

Abstract
Although a reduction in the high-frequency (HF) component of heart rate variability (HRV) is a major complication of diabetes and a risk factor for sudden death, its relationship to ventricular tachycardia (VT) is unknown. We developed a mouse model for the study of VT and its relationship to changes in HRV in the Akita type 1 diabetic mouse. Programmed ventricular stimulation of anesthetized mice demonstrated that Akita mice were more inducible for VT compared with wild-type mice: 78.6% versus 28.6% (P = 0.007). Optical mapping of perfused hearts demonstrated multifocal breakthroughs that occasionally gave rise to short-lived rotors consistent with focal initiation and maintenance of VT. Treatment of Akita mice with pravastatin, which had been previously shown clinically to decrease ventricular ectopy and to increase HRV, decreased the inducibility of VT: 36.8% compared with 75.0% with placebo treatment (P = 0.022). The HF fraction of HRV was reduced in Akita mice (48.6 ± 5.2% vs. 70.9 ± 4.8% in wild-type mice, P = 0.005) and was increased compared with placebo treatment in pravastatin-treated mice. Pretreatment of Akita mice with the muscarinic agonist carbamylcholine or the β-adrenergic receptor blocker propranolol decreased the inducibility of VT (P = 0.001). In conclusion, the increased inducibility of focally initiated VT and reduced HF fraction in Akita mice were partially reversed by both pravastatin treatment and pharmacologic reversal of parasympathetic dysfunction. In this new animal model for the study of the pathogenesis of VT in type 1 diabetes, pravastatin may play a role in the prevention of VT by attenuating parasympathetic dysfunction.
Keywords: type 1 diabetes, ventricular tachycardia, statins, secondary effects of diabetes
although diabetes mellitus is associated with an increase in cardiovascular mortality and sudden death (11), its relationship to ventricular tachycardia (VT) is not known. Risk factors for sudden death include clinical manifestations of parasympathetic dysfunction, such as a decreased high-frequency (HF), predominantly parasympathetic, component of heart rate (HR) variability (HRV) and increased dispersion of QT intervals (25, 27). Furthermore, analysis of HRV in a group of patients with a history of nonsustained VT demonstrated a reduction in parasympathetic activity in conjunction with an increase in sympathetic activity before the onset of nonsustained VT, further supporting the relationship between decreased parasympathetic tone and the development of arrhythmia (21). Patients with diabetes for 10 yr have an impaired response of the heart to parasympathetic stimulation characterized by a reduction in the HF component of HRV. The increase in the incidence of sudden death in diabetics has been associated, at least in part, with a decrease in parasympathetic responsiveness of the heart (1, 5). Finally, a study (32) of type 1 diabetics who died suddenly in their sleep (“dead in bed syndrome”) suggested that HRV analysis of diabetic patients who lacked clinical evidence of autonomic neuropathy often demonstrated decreased parasympathetic tone. Conversely, a study (26) in a canine model for myocardial infarction and arrhythmias suggested that parasympathetic stimulation was protective against ventricular arrhythmias.
The inducibility of VT in response to programmed ventricular stimulation has been associated with a predisposition to ventricular arrhythmias. The Multicenter Unsustained Tachycardia Trial was designed to evaluate the use of programmed ventricular stimulation to determine the efficacy of antiarrhythmic drug therapy in reducing the incidence of sudden death from arrhythmia in patients with coronary disease. This study (7) concluded that patients studied at least 6 wk after myocardial infarction, in whom sustained ventricular arrhythmias could not be induced, had a significantly lower risk of sudden death or cardiac arrest. In mice, Maguire et al. (19) used programmed ventricular stimulation using single, double, and triple extrastimulation techniques and burst pacing to stimulate VT. Thirty percent of mice studied were induced to VT with a mean duration of 1.6 s and a maximum of 24 s. The inducibility differed between strains and age and correlated best with a ventricular effective refractory period (VERP) of 44 ± 12 ms in inducible mice and 61 ± 16 ms in noninducible mice (19). Bursts of polymorphic VT have been demonstrated after programmed ventricular stimulation in a mouse model for hypertrophic cardiomyopathy in either the presence or absence of isoproterenol (4). A number of studies in mouse and rat models of chemically induced diabetes or diabetic db/db mice have demonstrated that rosiglitazone might play a therapeutic role in modifying infarct size and postreperfusion arrhythmias after ischemia and reperfusion (22), most often in Langendorf-perfused hearts (33). However, there is currently no established mouse model for the study of arrhythmogenesis as a complication of diabetes.
The Akita mouse is characterized by a point mutation in the proinsulin gene that interferes with insulin processing. This mouse demonstrates markedly decreased serum insulin levels, hypoplastic islets, islet cell destruction, and decreased pancreatic β-cells (17). The heterozygous Akita mice used in this study survive for up to a year and develop secondary effects of diabetes. We (23) have previously demonstrated that Akita mice exhibit marked parasympathetic dysfunction as determined by a blunting of the negative chronotropic response to the acetylcholine analog carbamylcholine. However, unlike HRV, the clinical significance of a decreased HR response to parasympathetic stimulation is less well established.
Here, we used programmed ventricular stimulation of the Akita mouse to determine whether type 1 diabetes was associated with a predisposition to VT. We used optical mapping of Langendorf-perfused hearts to determine the mechanism of initiation and propagation of VT in these mice. We further analyzed HRV in ECG recordings from these mice to determine whether an increased inducibility of VT was associated with a decrease in the HF fraction of HRV. Finally, retrospective studies (12, 16, 20, 28) have suggested that statins might exert an antiarrhythmic effect on the heart, whereas our data demonstrated decreased ventricular ectopy and increased parasympathetic activity as measured by an increase in the HF fraction in patients treated with pravastatin (30). Based on these findings, we determined whether pravastatin treatment of Akita mice attenuates the inducibility of VT and parasympathetic dysfunction and whether pharmacologically altering the balance between sympathetic and parasympathetic input to the heart mimicked the effect of pravastatin on the inducibility of VT in this mouse model.
