similar fashion to the imaging study as described earlier with the exception that lungs were not moved to the imaging platform. The flow of perfusate was stopped for 1 h by stopping the perfusion by switching off the pump. Immediately after the completion of the stop of flow period, the DHE and its oxidation products were extracted from 3-mm3 pieces of the lung as previously described (52). The pieces were frozen in liquid nitrogen and individually ground to a powder using a mortar and pestle resting in a bath of liquid nitrogen. The ground tissue was resuspended in 500 μl acetonitrile, sonicated at 4–6 watts for three cycles of 15 s each, and centrifuged for 10 min at 12,000 g. After the supernatant was removed to a separate tube, the pellet was retained for protein quantitation. The supernatant was concentrated by evaporation of acetonitrile in a SpeedVac in dark. HPLC analysis of the supernatant showed the 2-OHE peak; this was normalized to the total ethidium to quantitate superoxide production. A control experiment used a superoxide generating system xanthine/xanthine oxidase (X/XO) in the lung (in the presence of 400 U/ml catalase) to validate the detection of 2-OHE by HPLC. Since the 2-OHE product had to be separated from other oxidized DHE products, we chose a long duration of stop of flow period (1 h) to enable greater 2-OHE accumulation in lungs.
H2O2 Production by Whole Lung Using Intravascular H2O2-Specific Dye Amplex Red
ROS production in isolated lungs was monitored in the presence of NOX2 agonist angiotensin II (ANG II) (7, 26). Amplex Red (50 μM) plus horseradish peroxidase (HRP; 50 μg/ml) was added to the perfusate; this fluorophore does not permeate the cell membrane and thus detects extracellular H2O2. ANG II (50 μM) was added to the lung perfusate, aliquots of the perfusate were removed during 15- to 30-min intervals, fluorescence intensity of the samples was measured (excitation/emission, 545/610) using a spectrofluorimeter (Photon Technology, Birmingham, NJ), and H2O2 concentration was expressed as arbitrary fluorescence units (7, 26). ROS generation was measured with WT and PECAM-1−/− lungs; NOX2−/− lungs were used as a negative (no ROS) control.
Association of PECAM-1 and Caveolin
Immunolabeling.
Lung sections were treated with anti-caveolin-1 and anti-PECAM and visualized by fluorescence microscopy using a Zeiss LSM 510 Meta microscope coupled to a Chameleon Ultra mode-locked femtosecond pulse laser (set at 720 nm for 2-photon excitation of DAPI) and He-Ne and Ar lasers. Colocalization analysis was performed by the ImageJ program using colocalization plugin JACoP to calculate the standard colocalization coefficients (14). The extent of colocalization was measured by Pearson's coefficients, Overlap coefficient, and Mander's coefficients (28, 29).
Colocalization of PECAM-1 and caveolin-1 by Duolink proximal ligation assay.
This assay allows for visualization of protein-protein colocalization by fluorescence microscopy (Duolink; Olink, Uppsala, Sweden) using primary antibodies for each protein obtained from different species (7), in this case a polyclonal anti-caveolin antibody generated in rabbits and a monoclonal antibody to PECAM-1 generated in mice. Lungs isolated from WT mice were perfused to clear the tissue of blood, inflated and fixed with methanol-acetone, and treated with a blocking reagent followed by labeling of the endothelial cells by an antibody that is expressed in endothelial cells [anti-VE-cadherin conjugated to a FITC label (anti-VE-cadherin-FITC); 1:500]. After several washes, the primary antibodies to PECAM-1 and caveolin-1 were added. The secondary antibodies to rabbit and mouse IgG are each attached to a unique synthetic oligonucleotide; ligation causes the oligonucleotides on the two secondary antibodies to hybridize only if the epitopes of the two target proteins are in proximity (<40 nm). Following hybridization, amplification and labeling of the product by detection probes allow for the visualization of the interaction by fluorescence microscopy. In this case, a red signal indicated the interaction of caveolin-1 and PECAM-1. Duolink controls used to check nonspecific signal were 1) anti-PECAM-1/rabbit IgG, 2) anti-caveolin/mouse IgG, and 3) anti-caveolin/anti-PECAM-IgD1. The lung sections were also counterstained with a nuclear marker (DAPI) to enable visualization of endothelial cells in the sections.
Membrane fractionation of PMVEC.
Membranes were isolated from endothelial cells using previously described methods (4). For each experiment, WT mouse PMVEC were grown to confluence (in 150-mm cell culture dishes) removed, pooled, and lysed. The plasma membrane was isolated from postnuclear homogenate using a Percoll gradient (established by mixing the homogenate with 30% Percoll in tricene, followed by centrifugation at 77,000 g for 25 min). After centrifugation, the plasma membrane was clearly visible in the ultracentrifuge tube floating approximately one-half centimeter from the top of the tube. This band was collected, and subcellular fractionation on a sucrose gradient was carried out by loading this band onto a sucrose step gradient for overnight centrifugation at 87,400 g as previously described (4). Fractions were collected every 400 μl (11 fractions were collected), and protein was precipitated with 0.1% wt/vol deoxycholic acid in 100% wt/vol trichloroacetic acid. Proteins were run on a SDS-PAGE gel and immunoblotted for PECAM-1, caveolin-1, and the membrane marker flotillin using Odyssey Western blot analysis technique (Li-Cor, Omaha, NE). Secondary antibodies were IRDyeTM 800 goat anti-rabbit for the green channel and IRDyeTM 680 goat anti-mouse for the red channel. Blots were scanned by placing the membrane on the Odyssey color scanner, and the scanned images were converted to grayscale. All manipulations of contrast were done for the entire gel.
Caveolae immunoaffinity isolation.
Caveolae were isolated as described in our past reports (41a). Briefly, endothelial cells were scraped into ice-cold, detergent-free Tricene buffer (250 mM sucrose, 1 mM EDTA, and 20 mM Tricene pH 7.4) and centrifuged to precipitate nuclear material. The resulting supernatant was mixed with 30% Percoll in Tricene buffer and subjected to ultracentrifugation for 25 min (Beckman MLS50 rotor; 77,000 g, 4°C). The separated plasma membranes were collected, sonicated (3 × 30 s bursts), and incubated with anti-caveolin-1 conjugated goat anti-mouse IgG-coated magnetic beads for 1 h at 4°C. Bound material, representative of caveolae vesicles, was separated magnetically from unbound, noncaveolar membranes, subjected to SDS-PAGE and immunoblotted using indicated antibodies.
Functional interaction of PECAM-1 and caveolin-1.