MATERIALS AND METHODS
Animals
Akita type 1 diabetic mice (C57BL/6J-Ins2Akita) were obtained from Jackson Laboratories. The Akita mice used in this study demonstrated marked hyperglycemia (500 ± 28 mg/dl), and body weight was significantly lower in Akita mice compared with wild-type (WT) mice (Table 1). We (23) have previously demonstrated that there are no significant differences in electrolytes, acid-base balance, or left ventricular function between WT and Akita mice. In the present study, six protocols were undertaken. In the first protocol, the surface ECG and inducibility of VT were compared between 14 heterozygous male Akita C57BL/6J-Ins2Akita mice and 14 WT C57BL/6J male mice at the ages indicated (Table 1). In the second protocol, a set of similar-age Akita mice were randomized to normal saline (n = 16 mice) or pravastatin (20 mg·kg−1·day−1 ip, n = 19 mice) treatment for 7 days, and the inducibility of VT was determined. In the third protocol, HR was determined using implantable ECG transmitters in conscious active WT (n = 9) and Akita (n = 8) mice, and the HF fraction of the power spectrum was determined as previousy described. In the fourth protocol, Akita mice were randomized to normal saline (n = 10 mice) or pravastatin (20 mg·kg−1·day−1 ip, n = 10 mice) as previously described, and HRV was compared. In the fifth protocol, the surface ECG and inducibility of VT were compared between a group of Akita C57B/6J-Ins2Akita male mice randomized to pretreatment with 50 μg/kg carbamylcholine (n = 8 mice) or normal saline (8 mice) intraperitoneally 10 min before the study. Finally, the surface ECG and inducibility of VT in Akita mice (n = 4) were determined before and after treatment with 1 mg/kg propranolol. All animal protocols conformed with the Association for the Assessment and Accreditation of Laboratory Animal Care, with approvals from the Institutional Animal Care and Use Committee of Tufts Medical Center and Tufts University. Pravastatin was a gift of Bristol-Myers Squibb (Hopewell, NJ).
Table 1.
Mouse characteristics and surface ECG parameters
| ECG Characteristics |
||||||||
|---|---|---|---|---|---|---|---|---|
| Number of Mice | Weight, g | Glucose, mg/dl | Sinus cycle length, ms | PR, ms | QRS, ms | QT, ms | QTc, ms | |
| WT mice | 14 | 27.3 ± 1.1 | 163 ± 20 | 140.1 ± 3.8 | 37.2 ± 1.1 | 16.1 ± 0.5 | 46.9 ± 1.4 | 39.7 ± 1.2 |
| Akita mice | 14 | 21.8 ± 0.3† | 511 ± 28† | 133.8 ± 7.0 | 43.6 ± 1.5* | 19.9 ± 0.6† | 58.6 ± 3.2* | 50.7 ± 1.7† |
| Akita mice with placebo treatment | 16 | 21.6 ± 0.5 | 537 ± 17 | 142.9 ± 5.3 | 44.5 ± 1.4 | 19.3 ± 0.9 | 58.8 ± 2.6 | 49.2 ± 1.8 |
| Akita mice with statin treatment | 19 | 20.5 ± 0.3 | 540 ± 20 | 142.8 ± 4.5 | 45.9 ± 2.3 | 18.4 ± 0.5 | 51.2 ± 1.6‡ | 42.9 ± 1.3‡ |
Values are means ± SE.
P < 0.01 vs. wild-type (WT);
P < 0.001 vs. WT mice;
P < 0.01 vs. Akita mice with placebo treatment.
Intracardiac Electrophysiological Experiments
Mice were anesthetized with isoflurane (2% or 2.5%) in oxygen. A surface frontal plane ECG was obtained using 29-gauge electrodes placed subcutaneously in each limb. The right external jugular vein was exposed, and a 1.1-Fr electrophysiology octapolar catheter (EPR-800, Millar Instruments, Houston, TX) was inserted and advanced to the right ventricle using intracardiac electrogram guidance and pacing capture at low threshold to verify the intracardiac position.
Surface ECG and intracardiac electrogram recordings from mice at 12 ± 1 wk of age were sampled at 2,000 Hz using a PL3504 PowerLab 4/35 (ADInstruments). Surface ECG recordings were filtered at 0.01–100 Hz, and intracardiac electrogram recordings were filtered at 30–500 Hz. With the use of LabChart 7 (AD Instruments), all surface ECG measurements [cycle length (CL) and PR, QRS, and QT intervals] were performed with online calipers. QT was corrected to HR as follows: QTc = QT/ (6).
The stimulation protocol was performed using the FE180 Stimulus Isolator (AD Instruments) connected to the intracardiac octapolar catheter. VERP was determined using an eight-beat pacing drive train at 100 ms (VERP100) followed by single extrastimulation. Atrial and ventricular programmed stimulation consisted of a drive train of eight beats at 100 ms with up to three premature extrastimuli at progressively shorter CLs until refractoriness. Programmed stimulation was repeated at drive trains of 90 and 80 ms followed by burst stimulation for 5–10 s at three different intervals of 70, 60, and 50 ms. VT was defined as greater than or equal to four beats.
Optical Mapping
Akita and WT mice (2–6 mo old) were heparinized (0.5 U/g ip) and then anesthetized with a ketamine (1 g/ml) and xylazine (100 mg/ml) mixture injected intraperitoneally. The heart was rapidly excised through a thoracotomy and subsequently connected to a Langendorf perfusion system to be continuously perfused with warm oxygenated Tyrode solution (pH 7.4) with HEPES buffer bubbled with 100% O2. The temperature of the perfusate was maintained at 36 ± 1°C. The heart was placed in the well of a custom-made chamber maintained at 36 ± 1°C and allowed to equilibrate for 10 min. The potentiometric dye di-4-ANEPPS (Molecular Probes) was added to the perfusate as a bolus to achieve a final concentration of 10 μmol/l. To minimize signal distortion due to motion, excitation-contraction uncoupling was achieved with 5 μM blebbistatin (Cayman Chemicals). The excitation incident light was generated from an LED chip (Prizmatix) coupled to a 520 ± 40-nm band-pass filter (ThorLabs), and the emitted light was collected with a C-mount 25-mm fast video lens (Navitar), filtered with a 600-nm long-pass filter (ThorLabs), and focused onto the charge-coupled device chip of the recording camera (RedShirt Imaging) at 80 × 80 pixels and 85 μm/pixel. Fluorescence movies were acquired at 1 kHz. Fluorescent movie snapshots, phase movie snapshots, and pseudo-ECGs were generated as previously described (8) using custom-built software courtesy of the Jalife Lab (University of Michigan) running on the PV Wave platform. An octapolar pacing electrode was placed in the right ventricle via the pulmonary artery, and programmed ventricular stimulation was carried out as described above. Volume-conducted ECGs were recorded, and stimulation was delivered as described above (8). To induce ventricular arrhythmias, hearts were perfused with 0.1 μM isoproterenol, allowed to equilibrate for 20 min, and then subjected to programmed electrical stimulation.
Spontaneous ECG Monitoring and HRV Analysis in Conscious Mice
Mice were anesthetized with 1.5% isoflurane. An ECG signal wireless radiofrequency transmitter was implanted in a subcutaneous pocket in WT and Akita mice of the indicated age, and data were recorded at a sample rate of 5,000 Hz with the use of a telemetry receiver and an analog-to-digital acquisition system (Data Sciences). The ECG signal was analyzed using custom-built software. Beat-to-beat HR data were computed, and artifacts and nonsinus beats were removed after manual review. Composite HR plots and average HRs and duration of bradycardia were computed as previously described (22). For HRV analysis, R-wave detection and beat annotation were both manually reviewed as described above. All ectopic and postectopic beats and artifacts were removed and replaced with intervals interpolated from adjacent normal beats; segments were discarded where gaps accounted for >15% of the recording segment. Frequency-domain analysis was performed after construction of an instantaneous RR interval time series by resampling at 10 Hz. Power spectra of detrended 2-min segments were computed for frequency ranges of 0.5–1.5 Hz, designated as low-frequency (LF) power, and 1.5–5 Hz, designated as HF power. The HF fraction was computed as HF/(LF + HF). HF power has been shown to predominantly result from the parasympathetic modulation of HR with a small sympathetic contribution, whereas LF power has been shown to result from both the sympathetic and parasympathetic modulation of HR (15, 27a). HR and frequency-domain HRV parameters were computed for 2-min segments at the end of the baseline and propranolol phases where HR and frequency-domain parameters were relatively stationary and verified using Kalman smoothing and wavelet-based visualization in addition to fast Fourier transform-based spectrograms. Composite plots of the HF fraction were computed from fast Fourier transform power spectra over a 3-min sliding window of RR interval data, repeated every 10 s, and averaged to one HF fraction data point per minute per group (±SE). Given that β-adrenergic receptor inhibition blocks the sympathetic component of HRV, leaving the parasympathetic component relatively unopposed, we treated 13 mice with the β-adrenergic receptor blocker propranolol and computed the time course of the increase in the HF fraction after the injection of propranolol and computed the frequency-domain HRV parameters for 2-min segments at the end of the baseline and propranolol phases. The HF fraction increased from a mean of 39.65 ± 1.8 before propranolol to 59.3 ± 5.6% (n = 13, P = 0.008) at 15 min after propranolol, LF power decreased from 4.58 ± 1.01 to 2.49 ± 1.17 × 10−6 ms2/Hz (P = 0.074), and HF power decreased was from 2.17 ± 0.57 to 1.81 ± 0.70 × 10−6 ms2/Hz (P = 0.431). We further generated composite plots of the time course of the effect of propranolol on the HF fraction, LF power, HF power, and total power. The HF fraction increased with a time course similar to that for the decrease in LF power, whereas HF power decreased slightly (Fig. 1). Given that we compute the HF fraction as HF/(HF + LF), these findings support the conclusion that the increase in the HF fraction in response to propranolol is primarily due to a decrease in LF power resulting from sympathetic blockade. Hence, the increase of the HF fraction in the response of the mouse heart to propranolol is due to the parasympathetic modulation of HR that is not affected by sympathetic blockade (27a). In these experiments, we compared differences in the HF fraction after propranolol in WT and Akita mice and assessed the effect of pravastatin on parasympathetic dysfunction in the type 1 diabetic Akita mouse.
Fig. 1.
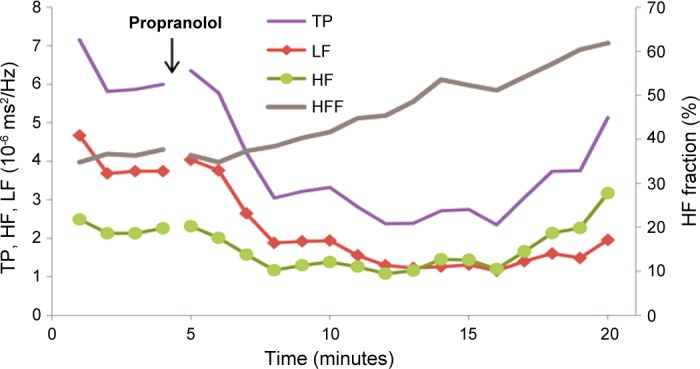
Time course of the changes in total power (TP), high-frequency fraction (HFF), and low-frequency (LF) power and high-frequency power (HF) components of the power spectrum in response to propranolol. ECGs were monitored in 4-mo-old male mice. Four days after mice had recovered from surgery, heart rate (HR) was recorded continuously for 5 min before and 15 min after the intraperitoneal injection of 1 mg/kg propranolol. Composite plots of TP (TP = HF + LF), HFF [HFF = HF/(HF + LF)], LF, and HF were obtained as described in materials and methods.
Statistical Analysis
Average values are expressed as means ± SE. Statistical differences between mean values were calculated by independent sample Student's t-test or Mann-Whitney U-test as appropriate (two sided). Normal distribution assumptions were verified using the Shapiro-Wilk test. Cross-tabulation analyses were performed using likelihood ratio χ2-tests and Goodman and Kruskal τ-tests. P values of <0.05 were considered significant.
RESULTS
Induced Ventricular Arrhythmias in WT Versus Akita Mice
Comparison of electrocardiographic properties.
Analysis of ECGs from WT and Akita mice demonstrated that HR as measured by sinus CL was not significantly different in WT and Akita mice. However, PR interval, QRS duration, QT interval, and QTc were significantly increased in Akita mice compared with WT mice (PR: 43.6 ± 1.5 vs. 37.2 ± 1.1 ms, P < 0.01; QRS: 19.9 ± 0.6 vs. 16.1 ± 0.5 ms, P < 0.001; QT: 58.6 ± 3.2 vs. 46.9 ± 1.49 ms, P < 0.01; and QTc: 50.7 ± 1.7 vs. 39.7 ± 1.2 ms, P < 0.001; Table 1). Increased QRS duration and QT interval have been shown to be associated with a predisposition to the development of ventricular arrhythmias (25, 26).
The inducibility of VT is markedly increased in Akita mice compared with WT mice.
We used programmed ventricular stimulation to compare the inducibility of VT in WT and Akita type 1 diabetic mice (Fig. 2). Akita mice were significantly more vulnerable to VT (78.6%, 11 of 14 mice) compared with WT (28.6%, 4 of 14 mice, P = 0.007; Fig. 3A). The mean number of episodes of VT was 0.36 ± 0.17 events/mouse in WT mice versus 10.5 ± 3.47 events/mouse in Akita mice (P = 0.001; Fig. 3B). Furthermore, of the 4 inducible WT mice, 3 mice were inducible only with burst pacing, whereas 10 of 11 Akita mice developed VT in response to S2 or S3, consistent with the increased predisposition of Akita mice to stimulation of VT (Table 2). Finally, of the WT mice that developed VT, the longest run of VT was 4–10 beats in 3 mice, and 1 mouse had a run of 11–30 beats. In contrast, six of the Akita mice had runs of 11–30 beats, and two mice had runs of >30 beats. These differences in the length and severity of VT were statistically significant (P = 0.013 by likelihood ratio χ2-test and P = 0.007 by Goodman and Kruskal τ-test; Fig. 3C). Thus, among those WT mice that were inducible, only short runs of VT could be elicited by burst pacing, whereas Akita mice demonstrated more prolonged runs of VT at less intense levels of stimulation. Interestingly, inducible Akita mice demonstrated shorter VERP (31.3 ± 1.7 ms) compared with noninducible Akita mice (41.6 ± 0.8 ms, P = 0.0035).
Fig. 2.