This was assessed by monitoring phosphorylation of PECAM in caveolin-1−/− cells with stop of flow. PMVEC from WT and caveolin-1 null lungs were either statically cultured (S) or kept for 24 h under flow. Flow was stopped for 1 h (stop of flow), cells were removed and lysed, and cell lysates were immunoblotted using an antibody to phospho-PECAM-1. PMVEC from lungs of PECAM-1−/− mice were used as controls. The phosphoTyr713PECAM antibody recognizes PECAM-1 phosphorylation at the Tyr 713 phosphorylation site and detects the phosphorylated PECAM band at approximatley 130–140 kDa. β-Actin was used as a protein loading control. Immunoblots were quantitated using National Institutes of Health ImageJ software.
Angiogenic Potential of PMVEC
The Matrigel plug assay is used as a test for angiogenesis in vivo. In this assay, PMVEC are introduced into cold liquid Matrigel and injected as a plug into nude mice. The solid gel permits penetration of host endothelium to form new blood vessels inside the plug. PMVEC (1 × 106), obtained after 72 h flow adaptation followed by 1 h of stopped flow, were mixed with 0.5 ml of growth factor-reduced Matrigel and injected subcutaneously into nude mice, using a B-D 26G1/2 needle so that the entire content can be delivered to form a single plug under the skin. After 5 days, the animals were killed; the plug was extracted from under the skin and washed in cold PBS to remove blood and cells sticking to the plug surface. The plugs were then analyzed for vessel formation by two methods. In one method, small pieces of the plug were stained with Alexa594-AcLDL that labels vascular structures. For the other method, the plugs were cryosectioned and stained with endothelial marker VE-cadherin (using anti-VE cadherin Ab) to outline endothelial structures. Five to eight sections were made from each plug. The vascularization within the plug reflects the extent to which blood vessels from the host entered the plug and is an index of the angiogenic signal emanating from the plug, i.e., from the endothelial cells initially present in the plug (3a).
Cessation or Stop of Pulmonary Blood Flow In Vivo in Live Mice
To attain stop of flow in vivo, 25-μm green fluorescent polystyrene microspheres were injected into the lung through the jugular vein. Twenty-four hours later the mice were killed and lungs sectioned and stained by hematoxylin and eosin. Sections were imaged at different magnifications.
VEGF Expression In Vivo
In separate experiments, animals were killed at 1, 4 and 7 days poststop of flow (as induced by beads) in the lung vessels. Lungs were removed and dehydrated by sequential sucrose treatment (10, 20 and 30% sucrose). The tissue was embedded in OCT blocks and longitudinal sections were immunostained for VEGF using anti-VEGF165 antibody (red fluorescence) and compared with nonspecific IgG. Untreated lungs from age matched mice were used as controls. All sections were imaged by confocal microscopy (BioRadiance 2000); images acquired were quantified for VEGF fluorescence by ImageJ software. Briefly, images were split into red (VEGF) and green (microbead) channels; the green channel was masked and the red used for quantitation (after threshold is set to measure only regions that were stained).
Electron Microscopy (Immunogold Staining)
Lungs from WT or PECAM-1−/− mice were isolated, perfused with 0.1 M Na cacodylate buffer, and fixed with 4% paraformaldehyde for 10 min. These were cut into pieces, and two tissue blocks (1 × 1 × 3 mm) were sampled from each lobe. The tissue blocks were further fixed by immersion in 4% paraformaldehyde for 4 h on ice, followed by a thorough wash with 0.1 M Na cacodylate buffer, permeabilized with 0.02% Triton-X-100 in PBS, and then blocked with 50 mM glycine in PBS and a mixture of 5% normal goat serum, 0.1% cool water fish skin gelatin, and 2% BSA in PBS. Following this, the tissue blocks were incubated overnight at 4°C with mouse anti caveolin primary antibody (1:50 dilution) and then incubated with an Ultra Small-Grade (0.8 nm) gold-labeled goat anti mouse secondary antibody (Electron Microscopy Sciences, Hatfield, PA) for 1 h. This was followed by fixation with 2% glutaraldehyde in Na Cacodylate buffer for 5 min and incubation with Aurion R-Gent SE-EM silver enhancement solution (Electron Microscopy Sciences) for 25 min. After further fixations in 1% osmium tetroxoide for 15 min and then 2% uranyl acetate for 10 min, the tissue blocks were dehydrated with graded acetone, embedded with EPON, and polymerized at 6°C for 24 h. The 85-um thick ultrathin sections were prepared with Leica Ultracut machine and imaged in JEM-100-CX electron microscope operating at 60 kV. Around 5–10 images were captured from each tissue block; caveolae were identified by their structure and counted for each cell. Data ware presented as the total number of the caveolar structures per field. The density of caveolae was determined by counting the number of caveolae on the P face in at least three different fields. The number of caveolae was expressed per square micrometer. Statistical analysis was performed using the two-tailed paired t-test.
Statistical Analysis
Values are means ± SD. Data sets were compared using a Student's t-test or ANOVA in SigmaStat (SPSS, San Jose, CA). Results were considered statistically significant at P < 0.05.
RESULTS
Endothelial Mechanosignaling-Induced ROS Production with Stopped Flow Is Compromised in Pulmonary Endothelium of PECAM-1−/− Lungs
The isolated lung in situ model allows for monitoring changes to the endothelium upon removal of shear, i.e., stop of flow; that ventilation is continued throughout the experiment ensures that the oxygen tension is unaffected so that the changes observed reflect the effects of the loss of flow component. ROS production (as monitored by DFF fluorescence) with stopped flow is observed in WT lungs but it is absent in lungs from NOX2−/− mice. This is consistent with our previous reports using other fluorescent probes and methods (31, 34, 50). Lungs from PECAM-1−/− mice showed lower ROS production compared with WT lungs (Fig. 1A). For imaging the DFF fluorescence in the lungs, focusing and image capture were carried out in reference to an independently captured image using an endothelial marker Alexa 594-conjugated AcLDL (data not shown). We attempted to limit the quantitation (of fluorescence intensity of H2DFFDA) to the signal arising from subpleural microvasculature. As is well known, AcLDL also labels certain nonendothelial cell types such as macrophages, leukocytes, etc.; however, in isolated perfused lungs completely cleared of blood this possibility is somewhat low. Image analysis of the intensity of the fluorescence signal with stopped flow showed that the relative increase in ROS in WT lungs was 40% over baseline, while in lungs from PECAM-1−/− mice it was ∼20% over baseline (Fig. 1B). A time course of images was captured in a single region of the lung to allow for measurement of ROS increase in the same area as a function of time.
Fig. 1.