Induction of ventricular tachycardia (VT) in a type 1 diabetic Akita mouse in response to programmed ventricular stimulation. Top and middle top: surface ECGs. Middle bottom and bottom: right ventricular (RV) and right atrial (RA) intracardiac electrograms (EG). This example shows a run of pacing-induced VT, which lasted for 1,854 ms. Programmed double extrastimulation (S1 at 100 ms × 8, S2 at 35 ms × 1, and S3 at 30 ms) induced VT that spontaneously terminated followed by a nonconducted P wave and conversion to normal sinus rhythm.
Fig. 3.

Inducibility and severity of VT in Akita and wild-type (WT) mice. A: comparison of the inducibility of VT in WT mice (4 of 14 mice) and Akita mice (11 of 14 mice, P = 0.007). B: mean number of runs of VT per mouse in WT mice (0.36 ± 0.17) versus Akita mice (10.5 ± 3.47, **P = 0.001). C: distribution of the severity of VT, plotted as the number of beats of VT, in WT and Akita mice (P = 0.013 by likelihood ratio χ2-test).
Table 2.
Level of stimulation at which ventricular tachycardia was first induced
| Singles: (S1 × 8) + S2 | Doubles: (S1 × 8) + S2S3 | Triples: (S1 × 8) + S1S2S3 | Burst Pacing: 70, 60, and 50 ms | |
|---|---|---|---|---|
| WT mice | 0 | 0 | 1 | 3 |
| Akita mice | 1 | 3 | 7 | 0 |
| Akita mice with placebo treatment | 1 | 7 | 4 | 0 |
| Akita mice with statin treatment | 1 | 4 | 1 | 1 |
Shown are numbers of mice that developed ventricular tachycardia in response to the indicated stimulation.
Optical Mapping of the Mechanism of Arrhythmogenesis of VT in the Akita Mouse
To elucidate the mechanism of ventricular arrhythmogenesis in the Akita mouse, we used optical mapping of isolated Langendorf-perfused hearts. Initially, Langendorf-perfused hearts from WT (n = 6) and Akita (n = 5) mice were found not to be inducible for VT in response to programmed ventricular stimulation. Although the in vivo experiments shown in Fig. 3 demonstrated the inducibility of VT in the Akita mouse heart, we (23) had previously demonstrated parasympathetic dysfunction in these mice. Given that denervated Langendorf-perfused hearts lacked autonomic input, we determined whether excess unopposed sympathetic stimulation might predispose these hearts to the inducibility of VT. We compared the effect of perfusion of hearts with a solution 0.1 μM isoproterenol on the inducibility of VT in hearts from WT and Akita mice. Five of five Akita mice were inducible for VT in response to burst pacing, with a mean average VT duration per mouse of 3.56 ± 1.49 s, whereas three of six WT mice developed short runs of VT (P =0.032 by likelihood ratio χ2-test), with an average duration of VT per mouse of 0.42 ± 0.81 s (P = 0.018 by Mann-Whitney U-test). These data suggested that the inducibility of VT in denervated Langendorf-perfused hearts from Akita mice was dependent on increased sympathetic stimulation.
For optical mapping experiments, hearts were perfused with 0.1 μM isoproterenol, and VT was induced in response to burst pacing. The data shown in Fig. 4 are from a typical experiment. This heart demonstrated a VERP100 of 40 ms, which decreased to 25 ms after continuous isoproterenol perfusion. VT was inducible 5 of 10 times in response to burst pacing at a CL of 25 ms (Fig. 4A). Optical mapping from this heart is shown here as nine snapshots of epicardial fluorescence at the time of initiation of each beat during a run of VT. Initial breakthrough sites (denoted by asterisks) occurred at different locations on the right and left ventricular free wall and the apex (Fig. 4B). The data shown in Fig. 5 demonstrated a similar pattern of multifocal breakthroughs, which occasionally gave rise to short-lived, unstable rotors. Figure 5A shows snapshots at different times (numbers in top right corners) from the phase movie. The colors correspond to the phases of the action potential, as shown in Fig. 5B, with the black line denoting the upstroke of the action potential. In the first three snapshots, focal breakthroughs are coexistent with an unstable rotor whose tip trajectory (the location of the rotational center over time) is delineated in Fig. 5D. Subsequently, the rotor terminates, and a new breakthrough emerges at the apex (5th snapshot). Two beats later (7th snapshot), another unstable rotor is initiated (tip trajectory in Fig. 5E). Subsequently (12th snapshot), the arrhythmia is maintained by multifocal discharges originating in the right and left ventricles. These data suggest a strong focal component in the initiation and maintenance of VT in this mouse model. Such epicardial breakthroughs are reminiscent of triggered activity at the level of the conduction system, as previously demonstrated by Cerrone et al. (8), that successfully propagate, activating the ventricular wall and finally emerging as breakthroughs on the epicardium.
Fig. 4.

Focal mechanism of VT in the Akita mouse. Langendorf-perfused Akita mouse hearts in a solution of 0.1 μM isoproterenol were optically mapped using di-4-ANEPPS as a voltage-sensitive dye and 5 μM blebbistatin to achieve contraction uncoupling and subjected to burst pacing as described in materials and methods. A: typical pattern of induction of VT from a volume-recorded ECG. B: nine snapshots (1–9) of epicardial fluorescence at the time of initiation of each beat in the run of VT superimposed on the anatomic image of the heart. *Initial breakthrough sites. LV, left ventricle. C: pseudo-ECG of the episode of tachyarrhythmia computed from the optical voltage measurements from the fluorescence movie. Scale bar = 1 mm. See Supplemental Movie M1 in the Supplemental Material.
Fig. 5.

Focal discharges giving rise to unstable rotors. A Langendorf-perfused Akita mouse heart was subjected to programmed ventricular stimulation as described in materials and methods. A: the first frame is the cartoon of the heart in the field of view. Subsequent snapshots (1–14) are taken from the phase movies, where the color-coded phases of the action potential are represented as indicated in B. Numbers in the top right corner of the snapshots denote the time, and the pink numbers in the bottom left corners are the beats indicated in the pseudo-ECG in C. Beats 1, 2, and 3 show the coexistence of a focal discharge in the LV free wall with three rotations of a meandering rotor, with the tip trajectory shown in D. Beat 5 is due to a focal discharge from the apex. Beat 6 is a wave that invades the field of view from the posterior wall of the heart followed by five rotations of an unstable rotor (beats 7–11), with the tip trajectory shown in E. These rotors coexist with multiple foci originating on the LV free wall and apex. Beats 12–14 arise due to focal discharges originating in the RV and LV. Curved arrows denote the rotation chirality. *Breakthrough sites. See Supplemental Movie M2.