Reactive oxygen species (ROS) in lungs with stop of flow. Imaging time course of DFF fluorescence in subpleural pulmonary microvasculature. A: isolated perfused lungs were labeled with H2DFFDA and stop of flow was initiated at 0 min. In the wild-type (WT) series, an alveolus (alv) and capillary lumen (lum) are indicated with dotted lines and an arrow, respectively. B: quantification of DFF fluorescence in vasculature (nonalveolar) space for 5.5–6.0 min of stop of flow. Data are means ± SE for n = 3–4 lungs in each group. *P < 0.05, compared with WT with stop of flow. NOX2, NADPH oxidase 2; PECAM-1, platelet endothelial cell adhesion molecule-1 (PECAM-1).
To identify the ROS species generated with stopped flow in the lung (since H2DFFDA is sensitive to H2O2 among other oxidants but not to O2·−), we used the superoxide specific dye DHE. First, the specificity of the probe for superoxide was tested using the superoxide generating system (X/XO). Isolated perfused lungs were prelabeled with DHE and treated with X/XO in the presence of catalase; lung extracts were analyzed by HPLC to confirm the probe had sufficient capacity for oxidation. The ratio of the specific superoxide product 2-OHE to the unreacted DHE from the same extract gives an indication of the superoxide levels without artifacts from unequal probe loading or variations in sample tissue size. We observed 2-OHE production post-X/XO addition from the peak; this peak is abolished upon pretreatment of lungs with superoxide dismutase (Fig. 2A). After the specificity of DHE for superoxide was established in our model, lungs from WT and NOX2−/− mice were labeled with DHE and subjected to 1 h of stopped flow; these lungs were then excised and DHE and its oxidation products were extracted. Our results (Fig. 2B) show that superoxide produced with cessation of flow in WT mice is significantly higher than that generated in NOX2−/− lungs exposed to the same conditions. Superoxide production in PECAM-1−/− lungs while significantly diminished compared with WT lungs is higher than that observed in NOX2−/− (a system that generates low superoxide). Sequential analysis of the superoxide-specific product 2-hydroxyethidium (2-OHE) and the unreacted DHE provided an intrinsic normalization of the amount of oxidation, avoiding artifacts from unequal probe loading or variation in tissue sample size. Analysis of the ratio of 2-OHE to DHE for at least three lungs indicated a significantly lower amount of superoxide in PECAM-1−/− lungs (0.22) to that in the WT lungs (0.34; Fig. 2C). The NOX2−/− lungs had the lowest amount of superoxide activity (0.10), which was approximately the same as the signal detected in samples from naïve (untreated) lungs (dashed line). This implies that a small amount of basal superoxide is generated in naïve lungs or that nonspecific fluorescence is detected in all samples.
Fig. 2.
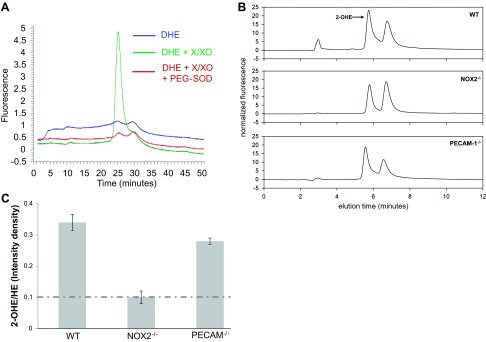
Superoxide produced in lungs with stop of flow [as detected by 2-hydroxyethidium (2-OHE)]. A: elution traces of 2-OHE, produced by xanthine/xanthine oxidase by HPLC. B: sample elution traces of 2-OHE in supernatant from lungs subjected to 1 h stop of flow (HE signal not shown). C: quantification of oxidized dihydroethidium. Dashed line shows 2-OHE in naïve (untreated) lungs. Data are ratio of peak density of 2-OHE over HE. Data are means ± SE for n > 3 lungs. * P < 0.05 compared with WT.
Effect of Loss of PECAM-1 on NOX2 Activation by ANG II (NOX2 Agonist)
We monitored NOX2 activation in lungs in situ in WT and PECAM-1−/− mice by treating lungs with a NOX2 agonist ANG II. NOX2 activation by ANG II (50 μM) was assessed by ROS generation in lungs as evidenced by oxidation of membrane impermeable dye Amplex Red (+HRP) at 15, 30, 45, and 60 min of recirculating perfusion (Fig. 3). Aliquots were removed at different periods of agonist induced NOX2 activation making this a suitable assay for monitoring ROS under continued perfusion. There was no detectable ROS production in the absence of added agonist in the lungs studied (WT, PECAM-1−/−, and NOX2−/−). Upon addition of ANG II to the perfusate, there was a linear increase in oxidized Amplex Red that was 8- to 10-fold greater than the basal rate indicating a constant rate of H2O2 production. There was no significant difference in ANG II-induced ROS production by PECAM-1−/− lungs; however, the ANG II-stimulated rate of H2O2 production was markedly diminished in the NOX2−/− lungs (as compared with WT) although it was slightly greater than untreated WT lungs. These data suggest that the NOX2 activation machinery is intact in PECAM-1−/− lungs.
Fig. 3.

PECAM-1−/− lungs show NOX2 activation upon agonist treatment. Lungs from WT and PECAM-1−/− mice were isolated and perfused in a recirculating system containing Amplex Red (25 μM) and horseradish peroxidase (25 μg/ml). ROS generation was measured in arbitrary fluorescence units (AFU) by Amplex Red oxidation at 15, 30, and 60 min of perfusion. ANG II when present was 50 μM. Numbers in parenthesis indicate the slope (change in AFU/min) calculated by least mean squares. Data are means ± SE from n = 3 lungs. *P < 0.05 vs. the corresponding control (no ANG II).
Association of PECAM-1 with Caveolin-1
Colocalization.
We have previously reported that caveolin-1 (the major coat protein of caveolae) plays a role in signaling with stop of flow; we observed that lungs from caveolin-1−/− mice showed significantly lower ROS production compared with WT (33, 45). To investigate whether caveolin-1 is involved in PECAM-1 signaling with stop of flow, we examined the location of PECAM-1 on plasma membrane. We checked the distribution of PECAM-1 by dual labeling for PECAM-1 and caveolin-1 by immunofluorescence of fixed lung sections as well as by monitoring PECAM-1 expression in caveolin-rich cell membrane fractions. Visual inspection of the colocalized signal (yellow channel) reveals regions of strong overlap separated by areas of little apparent or no overlap (Fig. 4, A–C). We carried out colocalization analysis by expressing the fraction of localization in each component of a dual color image by various colocalization coefficients. Pearson's coefficient, a standard pattern recognition for matching one image with another, measures the degree of overlap between two patterns independent of the intensity of the signal. This involves subtracting the average values from the original values. Pearson's coefficient indicates a 57% colocalization. The overlap coefficient, also independent of signal intensity caused by differential labeling of the fluorophores or photobleaching or different setting of the amplifiers, shows a 70% correlation between PECAM-1 and caveolin-1. The Mander's coefficients are proportional to the amount of fluorescence of the colocalizing objects in each component of the image relative to the total fluorescence in that component. The Mander's coefficients indicate that 71% of the PECAM-1 was colocalized with the caveolin-1, while 87% of the caveolin-1 was colocalized with the PECAM-1. Overall, the colocalization analysis shows a statistically significant correlation in the signals from caveolin-1 and PECAM-1 (Fig. 4D).
Fig. 4.

PECAM-1 associates with caveolin-1. A: immunofluorescent labeling of PECAM-1 (red) and caveolin-1 (green) in lung tissue sections from WT mice. Scale bar = 50 μm. B: nonimmune IgG control. C: magnification of boxed regions of A. Scale bar = 10 μm. D: quantitative colocalization analysis of PECAM-1 and caveolin-1 for Mander's coefficients (M1 for fraction of PECAM-1 colocalizing with caveolin-1, and M2 for fraction of caveolin-1 colocalizing with PECAM-1), overlap coefficient (O.C.), and Pearson's coefficient (P.C.). Data are presented as means ± SE of 10–15 images from n = 3–4 lungs. E: Duolink to evaluate association of PECAM with caveolin-1 in lung sections. Red fluorescent dots indicate interaction i.e., proximity (<40 nm) of the 2 proteins. Green indicates endothelial cells immunostained for VE-cadherin (by anti-VE-cadherin-FITC) and blue is the nuclear stain (DAPI). The lung section shows part of a pulmonary vessel; bottom: magnification of the boxed region. Scale bar = 10 μm. For all experiments, Duolink controls used to check nonspecific signal showed no fluorescence (data not shown). F: flow-adapted cells subjected to stop of flow (SF) and statically cultured cells [WT and caveolin-1−/− pulmonary microvascular endothelial cells (PMVEC)] (S) and PECAM-1−/− cells were immunoblotted for phospho-PECAM using an Ab that detects Tyr 713-PECAM-1. Top: phospho-PECAM band at approximately 130–140 kDa. Bottom: β-actin band to show equal protein loading across lanes. G: quantitation of immunoblots by using ImageJ software. Lanes were outlined and the intensity of pixels in the entire lane obtained. Within each lane, the software was used for quantification of the intensity of the band at ∼130 kDa. Data are means ± SE from 3 separate blots (experiments). *P < 0.001 compared with all other conditions.
Next, we used the Duolink procedure to evaluate the interaction of caveolin-1 and PECAM-1. For the lung sections, we chose part of a pulmonary vessel. We counterstained the lung sections for endothelial cells using anti-VE-cadherin (green) and nuclei using nuclear marker DAPI (blue) to observe cells/endothelial cells in each field (Fig. 4E). Magnification of the image shows that along the pulmonary vessel red fluorescent dots appear in cells that have green fluorescence (Fig. 4E). Each dot represents a single molecular interaction between the two proteins PECAM-1 and caveolin. For each experiment, the nonspecific Duolink signal was monitored by controls using anti-PECAM-1/rabbit IgG, anti-caveolin-1/mouse IgG, and anti-PECAM-IgG1/anti-caveolin. These showed no fluorescence dots thus ruling out any nonspecific interactions (data not shown).
The functional nteraction of PECAM-1 and caveolin-1 was evaluated by assessing phosphorylation of PECAM-1 in the presence and absence of caveolin-1, using an antibody against a peptide derived from PECAM-1. With this antibody, phosphorylated PECAM-1 (when phosphorylated at tyrosine713) is detected as a band at ∼130 kDa. With mechanical stimuli, PECAM-1 has been reported to be phosphorylated (at Tyr 713) (9, 39). We observed a band at approximately 130–140kDa in WT cells that was not observed in PECAM-1−/− cell lysate. There was increase in phospho-PECAM in flow-adapted PMVEC subjected to stop of flow compared with static cells (Fig. 4F). This band was very weak in cells that lacked caveolin-1 (Fig. 4, F and G). Thus caveolin-1 (and caveolae) are required for the functional activation of PECAM-1.
To check of PECAM-1 was resident in caveolin-1-rich light density membrane fractions, we assessed PECAM-1 expression in plasma membrane fractions of PMVEC (Fig. 5A). Immunoblotting these membrane fractions show enrichment for caveolin-1 in fractions 5–8, consistent with previous reports of the distribution of caveolin-1 in endothelium (17); PECAM-1 expression was observed only in caveolin-1-rich fractions. The heavier fractions contain membranes in varying amounts as seen by the expression of plasma membrane marker flotillin. Next, we checked PECAM expression in caveolar vesicles isolated from plasma membranes of mouse PMVEC. Caveolae showed high PECAM-1 levels compared with noncaveolar membranes (i.e., membrane that is not bound to the caveolin-1-coated magnetic beads), which showed low to no PECAM expression (Fig. 5B). In our experiments, noncaveolar fraction showed a caveolin-1 signal, as larger vesicles with caveolin-1 are often not accessible to the immunoaffinity pull down assay and thus remain unbound to the magnetic beads. Because of this, there is also a faint PECAM-1 band in the noncaveolar fraction.
Fig. 5.

PECAM-1 is enriched in caveolar fractions. A: plasma membrane from mouse PMVEC was isolated from post nuclear homogenate using a Percoll gradient shows PECAM-1 expression in the caveolin-1-enriched fractions (fractions 5–8). All lanes were immunoblotted with membrane marker flotillin. B: caveolae obtained from plasma membranes of mouse PMVEC after Percoll gradient centrifugation and sonication. Intact caveolae vesicles were separated from other membrane domains (noncaveolar) by immunoaffinity methodology using magnetic beads. Noncaveolar fraction shows a caveolin-1 signal as larger vesicles with caveolin-1 are often not pulled down by this immune-assay and remain unbound to the magnetic beads. Proteins from each fraction were separated by SDS-PAGE, transferred to nitrocellulose membranes and immunoblotted using anti-PECAM and caveolin-1 antibodies.
Effect of Loss of PECAM-1 on Caveolin-1 Expression
Since PECAM-1 and caveolin-1 were functionally associated, could lack of PECAM-1 affect caveolin-1 expression and caveolae? Our investigations revealed that there were no differences in WT or PECAM-1−/− mice in expression of caveolin-1 in mouse lung homogenates (Fig. 6, A and B). Ultrathin thawed cryosections were fixed, cryoprotected, and immunolabeled for caveolin-1. Transmission electron microscopy images showed caveolae as flask-shaped profiles located beneath the plasma membrane. The mean density of caveolae in WT lungs was not significantly different from PECAM-1−/− lungs. Thus the lack of PECAM-1 did not alter the number of caveolae or the expression of caveolin-1 in mouse lung endothelium (Fig. 6, C and D).