Effect of Pravastatin Treatment on Akita Mice: Pravastatin Treatment Decreases the Inducibility of VT in Akita Mice
Given retrospective studies (12, 16, 20, 28) suggesting that statins exert an antiarrhythmic effect on the heart and our previous data (30) demonstrating decreased ventricular ectopy in statin-treated patients, we determined the effect of pretreatment of Akita mice with pravastatin on the inducibility of VT. Pravastatin pretreatment of Akita mice (20 mg·kg−1·day−1 ip) had no effect on HR and PR interval but decreased QT interval from 58.8 ± 2.6 ms in placebo-treated mice to 51.2 ± 1.6 ms (P < 0.01) and QTc from 49.2 ± 1.8 ms in placebo-treated mice to 42.9 ± 1.3 ms (P < 0.01), partially reversing the increase in QT and QTc observed in untreated Akita mice (Table 1). Interestingly, Akita mice pretreated with pravastatin were less inducible for VT (36.8%, 7 of 19 mice) compared with placebo-treated Akita mice, in which 75.0% (12 of 16 mice, P = 0.022; Fig. 6A) were inducible. Similarly, the mean number of episodes of VT in placebo-treated Akita mice was 9.81 ± 2.63 versus 4.11 ± 1.73 events/mouse for statin-treated Akita mice (P = 0.041; Fig. 6B). Finally, of the placebo-treated mice that developed VT, the longest run of VT was 4–10 beats in 4 mice, 3 mice had a run of 11–30 beats, and 5 mice had >30 beats. In contrast, one of the Pravastatin treated Akita mice had 4–10 beats of VT, 4 had 11–30 beats and 2 mice had runs of >30 beats. These differences in the length and severity of VT were statistically significant (P = 0.035 by Kruskal τ-test; Fig. 6C). There was no difference in the level of stimulation required to elicit VT in these two groups (Table 2).
Fig. 6.

Inducibility and severity of VT in placebo- and pravastatin-treated Akita mice. Mice were treated with placebo or pravastatin for 7 days. A: comparison of the inducibility of VT in placebo-treated (12 of 16 mice) versus pravastatin-treated (7 of 19 mice) Akita mice. B: mean number of runs of VT per mouse in placebo-treted (9.81 ± 2.63) and pravastatin-treated (4.11 ± 1.73, *P = 0.041) Akita mice. C: distribution of the severity of VT, plotted as the number of beats, in placebo- and pravastatin-treated Akita mice (P = 0.035 by Kruskal τ-test).
Effect of Pravastatin Treatment on HRV in Conscious Mice: Akita Mice Demonstrate a Decreased HF Fraction of HRV, Which Is Reversed by Pravastatin
To determine whether the inducibility of VT in the Akita mouse was associated with parasympathetic dysfunction and whether pretreatment with pravastatin reversed parasympathetic dysfunction, we computed the HF fraction of the HRV power spectrum [HF/(HF + LF)] in WT and Akita mice. Given that β-adrenergic receptor inhibition blocks the sympathetic component of HRV, decreasing LF power and leaving the parasympathetic component relatively unopposed, we compared the increase in the HF fraction after the injection of the β-adrenergic receptor blocker propranolol in Akita and WT mice. The HF fraction measured 15 min after propranolol injection was significantly lower in Akita mice compared with WT mice (48.6 ± 5.2% compared with 70.9 ± 4.8%, respectively, n = 10, P = 0.005; Fig. 7A). To further determine whether parasympathetic signaling was decreased in the Akita mouse, we compared the increase in HR in WT and Akita mice after parasympathetic blockade by the M2 muscarinic receptor antagonist atropine. HR was recorded for 1 h before and after treatment with atropine (0.5 mg/kg). The mean HR in WT mice increased from 569 ± 33.6 beats/min before atropine treatment to 740.3 ± 13.7 beats/min after atropine, or 32.6 ± 6.5% (n = 8, P < 0.05). The mean HR in Akita mice increased from 589.5 ± 8.6 to 689 ± 6.5 beats/min, or 16.9 ± 1.5% (n = 8, P < 0.05; Fig. 7C). Resting HRs were not different between WT and Akita mice. The percent increase in HR was significantly lower in Akita mice compared with WT mice (P = 0.01), consistent with parasympathetic dysfunction in the Akita mouse.
Fig. 7.

Comparison of the HFF of HR variability in WT and Akita mice and in placebo- and pravastatin-treated Akita mice. ECG was monitored in male mice as described in materials and methods. Four days after mice had recovered from surgery, HR was recorded continuously from 5 min before the injection of 1 mg/kg propranolol for 20 min. ECG transmitters were implanted in a second set of Akita mice, and the HR response to atropine was determined before and after treatment with placebo or 20 mg/kg pravastatin for 7 days. A: quantitation of HFF at 15–20 min after propranolol injection in WT mice (70.9 ± 4.8%), Akita mice (48.6 ± 5.2%, n = 10, *P = 0.005), placebo-treated Akita mice (59.1 ± 4.2%, n = 8), and pravastatin-treated Akita mice (71.0 ± 2.7%, n = 9, #P = 0.027). There was no significant difference in HFF between Akita and placebo-treated Akita mice. Baseline HFF was not significantly different in the two groups. B: comparison of group-wise averaged (±SE) composite plots of HFF before and over the time course of the propranolol phase in placebo- and pravastatin-treated Akita mice. *P < 0.01. C: relative increase in HR in WT and Akita mice in response to muscarinic inhibition with atropine. *P = 0.01. Statistical comparisons were carried out using Student's t-test. D: relative increased in the HR response to atropine before and after pravastatin treatment. *P = 0.016. Statistical comparisons were carried out using a paired t-test. Results are reported as means ± SE.
To determine the effect of pravastatin on HRV, Akita mice were treated with either placebo or pravastatin, and the increase in the HF fraction in response to propranolol was determined. Fifteen minutes after propranolol injection, the HF fraction was significantly higher in pravastatin-treated Akita mice compared with placebo-treated Akita mice (71.0 ± 2.7%, n = 9, vs. 59.1 ± 4.2%, n = 8, respectively, P = 0.027; Fig. 7A). To further establish the difference in the HF fraction in placebo- versus pravastatin-treated Akita mice, we compared the time course of the increase in the HF fraction after the injection of propranolol in placebo- and pravastatin-treated Akita mice. The time course of the increase in the HF fraction was significantly faster and reached a higher level in pravastatin-treated mice (Fig. 7B).
To further support the conclusion that pravastatin treatment of Akita mice increased parasympathetic signaling, we compared the increase in HR in Akita mice in response to atropine before and after pravastatin treatment. Mean resting HRs before and after pravastatin were not significantly different (550 ± 32.4 and 596.7 ± 13.5 beats/min, respectively). The mean HR increased by 101.5 ± 13.46 beats/min, or 17 ± 2.6%, before atropine treatment to 175.2 ± 8.19 beats/min, or 34 ± 4.0%, after atropine (n = 4, P = 0.009; Fig. 7D). The difference in the percent change was also significant (P = 0.016).