Fig. 6.
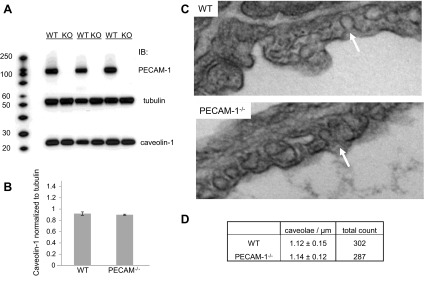
Lack of PECAM-1 does not affect expression of caveolin-1 and number of caveolae. A: immunoblot (IB) for caveolin-1 in lung homogenate from WT and PECAM-1−/− mice. B: densitometry analysis of immunoblot data from A normalized to tubulin. C: transmission electron microscopy (TEM) images of caveolae in pulmonary capillaries of WT and PECAM-1−/− mice. Arrows indicate representative caveolae from the capillary lumen. D: quantification of caveolae density from 5–10 TEM images (caveolae number per μm of endothelium) per lung. Data are expressed as means ± SE from n = 3 lungs per genotype.
Mechanosignaling via PECAM-1 Correlates with Neovascularization In Situ
To understand the functional role for ROS produced with stop of flow, we assessed the angiogenic potential of cells subjected to stop of flow (72 h under flow followed by stop of flow). Cells were mixed in Matrigel and were injected as subcutaneous plugs into nude mice. Five days later plugs containing flow-adapted cells subjected to stop of flow or statically cultured cells were excised and treated with the endothelial marker Alexa594-AcLDL. Visible vessel formation was observed that was significantly greater in flow-adapted WT compared with PECAM-1 −/− cells cultured under similar conditions (Fig. 7, A and B). Cryosections of the plugs showed staining with anti-VE cadherin indicating endothelium within the plugs (Fig. 7, C and D). Statically cultured cells from both WT and PECAM null lungs showed low neovascularization.
Fig. 7.
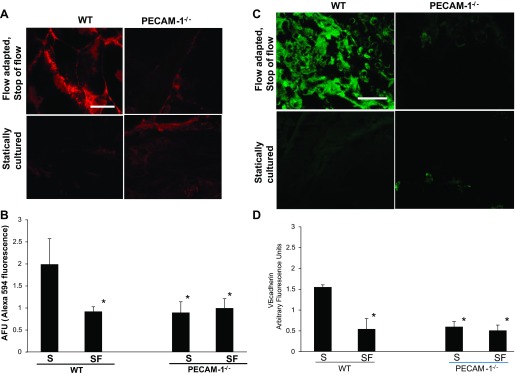
PECAM-1 participates in neovascularization within subcutaneous Matrigel plugs. Matrigel mixed with cells (flow-adapted cells subjected to stop of flow or statically cultured) derived from wild type or PECAM-1−/− lungs were injected as a plug subcutaneously into the left and right flank of nude mice. The plugs were removed 5 days later. A: section of the plugs was incubated in Alexa594-acetylated low-density lipoprotein (AcLDL) and imaged by confocal microscopy, Scale bar = 20 μm. B: quantitation of the Alexa594 fluorescence from 3–5 fields for each plug. Data are means ± SD of 3–4 independent experiments. C: rest of the plug was fixed in 4% paraformaldehyde, sectioned and stained for VE-cadherin (using anti-VE cadherin) to label endothelial cells. Scale bar = 50 μm. D: quantitation of VE-cadherin from the arbitrary fluorescence intensity of 5–8 sections from each plug. Three fields from each section were imaged. Data are means ± SE of 3–4 independent experiments. *P < 0.05, as compared WT cells that were flow adapted and subjected to stop of flow.
Lack of PECAM-1 and Neutrophil Recruitment and VEGF Expression with Stopped Flow in the Lung In Vivo
What are the consequences of mechanosignaling with stop of flow in the lung? To investigate this, we occluded lung vessels in vivo. This, of course, is not comparable to stop of flow in vitro or in situ and is closer to a microemboli model; nevertheless, it represents obstructed flow in an in vivo setting. We injected 25-μm beads into the lungs of WT, PECAM-1−/−, and NOX2−/− mice; 24 h later the mice were killed and the lungs were fixed and sectioned. Hematoxylin and eosin staining showed that that the infiltration of polymorphonuclear neutrophils that occurs 24 h after obstruction of blood flow in WT mice is significantly compromised in the null mice (Fig. 8). The polymorphonuclear neutrophils were clustered in regions around the beads (Fig. 8). We also checked VEGF expression in WT lungs at 1–7 days poststop of blood flow. At day 4 poststopped flow, VEGF expression increased significantly compared with untreated lungs; this decreased by day 7 (data not shown). We thus monitored VEGF expression at day 4 across the three types of lungs, i.e., WT, PECAM-1−/−, and NOX2−/−. Lack of PECAM-1 and NOX2 (and thus mechanosignaling-induced ROS production) abolished the increase in VEGF expression that was observed in WT lungs pointing to a role for PECAM-1 induced NOX2 activation in triggering an angiogenic signal with obstruction of blood flow (Fig. 9, A and B).
Fig. 8.

PECAM-1 signaling participates in neutrophil recruitment postocclusion in lungs in vivo; 25-μm microspheres were injected into the jugular vein of WT, PECAM-1−/−, and NOX2−/− mice. Mice were killed 24 h later, and lungs were sectioned and stained by hematoxylin and eosin. Images were acquired under ×10 and ×60 objectives. Bottom: magnification of boxed region.
Fig. 9.

Expression of vascular endothelial growth factor (VEGF) in the lung is PECAM-1 and NOX2 dependent. A: 25-μm polystyrene beads (green fluorescent) were injected into the mice; mice were killed 4 days later, sectioned and stained for VEGF. B: quantitation of the fluorescence intensity from the microscopy images, using 8–10 fields per lung. Data are means ± SE from n = 3 independent experiments. Unt, untreated lungs; WT (IgG), IgG controls. *P < 0.05, compared with knockouts.