Effect of Pharmacological Alterations in Autonomic Balance in the Akita Mouse on the Inducibility of VT
Direct stimulation of the muscarinic receptor by carbamylcholine protects against the inducibility of VT in the Akita mouse.
The finding of increased inducibility of VT and decreased HF fraction in Akita mice and the partial reversal of these findings by pravastatin were consistent with the hypothesis that parasympathetic dysfunction increases the inducibility of VT in Akita mice. To further test this hypothesis, we carried out programmed ventricular stimulation in Akita mice pretreated with the muscarinic agonist carbamylcholine. Two groups of Akita mice were treated with either 50 μg/kg carbamylcholine or normal saline intraperitoneally. Mice were subjected to the standard protocol of S1, S2, and S3 stimulation, which was completed in <55 min after injection with propranolol, and the surface ECG and intracardiac electrogram were recorded. Of the eight Akita mice treated with carbamylcholine, none were inducible, whereas of the eight mice pretreated with saline, six mice were inducible for VT and two mice were not (P = 0.007; Fig. 8A). The mean number of episodes of VT in saline-treated mice was 4.87 ± 1.31 events/mouse compared with none in untreated Akita mice (P = 0.007).
Fig. 8.

Effect of carbamylcholine and/or propranolol on the inducibility of VT in Akita mice. A: mice were pretreated for 10 min with either placebo or carbamylcholine followed by programmed ventricular stimulation, and the inducibility of VT was determined. Whereas six of eight placebo-treated Akita mice were inducible for VT, none of the eight carbamylcholine-treated Akita mice were inducible (P = 0.007). B: the inducibility of VT in four Akita mice was determined, followed by the injection of 1 mg/kg propranolol, and programmed ventricular stimulation was repeated.
To determine whether a muscarinic antagonist might increase the incidence of inducible VT, we determined the effect of atropine on the inducibility of VT in WT mice in which <30% of mice were inducible. Four WT mice were treated with 0.5 mg/kg atropine intraperitoneally and subjected to programmed ventricular stimulation after 15 min. Three mice were not inducible, but the fourth mouse developed a single run of four beats of VT, consistent with the conclusion that atropine had little effect on the inducibility of VT. One possible explanation for these findings is that isoflurane and many other general anesthetics have been shown to blunt sympathetic responses in the heart (9). This conclusion was supported by the finding that in the presence of isoflurane anesthesia, atropine had no effect on resting HR while resting HR decreased from 588.1 ± 9.4 beats/min in conscious mice (n = 8) to 447.1 ± 10.8 beats/min in mice treated with 2.0% isoflurane (n = 14, P < 0.001) to 410 ± 10.18 beats/min in mice treated with 2.5% isoflurane (n = 12, P < 0.05 compared with the 2.0% isoflurane-treated group). To determine the effect of isoflurane on the inducibility of VT, we compared the response of mice treated with either 2% or 2.5% isoflurane to programmed ventricular stimulation. At 2% isoflurane, 10 of 10 male Akita mice were inducible for VT in response to extrastimulation, whereas at 2.5% isoflurane, 4 of 7 mice were inducible (P = 0.012 by likelihood ratio χ2-test); 2 of these mice had only short runs of VT and only in response to burst pacing. The decrease in both resting HR and the inducibility of VT in response to isoflurane anesthesia are consistent with the conclusion that isoflurane suppresses the inducibility of VT via the inhibition of sympathetic stimulation of the heart and further supports the conclusion that excess sympathetic input in the Akita heart predisposes the mouse to inducible VT.
Inhibition of β-adrenergic receptors by propranolol decreases the inducibility of VT in the Akita mouse.
If autonomic imbalance plays a role in the inducibility of VT in the Akita mouse and isoflurane suppresses inducibility via interference with sympathetic stimulation, then inhibition of sympathetic input by the β-adrenergic receptor blocker propranolol should also decrease the incidence of VT. The inducibility of VT was compared before and after injection of propranolol (1 mg/kg) in four Akita mice. All four mice were inducible for VT before propranolol. After injection, HR decreased by 19 ± 3.5% from 402 ± 22 to 325 ± 18 beats/min (P < 0.021), and three of four mice were not inducible for VT (Fig. 8B).
DISCUSSION
The present experiments demonstrate that type 1 diabetes in the Akita mouse is associated with both the inducibility of VT and a significantly reduced parasympathetic component of HRV, both of which are risk factors for cardiovascular disease and sudden death. Up to 80% of C57/BL6 mice with the Akita mutation were inducible for VT compared with up to 30% of WT littermates. More importantly, the mean number of events/mouse was nearly 30 times higher in Akita mice compared with WT mice, and VT in WT mice was limited to runs of 4–10 beats compared with Akita mice, which demonstrated runs of 11–30 or >30 beats. We further demonstrated that Akita mice not only had a decrease in the HR response to parasympathetic stimulation, as previously reported (23), but also demonstrated a decreased HF component of the HRV power spectrum as measured by the HF fraction. This parasympathetic dysfunction was further demonstrated by the finding that the increase in HR in response to inhibition of the muscarinic receptor with atropine was significantly greater in WT mice compared with Akita mice. We (30) have previously presented data that suggested that patients treated with pravastatin appeared to demonstrate decreased ventricular irritability in parallel with an increase in parasympathetic responsiveness. Here, we demonstrated that pravastatin not only decreased the inducibility of VT but also markedly decreased the mean number of events per mouse and the severity of VTs. Interestingly, pravastatin treatment of Akita mice also significantly increased the HF fraction compared with placebo treatment and significantly increased the HR response to atropine treatment, suggesting that the reversal of the parasympathetic dysfunction in the diabetic heart in response to pravastatin might play a role in the decreased inducibility of VT. Although HRV reflects the response of the heart to autonomic input from the central nervous system via vagal and sympathetic nerves, the role of autonomic imbalance in the inducibility of VT in the Akita mouse was further supported by the finding that treatment of the Akita mouse with carbamylcholine, which directly activates the parasympathetic response at the level of the M2 muscarinic receptor in the heart muscle, thus bypassing vagal innervation (23), protected the heart from the inducibility of VT. Furthermore, β-adrenergic blockade of sympathetic input to the heart by propranolol also attenuated the inducibility of VT in the Akita mouse. These data support the conclusion that autonomic imbalance in the Akita diabetic heart might play a role in the predisposition to VT and that pravastatin protects the heart from the inducibility of VT by increasing parasympathetic function.
We and others (10, 23) have demonstrated that Akita type 1 diabetic mice develop secondary effects of diabetes, including abnormalities of nerve conduction and the parasympathetic dysfunction of diabetic autonomic neuropathy. The increased incidence of sudden death in the diabetic population and data supporting the protective effect of parasympathetic stimulation from the development of ventricular arrhythmias (11, 25) suggested that diabetics with parasympathetic dysfunction might have a predisposition to the development of VT. To test this hypothesis in an animal model, we studied the effect of programmed ventricular stimulation in type 1 diabetic Akita mice, which, as we (23) have previously demonstrated, have parasympathetic dysfunction, as measured by a blunted HR response to carbamylcholine. The inducibility of VT in WT mice has been previously demonstrated in 2–40% of mice depending on strain, with C57/BL6 mice being the least inducible (2%) and FVB mice the most prone to inducibility (43%) (14, 19). Hence, the finding of close to 80% inducibility in the C57/BL6 Akita mouse was highly significant. We (23) have previously demonstrated that there were no significant differences in pH, Pco2, electrolytes, anion gap, and left ventricular function between Akita and WT mice. We further demonstrated that there were no significant differences in left ventricular dimensions and left ventricular function in Akita and WT mice, thus ruling out the possibility that differences in inducibility might be due to metabolic abnormalities and impaired left ventricular function in Akita mice.
We have previously demonstrated that pravastatin treatment decreased the incidence of premature ventricular contractions, couplets, and short runs of nonsustained VT in a population of subjects with otherwise structurally normal hearts who demonstrated lipid profiles and risk factors that met the criteria for statin therapy. This decrease in spontaneous ventricular arrhythmias correlated with an increase in parasympathetic modulation of HR, as measured by changes in the peak HF fraction. While the spontaneous occurrence of premature ventricular contractions, couplets, and short runs of nonsustained VT in a structurally normal heart lack prognostic significance, it does reflect the level of excitability of the ventricle (18). The finding that pravastatin treatment of the Akita mouse markedly decreased the inducibility of VT while increasing parasympathetic responsiveness as measured by the HF fraction of HRV further supported the hypothesis that pravastatin might decrease excitability via an effect on the parasympathetic response of the heart. Several retrospective studies (20, 28) have suggested that statins might exert an antiarrhythmic effect on the heart. The Multicenter Autonomic Defibrillator Implantation Trial II demonstrated that statin use in patients with implantable cardioverter-defibrillators was associated with a decrease in the end points of VT or ventricular fibrillation or cardiac death (28). In a group of 78 patients with coronary artery disease and ventricular arrhythmias treated with implantable cardioverter-defibrillators, 22% of the patients treated with lipid-lowering drugs had a recurrence of arrhythmia compared with 57% of patients not treated with lipid-lowering drugs (12). The interpretation of these studies is complicated by the presence of ischemic heart disease in the patient population. However, the Defibrillators in Nonischemic Cardiomyopathy Treatment Evaluation study (16) suggested that statin use was associated with a 78% decrease in mortality, which was due in part to a reduction in arrhythmic death.
Optical mapping of the mechanism for the development of inducible VT in the type 1 diabetic Akita heart demonstrated that the VTs originated predominately as focal discharges, with initial breakthrough sites occurring at different locations on the right and left ventricular free walls and the apex. These focal discharges occasionally gave rise to unstable rotors, which also suggested that VT in the Akita mouse heart might be maintained by interplay between focal discharges and unstable rotors. These epicardial breakthroughs could result from propagated triggered activity in the conduction system, which successfully activates the ventricles and finally emerges as breakthroughs on the epicardium, as previously described (8). Furthermore, experimental and numerical simulations (3, 8, 29) have shown that VT resulting from triggered activity is mainly focal and involves the specialized conduction system as the initial site of initiation due to the cable-like structure of the His-Purkinje system. This cable-like geometry would necessarily lead to a smaller electrical load on Purkinje cells compared with ventricular myocytes in the three-dimensional myocardium. The smaller sink to source mismatch of Purkinje fibers would increase the safety of propagation of a triggered beat compared with the working myocardium. The Purkinje system has been shown to be innervated by both sympathetic and parasympathetic neurons (31). We (23) have previously demonstrated a marked decrease in the ACh-sensitive inward rectifier K+ current in atrial myocytes from the Akita mouse. Such an abnormality of the ACh-sensitive inward rectifier K+ current in the Purkinje system could result in a decrease in resting membrane potential, facilitating the occurrence of triggered activity and therefore predisposing to the initiation of VT.
The increased QRS duration and QT intervals in the Akita versus WT mouse might play a role in the predisposition to arrhythmias. This conclusion is further supported by the finding that pravastatin treatment, which attenuated the inducibility of VT and reversed the abnormality of HRV, partially decreased QT interval and had a small effect on QRS duration. Data from several sources (13, 24–26) support the conclusion that parasympathetic stimulation protects the heart from arrhythmia in both a canine model for ventricular pacing after myocardial infarcion and in studies in which parasympathetic responsiveness in rats was attenuated by chronic treatment with carbamylcholine or pertusiss toxin. Isolated papillary muscles from these rats demonstrated marked sensitization to the arrhythmogenic effects of isoproterenol or forskolin. Further elucidation of the mechanism by which pravastatin protects the heart from the induction of VT should offer new therapeutic targets in the treatment and prevention of arrhythmias and sudden death in the diabetic population.1
GRANTS
This work was supported by National Institutes of Health Grants R21-DK-079622 (to H-J. Park), RO1-HL-074876 (to J. B. Galper), HL-069770 (to H. Jin and A. Albano), and R00-HL-105574 (to S. F. Noujaim) as well as by American Heart Association Grant 13SDG17050021 (to H. Jin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.R., H.J., C.M.W., H.-J.P., M.S.L., S.F.N., and J.B.G. conception and design of research; M.R., H.J., A.A., M.J.A., Y.Z., and S.F.N. performed experiments; M.R., H.J., C.M.W., A.A., Y.Z., H.-J.P., S.F.N., and J.B.G. analyzed data; M.R., H.J., C.M.W., A.A., H.-J.P., M.S.L., S.F.N., and J.B.G. interpreted results of experiments; M.R., H.J., C.M.W., A.A., S.F.N., and J.B.G. prepared figures; M.R., H.J., C.M.W., S.F.N., and J.B.G. drafted manuscript; M.R., H.J., M.S.L., S.F.N., and J.B.G. edited and revised manuscript; M.R., H.J., A.A., H.-J.P., M.S.L., S.F.N., and J.B.G. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Bristol Myers Squibb for the gift of pravastatin.
Footnotes
Supplemental Material for this article is available at the American Journal of Physiology-Heart and Circulatory Physiology website.