DISCUSSION
Mechanosignaling associated with blood flow has hitherto involved studying cellular responses with onset of shear or start of flow. In a departure from this paradigm, we investigated shear from the point of removal of shear on endothelial cells, a condition that has physiological similarity with vascular obstruction. In general, stop of flow involves 1) compromised oxygen delivery to tissue, and 2) alteration of the mechanical component of blood flow. We established models where stop of flow did not compromise oxygen delivery and nutrient supply; using these models we observed that abrupt cessation of flow causes ROS production via activation of endothelial cell NADPH oxidase (34, 35, 50). We discovered elements of the cascade, viz. K+ channel depolarization and phosphatidylinositol 3-kinase/AKT activation that participated in NOX2 assembly (6, 50). However, the mechanosensing elements on the endothelium that “sensed” the changes in tension on the membrane with stop of flow remained unknown.
Here we investigate the initial element(s) that participate in sensing shear in the pulmonary endothelium. Several reports show a role for PECAM-1 in sensing physical force (9, 39, 42). By virtue of its localization at cell-cell junctions and due to its cytoskeletal linkage (downstream of PECAM-1 expression is the αvβ3-integrin signal), PECAM-1 on the pulmonary endothelium can be hypothesized to be part of a sensory complex that senses stop of flow.
In this study, mechanosensing and the attendant signaling were monitored via ROS production; we were thus judicious in the selection of dyes to be used. Instead of the more extensively used chlorinated fluorescein derivatives such as H2DCFDA (which are notorious for photo enhancement of the fluorescence signal), we used the fluoro compound H2DFFDA. This fluorinated derivative exhibits improved photostability, and the carboxy group in H2DFFDA improves cell retention. As H2DFFDA oxidation occurs predominantly via H2O2, we used in separate experiments, a second dye DHE, to identify if superoxide was produced with stop of flow. DHE is reported to react with superoxide to form 2-OHE. It also forms other products, but 2-OHE is the superoxide specific adduct. As 2-OHE had to be obtained from the lung extract by HPLC, its separation from other DHE products and subsequent detection necessitated exposing the lungs to longer periods of stopped flow (∼1 h) to enable greater 2-OHE accumulation in lungs. In contrast, confocal imaging of DFF in intact lungs could detect small changes in fluorescence due to the sensitivity of microscopy. In addition, visualization was aided by pseudocolor and thus DFF fluorescence could be seen within 1–5 min of stopped flow. Despite differences in the period of stopped flow in the lungs, both these dyes showed that lack of PECAM-1 reduced the response to stop of flow (in terms of ROS production) by the pulmonary endothelium. Lungs from mice with knockout of PECAM-1 produced ROS that was intermediate between WT and NOX2−/− lungs. This indicates that PECAM-1 is possibly part of a complex or part of a family of sensors that participates in signaling with loss of flow and that its loss abrogates but does not completely abolish the signaling with stop of flow. This signaling involves the eventual activation of NOX2; thus lack of NOX2 causes very low to no ROS production as we have shown here and reported earlier (6, 50).
NOX2 activation involves assembly of its cytosolic and membrane components; incomplete assembly can alter ROS production. We thus checked NOX2 activation in WT and PECAM-1−/− lungs treated with ANG II. For this we used another H2O2-sensitive dye Amplex Red that differs from DFF in that it monitors ROS (H2O2) production only in the vessel lumen (as it a membrane impermeable dye). The advantage of this assay is that NOX2 activation (and ROS) can be assessed over multiple time points from the same lung by removing aliquots of the perfusate at different time points postagonist stimulation (7). This assay is suitable for studying the total H2O2 produced in whole lungs under continuous perfusate flow but not under stop of flow. For stop of flow, the pump would have to be turned off; perfusate retrieval would necessitate restart of flow (reperfusion), thus introducing the confounding effects of reperfusion induced ROS into the measurement. We observed that ANG II-treated PECAM-1−/− lungs showed similar ANG II-induced NOX2 activation (ROS production) comparable with WT lungs. Thus NOX2 activation machinery is intact in PECAM-1−/− lungs. The diminution of ROS in PECAM-1 null lungs as observed in Figs. 1 and 2 is thus specific to stop of flow induced ROS production and is not a generalized negative effect on NOX2 activation in PECAM-1−/− mice.
Our earlier studies on stop of flow in lungs in situ had shown a role for caveolin-1 in mechanosignaling (33, 45). Caveolae are flask-like invaginations of the plasma membrane; these invaginations are lipid-rich cellular domains that concentrate plasma membrane proteins and lipids. Caveolae serve as signaling platforms for receptors, kinases, and several proteins all of which recognize external stimuli and transduce signals to modulate cellular activity. Among the proteins reportedly located within caveolae are NOX proteins and endothelial nitric oxide synthase (16, 18, 21, 53). Our colocalization studies indicate that most of cellular PECAM-1 was located in caveolae. This was also reported in mouse lung development studies where patterns of caveolin-1 expression overlapped with PECAM-1 expression (40). How does caveolin-1 status affect mechanosignaling that occurs via PECAM-1? We reported earlier that mice with knockout of caveolin-1 (i.e., lacking caveolin-1 and thus caveolae) showed a complete abolition of ROS with stop of flow in lungs in situ (33); however, PECAM-1−/− lungs show a diminution in ROS production. Thus lack of caveolae (and therefore a presumable loss of a signaling scaffold or platform comprising PECAM-1 and NOX2) completely prevented mechanosignaling (33), while lack of PECAM-1 merely reduced it. We also observed phosphorylation of PECAM-1 in (flow adapted) WT cells upon stop of flow. This did not occur in caveolin-1 null cells, indicating the requirement of caveolin-1 (and caveolae) in PECAM-1 activation. Studies elsewhere have also shown that PECAM-1 and caveolin-1 are functionally linked in several signaling processes including those participating in vasculogenesis (40).
Does loss of PECAM-1 affect caveolin-1 expression and caveolar number? In other words, does the diminution in mechanosignaling in PECAM-1−/− arise from altered caveolar expression? Our investigations revealed that caveolin-1 expression or numbers of caveolae are not significantly reduced in PECAM-1−/− lungs compared with WT, indicating that PECAM-1 (as part of the caveolar complex) functions as a mechanosensing platform that initiates signaling with loss of shear. Of course, PECAM-1 is known to be distributed heavily at cell-cell contacts while caveolae are found both at the luminal surface and cell borders. Presumably, the subset of PECAM-1 (and caveolae) that is on the luminal surface would play a major role in shear sensing compared with that at the cell contacts.