REFERENCES
- 1.Aronson D. Pharmacologic modulation of autonomic tone: implications for the diabetic patient. Diabetologia 40: 476–481, 1997. [DOI] [PubMed] [Google Scholar]
- 3.Baher AA, Uy M, Xie F, Garfinkel A, Qu Z, Weiss JN. Bidirectional ventricular tachycardia: ping pong in the His-Purkinje system. Heart Rhythm 8: 599–605, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berul CI. Electrophysiological phenotyping in genetically engineered mice. Physiol Genomics 13: 207–216, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Brown DW, Giles WH, Greenlund KJ, Valdez R, Croft JB. Impaired fasting glucose, diabetes mellitus, and cardiovascular disease risk factors are associated with prolonged QTc duration. Results from the Third National Health and Nutrition Examination Survey. J Cardiovasc Risk 8: 227–233, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Brunner M, Kodirov SA, Mitchell GF, Buckett PD, Shibata K, Folco EJ, Baker L, Salama G, Chan DP, Zhou J, Koren G. In vivo gene transfer of Kv1.5 normalizes action potential duration and shortens QT interval in mice with long QT phenotype. Am J Physiol Heart Circ Physiol 285: H194–H203, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Buxton AE, Lee KL, Hafley GE, Wyse DG, Fisher JD, Lehmann MH, Pires LA, Gold MR, Packer DL, Josephson ME, Prystowsky EN, Talajic MR. Relation of ejection fraction and inducible ventricular tachycardia to mode of death in patients with coronary artery disease: an analysis of patients enrolled in the multicenter unsustained tachycardia trial. Circulation 106: 2466–2472, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 101: 1039–1048, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaves AA, Weinstein DM, Bauer JA. Non-invasive echocardiographic studies in mice: influence of anesthetic regimen. Life Sci 69: 213–222, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Choeiri C, Hewitt K, Durkin J, Simard CJ, Renaud JM, Messier C. Longitudinal evaluation of memory performance and peripheral neuropathy in the Ins2C96Y Akita mice. Behav Brain Res 157: 31–38, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Curb JD, Rodriguez BL, Burchfiel CM, Abbott RD, Chiu D, Yano K. Sudden death, impaired glucose tolerance, and diabetes in Japanese American men. Circulation 91: 2591–2595, 1995. [DOI] [PubMed] [Google Scholar]
- 12.De Sutter J, Tavernier R, De Buyzere M, Jordaens L, De Backer G. Lipid lowering drugs and recurrences of life-threatening ventricular arrhythmias in high-risk patients. J Am Coll Cardiol 36: 766–772, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Eschenhagen T, Mende U, Diederich M, Hertle B, Memmesheimer C, Pohl A, Schmitz W, Scholz H, Steinfath M, Bohm M, Michel MC, Brodde OE, Raap A. Chronic treatment with carbachol sensitizes the myocardium to cAMP-induced arrhythmia. Circulation 93: 763–771, 1996. [DOI] [PubMed] [Google Scholar]
- 14.Fabritz L, Kirchhof P, Franz MR, Eckardt L, Monnig G, Milberg P, Breithardt G, Haverkamp W. Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol 98: 25–32, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Gehrmann J, Hammer PE, Maguire CT, Wakimoto H, Triedman JK, Berul CI. Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol 279: H733–H740, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Goldberger JJ, Subacius H, Schaechter A, Howard A, Berger R, Shalaby A, Levine J, Kadish AH. Effects of statin therapy on arrhythmic events and survival in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol 48: 1228–1233, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Kayo T, Koizumi A. Mapping of murine diabetogenic gene mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and beta cell development during the perinatal period. J Clin Invest 101: 2112–2118, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerman BM, Mittal S, Iwai S. Ventricular Tachycardia in Structually Normal Hearts. Philadephia, PA: Saunders, 2004, p. 668–682. [Google Scholar]
- 19.Maguire CT, Wakimoto H, Patel VV, Hammer PE, Gauvreau K, Berul CI. Implications of ventricular arrhythmia vulnerability during murine electrophysiology studies. Physiol Genomics 15: 84–91, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell LB, Powell JL, Gillis AM, Kehl V, Hallstrom AP. Are lipid-lowering drugs also antiarrhythmic drugs? An analysis of the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial. J Am Coll Cardiol 42: 81–87, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Osaka M, Saitoh H, Sasabe N, Atarashi H, Katoh T, Hayakawa H, Cohen RJ. Changes in autonomic activity preceding onset of nonsustained ventricular tachycardia. Ann Noninvasive Electrocardiol 1: 3–11, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Park HJ, Georgescu SP, Du C, Madias C, Aronovitz MJ, Welzig CM, Wang B, Begley U, Zhang Y, Blaustein RO, Patten RD, Karas RH, Van Tol HH, Osborne TF, Shimano H, Liao R, Link MS, Galper JB. Parasympathetic response in chick myocytes and mouse heart is controlled by SREBP. J Clin Invest 118: 259–271, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park HJ, Zhang Y, Du C, Welzig CM, Madias C, Aronovitz MJ, Georgescu SP, Naggar I, Wang B, Kim YB, Blaustein RO, Karas RH, Liao R, Mathews CE, Galper JB. Role of SREBP-1 in the development of parasympathetic dysfunction in the hearts of type 1 diabetic Akita mice. Circ Res 105: 287–294, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz PJ, Billman GE, Stone HL. Autonomic mechanisms in ventricular fibrillation induced by myocardial ischemia during exercise in dogs with healed myocardial infarction. An experimental preparation for sudden cardiac death. Circulation 69: 790–800, 1984. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation 85: I77–I91, 1992. [PubMed] [Google Scholar]
- 26.Schwartz PJ. Autonomic Modulation of Cardiac Arrhythmias. Philadelphia, PA: Saunders, 2000, p. 300–314. [Google Scholar]
- 27.Stettler C, Allemann S, Juni P, Cull CA, Holman RR, Egger M, Krahenbuhl S, Diem P. Glycemic control and macrovascular disease in types 1 and 2 diabetes mellitus: meta-analysis of randomized trials. Am Heart J 152: 27–38, 2006. [DOI] [PubMed] [Google Scholar]
- 27a.Task Force of the European Society of Cardiology, North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 93: 1043–1065, 1996. [PubMed] [Google Scholar]
- 28.Vyas AK, Guo H, Moss AJ, Olshansky B, McNitt SA, Hall WJ, Zareba W, Steinberg JS, Fischer A, Ruskin J, Andrews ML. Reduction in ventricular tachyarrhythmias with statins in the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II. J Am Coll Cardiol 47: 769–773, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15: 380–383, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welzig CM, Park HJ, Naggar J, Confalone D, Rhofiry J, Shea J, Karas RH, Estes NAM, 3rd, Galper JB. Differential effects of statins (pravastatin or simvastatin) on ventricular ectopic complexes. Am J Cardiol 105: 1112–1117, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Wendt DJ, Martins JB. Autonomic neural regulation of intact Purkinje system of dogs. Am J Physiol Heart Circ Physiol 258: H1420–H1426, 1990. [DOI] [PubMed] [Google Scholar]
- 32.Weston PJ, Gill GV. Is undetected autonomic dysfunction responsible for sudden death in type 1 diabetes mellitus? The “dead in bed” syndrome revisited. Diabet Med 16: 626–631, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XJ, Xiong ZB, Tang AL, Ma H, Ma YD, Wu JG, Dong YG. Rosiglitazone-induced myocardial protection against ischaemia-reperfusion injury is mediated via a phosphatidylinositol 3-kinase/Akt-dependent pathway. Clin Exp Pharmacol Physiol 37: 156–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.