ROS generated with stop of flow in vitro has been reported by us to drive cell proliferation (33–35). To understand if this cell proliferation is random or leads to formation of new vessels, we investigated the angiogenic potential of flow-adapted cells subjected to stop of flow. The subcutaneous Matrigel plug model is a standard assay for detecting angiogenesis in situ. Matrigel matrix is used as this is similar to the natural environment of a basement membrane for endothelial cells and vascularization in the plugs (the plug itself is growth factor free) is caused by angiogenic signals emanating from inside the plug (3a). We have reported earlier that when WT pulmonary endothelial cells subjected to flow adaptation and stop of flow are mixed with Matrigel and injected into the flanks of nude mice, vascularization within the plug was observed after 5 days. This was not observed with cells that did not generate ROS with stopped flow, i.e., NOX2−/− cells (3a). This indicates that angiogenic signals emanating from the endothelial cells in the plug recruit endothelial cells from the host animal and form blood vessels within the plug (3a). Thus lack of vascularization indicated that the endothelial cells within the plug have low to no angiogenic potential. Vascularization within the plugs was assessed by staining pieces of the plug or selected regions of histological sections for vascular markers. We observed that flow-adapted WT cells subjected to stop of flow showed greater angiogenic potential compared with the statically cultured cells and to PECAM-1−/− cells (both flow adapted and static). In fact plugs with PECAM-1−/− cells did not shown neovessel structures within the plug as WT did, although they did show some staining for endothelial markers. Overall the Matrigel plug studies indicate that ROS generated with stop of flow modulates an angiogenic response.
Our data indicate that PECAM-1-induced mechanotransduction participates in vascularization in pulmonary endothelial cells; however, pulmonary obstruction in vivo (attained by ligation of the pulmonary artery) has been reported to lead to ROS-dependent systemic angiogenesis, with no vascularization within the pulmonary vasculature (36, 44). We reasoned that this was not due to the lack of an angiogenic signal in the pulmonary endothelial cells per se (as our data showed increased vascularization in plugs that was PECAM-1 dependent) but due to the unresponsiveness of the pulmonary artery. The pulmonary artery is not proangiogenic, except in rare cases where new vessels develop as a secondary source of perfusion in some lung carcinomas (32). We thus sought to achieve in vivo occlusion of smaller vessels by injecting 25-μm polystyrene beads via jugular vein. Hematoxylin and eosin staining of lung sections showed neutrophil accumulation in lungs of WT mice; that neutrophils were significantly lower in NOX2−/− pointed to a role for NOX2 activation in driving neutrophil influx. Low neutrophil infiltration in PECAM-1−/− lungs implies a role for PECAM mechanosignaling in neutrophil recruitment into the lungs. However, PECAM-1 is also a multifunctional protein that serves as an adhesion molecule to regulate leukocyte trafficking. Indeed a role for PECAM-1 in neutrophil recruitment with ischemic insult and other injury models has been reported earlier (3, 24, 43). Blockade of PECAM-1 inhibited neutrophil transmigration and significantly reduced necrosis in a feline model of myocardial ischemia reperfusion (38). However, these studies focused on transmigration of the available neutrophils, whereas our interest was to examine PECAM-1-induced signaling in recruiting neutrophils. Homing of neutrophils is a very complex process and is reported to occur via a signaling cascade in which adhesion molecules, chemokines, and chemoattractants are expressed in a spatially and temporally controlled manner (2, 15, 41). Our data suggest a link between endothelial PECAM-1 and neutrophil recruitment: the lack of neutrophils in NOX2−/− mice point to a role for ROS (and mechanosignaling), although the trafficking effect of PECAM-1 possibly plays a role as well.
Is there an angiogenic signal postbead-induced stop of flow in the pulmonary vasculature in vivo? Also, what is the initiating signal for vascularization with the removal of flow stimulus? VEGF is well established to regulate angiogenesis; indeed, it is one of the most potent angiogenic factors identified so far that stimulates endothelial cell proliferation, migration, and tube formation. Our inspection of lungs postocclusion at 1, 4, and 7 days after bead insertion, revealed an increase in VEGF at day 4 in WT, which was reduced to the levels of untreated lungs by day 7. In both PECAM-1−/− and NOX2−/− lungs, there was no increase in VEGF, indicating that PECAM-1 and NOX2 mediate VEGF production.
Taken together our data indicate that PECAM-caveolae mechanosensing machinery on the endothelium is able to sense the changes in membrane tension with stop of flow; this triggers a signaling cascade that leads to ROS production. ROS propagates signaling that participate in neutrophil recruitment and angiogenic processes in the region of obstructed flow. We posit that these signals are for vascularization to restore the impeded blood flow.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-075587 and NIH-T32-HL-07748-17 and a McCabe Foundation Award.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.N., H.W., N.H., J.-Q.T., K.J.Y., E.M.S., K.M.D., and M.H. performed experiments; J.N. and S.C. analyzed data; J.N., V.R., H.M.D., and S.C. interpreted results of experiments; J.N., H.W., and S.C. prepared figures; J.N. and S.C. drafted manuscript; V.R., H.M.D., A.B.F., and S.C. approved final version of manuscript; A.B.F. and S.C. conception and design of research; S.C. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Chandra Dodia for technical support and acknowledge helpful comments from Dr. Steven Albelda, Department of Medicine, University of Pennsylvania Perelman School of Medicine.
This work was presented in part at the 2011 Experimental Biology meeting in Washington D.C, 2012 Experimental Biology meeting in San Diego, CA.
REFERENCES
- 1.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res 83: 730–737, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 46: 566–572, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J Exp Med 179: 1059–1064, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Browning E, Wang H, Hong N, Yu K, Buerk DG, DeBolt K, Gonder D, Sorokina EM, Patel P, De Leon DD, Feinstein SI, Fisher AB, Chatterjee S. Mechanotransduction drives post ischemic revascularization through KATP channel closure and production of reactive oxygen species. Antioxid Redox Signal 2013. July 31 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlile-Klusacek M, Rizzo V. Endothelial cytoskeletal reorganization in response to PAR1 stimulation is mediated by membrane rafts but not caveolae. Am J Physiol Heart Circ Physiol 293: H366–H375, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol 285: C959–C967, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol 302: H105–H114, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, Fisher AB. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem 286: 11696–11706, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 13: 633–644, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Chiu YJ, McBeath E, Fujiwara K. Mechanotransduction in an extracted cell model: Fyn drives stretch- and flow-elicited PECAM-1 phosphorylation. J Cell Biol 182: 753–763, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res 101: 723–733, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Dong QG, Bernasconi S, Lostaglio S, De Calmanovici RW, Martin-Padura I, Breviario F, Garlanda C, Ramponi S, Mantovani A, Vecchi A. A general strategy for isolation of endothelial cells from murine tissues. Characterization of two endothelial cell lines from the murine lung and subcutaneous sponge implants. Arterioscler Thromb Vasc Biol 17: 1599–1604, 1997 [DOI] [PubMed] [Google Scholar]
- 12.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol 12: 308–319, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan GS, Andrew DP, Takimoto H, Kaufman SA, Yoshida H, Spellberg J, de la Pompa JL, Elia A, Wakeham A, Karan-Tamir B, Muller WA, Senaldi G, Zukowski MM, Mak TW. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol 162: 3022–3030, 1999 [PubMed] [Google Scholar]
- 14.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–C742, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan J. TLR cross-talk mechanism of hemorrhagic shock-primed pulmonary neutrophil infiltration. Open Crit Care Med J 2: 1–8, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García-Cardeña G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA 93: 6448–6453, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gosens R, Stelmack GL, Dueck G, Mutawe MM, Hinton M, McNeill KD, Paulson A, Dakshinamurti S, Gerthoffer WT, Thliveris JA, Unruh H, Zaagsma J, Halayko AJ. Caveolae facilitate muscarinic receptor-mediated intracellular Ca2+ mobilization and contraction in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 293: L1406–L1418, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 677–683, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Hove JR, Köster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421: 172–177, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J 20: 811–827, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Insel PA, Patel HH. Membrane rafts and caveolae in cardiovascular signaling. Curr Opin Nephrol Hypertens 18: 50–56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10: 63–73, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem 279: 12001–12004, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Korthuis RJ, Gute DC. Adhesion molecule expression in postischemic microvascular dysfunction: activity of a micronized purified flavonoid fraction. J Vasc Res 36, Suppl 1: 15–23, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Krump-Konvalinkova V, Bittinger F, Unger RE, Peters K, Lehr HA, Kirkpatrick CJ. Generation of human pulmonary microvascular endothelial cell lines. Lab Invest 81: 1717–1727, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Lee I, Dodia C, Chatterjee S, Zagorski J, Mesaros C, Blair IA, Feinstein SI, Jain M, Fisher AB. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J Pharmacol Exp Ther 345: 284–296, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucitti JL, Jones EA, Huang C, Chen J, Fraser SE, Dickinson ME. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134: 3317–3326, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103: 857–862, 1992 [DOI] [PubMed] [Google Scholar]
- 29.Manders EM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169: 375–382, 1993 [DOI] [PubMed] [Google Scholar]
- 30.Manevich Y, Al-Mehdi A, Muzykantov V, Fisher AB. Oxidative burst and NO generation as initial response to ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol 280: H2126–H2135, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Matsuzaki I, Chatterjee S, Debolt K, Manevich Y, Zhang Q, Fisher AB. Membrane depolarization and NADPH oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol 288: H336–H343, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Milne EN, Zerhouni EA. Blood supply of pulmonary metastases. J Thorac Imaging 2: 15–23, 1987 [DOI] [PubMed] [Google Scholar]
- 33.Milovanova T, Chatterjee S, Hawkins BJ, Hong N, Sorokina EM, Debolt K, Moore JS, Madesh M, Fisher AB. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta 1783: 1866–1875, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milovanova T, Chatterjee S, Manevich Y, Kotelnikova I, Debolt K, Madesh M, Moore JS, Fisher AB. Lung endothelial cell proliferation with decreased shear stress is mediated by reactive oxygen species. Am J Physiol Cell Physiol 290: C66–C76, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Milovanova T, Manevich Y, Haddad A, Chatterjee S, Moore JS, Fisher AB. Endothelial cell proliferation associated with abrupt reduction in shear stress is dependent on reactive oxygen species. Antioxid Redox Signal 6: 245–258, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Mitzner W, Lee W, Georgakopoulos D, Wagner E. Angiogenesis in the mouse lung. Am J Pathol 157: 93–101, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monshausen GB, Gilroy S. Feeling green: mechanosensing in plants. Trends Cell Biol 19: 228–235, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Murohara T, Delyani JA, Albelda SM, Lefer AM. Blockade of platelet endothelial cell adhesion molecule-1 protects against myocardial ischemia and reperfusion injury in cats. J Immunol 156: 3550–3557, 1996 [PubMed] [Google Scholar]
- 38a.Nakada MT, Amin K, Christofidou-Solomidou M, O'Brien CD, Sun J, Gurubhagavatula I, Heavner GA, Taylor AH, Paddock C, Sun QH, Zehnder JL, Newman PJ, Albelda SM, DeLisser HM. Antibodies against the first Ig-like domain of human platelet endothelial cell adhesion molecule-1 (PECAM-1) that inhibit PECAM-1-dependent homophilic adhesion block in vivo neutrophil recruitment. J Immunol 164: 452–462, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Osawa M, Masuda M, Harada N, Lopes RB, Fujiwara K. Tyrosine phosphorylation of platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31) in mechanically stimulated vascular endothelial cells. Eur J Cell Biol 72: 229–237, 1997 [PubMed] [Google Scholar]
- 40.Ramirez MI, Pollack L, Millien G, Cao YX, Hinds A, Williams MC. The alpha-isoform of caveolin-1 is a marker of vasculogenesis in early lung development. J Histochem Cytochem 50: 33–42, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome - how neutrophils migrate into the lung. Crit Care 8: 453–461, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41a.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Recruitment of endothelial caveolae into mechanotransduction pathways by flow-conditioning in vitro. Am J Physiol Heart Circ Physiol 285: H1720–H1729, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Vaporciyan AA, DeLisser HM, Yan HC, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science 262: 1580–1582, 1993 [DOI] [PubMed] [Google Scholar]
- 44.Wagner EM, Petrache I, Schofield B, Mitzner W. Pulmonary ischemia induces lung remodeling and angiogenesis. J Appl Physiol 100: 587–593, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Wei Z, Al-Mehdi AB, Fisher AB. Signaling pathway for nitric oxide generation with simulated ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol 281: H2226–H2232, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Wei Z, Costa K, Al-Mehdi AB, Dodia C, Muzykantov V, Fisher AB. Simulated ischemia in flow-adapted endothelial cells leads to generation of reactive oxygen species and cell signaling. Circ Res 85: 682–689, 1999 [DOI] [PubMed] [Google Scholar]
- 47.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol 187: 4809–4817, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiang M, Yin L, Li Y, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock activates lung endothelial reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase via neutrophil NADPH oxidase. Am J Respir Cell Mol Biol 44: 333–340, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q, Chatterjee S, Wei Z, Liu WD, Fisher AB. Rac and PI3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxid Redox Signal 10: 679–689, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am J Physiol Lung Cell Mol Physiol 289: L954–L961, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vásquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med 34: 1359–1368, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc 3: 8–21, 2008 [DOI] [PubMed] [Google Scholar]
- 53.Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol 25: 1824–1830, 2005 [DOI] [PubMed] [Google Scholar]